
Genes, Gender, Environment, and Novel Functions of Estrogen Receptor Beta in the Susceptibility to Neurodevelopmental Disorders



{kind=link}
Abstract
:1. Introduction
2. Chromosomal Effects: SRY
3. Estrogen Signaling during Brain Development
4. A New Role for ERβ in Epigenetic Remodeling
5. Endocrine Disruptors and Neurodevelopment
6. Sex Hormones and Dyslexia Susceptibility
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gillies, G.E.; McArthur, S. Estrogen actions in the brain and the basis for differential action in men and women: A case for sex-specific medicines. Pharmacol. Rev. 2010, 62, 155–198. [Google Scholar] [CrossRef] [PubMed]
- Loke, H.; Harley, V.; Lee, J. Biological factors underlying sex differences in neurological disorders. Int. J. Biochem. Cell Biol. 2015, 65, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.; Launer, L.J.; Dewey, M.E.; Letenneur, L.; Ott, A.; Copeland, J.R.; Dartigues, J.F.; Kragh-Sorensen, P.; Baldereschi, M.; Brayne, C.; et al. Gender differences in the incidence of ad and vascular dementia: The eurodem studies. Eurodem incidence research group. Neurology 1999, 53, 1992–1997. [Google Scholar] [CrossRef] [PubMed]
- Baldereschi, M.; Di Carlo, A.; Rocca, W.A.; Vanni, P.; Maggi, S.; Perissinotto, E.; Grigoletto, F.; Amaducci, L.; Inzitari, D. Parkinson’s disease and parkinsonism in a longitudinal study: Two-fold higher incidence in men. Ilsa working group. Italian longitudinal study on aging. Neurology 2000, 55, 1358–1363. [Google Scholar] [CrossRef] [PubMed]
- Rocca, W.A.; Amaducci, L.A.; Schoenberg, B.S. Epidemiology of clinically diagnosed Alzheimer’s disease. Ann. Neurol. 1986, 19, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Ruitenberg, A.; Ott, A.; van Swieten, J.C.; Hofman, A.; Breteler, M.M. Incidence of dementia: Does gender make a difference? Neurobiol. Aging 2001, 22, 575–580. [Google Scholar] [CrossRef]
- Van Den Eeden, S.K.; Tanner, C.M.; Bernstein, A.L.; Fross, R.D.; Leimpeter, A.; Bloch, D.A.; Nelson, L.M. Incidence of Parkinson’s disease: Variation by age, gender, and race/ethnicity. Am. J. Epidemiol. 2003, 157, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Hallmayer, J.; Cleveland, S.; Torres, A.; Phillips, J.; Cohen, B.; Torigoe, T.; Miller, J.; Fedele, A.; Collins, J.; Smith, K.; et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 2011, 68, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Larsson, H.; Hultman, C.M.; Reichenberg, A. The familial risk of autism. JAMA 2014, 311, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Cui, J.; Shen, Y. Brain sex matters: Estrogen in cognition and Alzheimer’s disease. Mol. Cell. Endocrinol. 2014, 389, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Kuo, Y.; Cherng, C.G.; Chen, H.H.; Hsu, C.H. Ovarian hormones do not attenuate methamphetamine-induced dopaminergic neurotoxicity in mice gonadectomized at 4 weeks postpartum. Neuroendocrinology 2002, 75, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Phoenix, C.H.; Goy, R.W.; Gerall, A.A.; Young, W.C. Organizing action of prenatally administered testosterone propionate on the tissues mediating mating behavior in the female guinea pig. Endocrinology 1959, 65, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Hines, M. Neuroscience and intersex. Psychologist 2004, 17, 455–458. [Google Scholar]
- McEwen, B. Estrogen actions throughout the brain. Recent Prog. Horm. Res. 2002, 57, 357–384. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S.; Alves, S.E. Estrogen actions in the central nervous system. Endocr. Rev. 1999, 20, 279–307. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, D. Hormone-driven mechanisms in the central nervous system facilitate the analysis of mammalian behaviours. J. Endocrinol. 2005, 184, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Schaafsma, S.M.; Pfaff, D.W. Etiologies underlying sex differences in autism spectrum disorders. Front. Neuroendocrinol. 2014, 35, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Dewing, P.; Shi, T.; Horvath, S.; Vilain, E. Sexually dimorphic gene expression in mouse brain precedes gonadal differentiation. Brain Res. Mol. Brain Res. 2003, 118, 82–90. [Google Scholar] [CrossRef]
- Kopsida, E.; Stergiakouli, E.; Lynn, P.M.; Wilkinson, L.S.; Davies, W. The role of the y chromosome in brain function. Open Neuroendocrinol. J. 2009, 2, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, J.B. Gender-specific steroid metabolism in neural differentiation. Cell. Mol. Neurobiol. 1997, 17, 603–626. [Google Scholar] [CrossRef] [PubMed]
- Koopman, P.; Munsterberg, A.; Capel, B.; Vivian, N.; Lovell-Badge, R. Expression of a candidate sex-determining gene during mouse testis differentiation. Nature 1990, 348, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, A.H.; Berta, P.; Palmer, M.S.; Hawkins, J.R.; Griffiths, B.L.; Smith, M.J.; Foster, J.W.; Frischauf, A.M.; Lovell-Badge, R.; Goodfellow, P.N. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 1990, 346, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.A.; Davies, D.C. The control of sexual differentiation of the reproductive system and brain. Reproduction 2007, 133, 331–359. [Google Scholar] [CrossRef] [PubMed]
- Czech, D.P.; Lee, J.; Correia, J.; Loke, H.; Moller, E.K.; Harley, V.R. Transient neuroprotection by sry upregulation in dopamine cells following injury in males. Endocrinology 2014, 155, 2602–2612. [Google Scholar] [CrossRef] [PubMed]
- Czech, D.P.; Lee, J.; Sim, H.; Parish, C.L.; Vilain, E.; Harley, V.R. The human testis-determining factor sry localizes in midbrain dopamine neurons and regulates multiple components of catecholamine synthesis and metabolism. J. Neurochem. 2012, 122, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Dewing, P.; Chiang, C.W.; Sinchak, K.; Sim, H.; Fernagut, P.O.; Kelly, S.; Chesselet, M.F.; Micevych, P.E.; Albrecht, K.H.; Harley, V.R.; et al. Direct regulation of adult brain function by the male-specific factor sry. Curr. Biol. CB 2006, 16, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Harley, V.R. The male fight-flight response: A result of sry regulation of catecholamines? BioEssays News Rev. Mol. Cell. Dev. Biol. 2012, 34, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Spence, R.D.; Voskuhl, R.R. Neuroprotective effects of estrogens and androgens in cns inflammation and neurodegeneration. Front. Neuroendocrinol. 2012, 33, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, M.D.; Maraganore, D.M.; Bower, J.H.; McDonnell, S.K.; Peterson, B.J.; Ahlskog, J.E.; Schaid, D.J.; Rocca, W.A. Hysterectomy, menopause, and estrogen use preceding Parkinson’s disease: An exploratory case-control study. Mov. Disord. 2001, 16, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Currie, L.J.; Harrison, M.B.; Trugman, J.M.; Bennett, J.P.; Wooten, G.F. Postmenopausal estrogen use affects risk for Parkinson disease. Arch. Neurol. 2004, 61, 886–888. [Google Scholar] [CrossRef] [PubMed]
- Tsang, K.L.; Ho, S.L.; Lo, S.K. Estrogen improves motor disability in parkinsonian postmenopausal women with motor fluctuations. Neurology 2000, 54, 2292–2298. [Google Scholar] [CrossRef] [PubMed]
- Vannier, B.; Raynaud, J.P. Effect of estrogen plasma binding on sexual differentiation of the rat fetus. Mol. Cell. Endocrinol. 1975, 3, 323–337. [Google Scholar] [CrossRef]
- Swartz, S.K.; Soloff, M.S. The lack of estrogen binding by human alpha-fetoprotein. J. Clin. Endocrinol. Metab. 1974, 39, 589–591. [Google Scholar] [CrossRef] [PubMed]
- Petra, P.H.; Woodcock, K.T.; Orr, W.R.; Nguyen, D.K.; Sui, L.M. The sex steroid binding protein (SBP or SHBG) of human plasma: Identification of Tyr-57 and Met-107 in the steroid binding site. J. Steroid Biochem. Mol. Biol. 2000, 75, 139–145. [Google Scholar] [CrossRef]
- Wallen, K. Hormonal influences on sexually differentiated behavior in nonhuman primates. Front. Neuroendocrinol. 2005, 26, 7–26. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, N.; Andersson, S.; Lathe, R.; Fan, X.; Alonso-Magdalena, P.; Schwend, T.; Nalvarte, I.; Warner, M.; Gustafsson, J.A. Spatiotemporal dynamics of the expression of estrogen receptors in the postnatal mouse brain. Mol. Psychiatry 2009, 14, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Handa, R.J.; Pak, T.R.; Kudwa, A.E.; Lund, T.D.; Hinds, L. An alternate pathway for androgen regulation of brain function: Activation of estrogen receptor beta by the metabolite of dihydrotestosterone, 5alpha-androstane-3beta,17beta-diol. Horm. Behav. 2008, 53, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Cochran, R.C.; Schuetz, A.W.; Ewing, L.L. Age-related changes in conversion of 5 alpha-androstan-17 beta-ol-3-one to 5 alpha-androstane-3 alpha,17 beta-diol and 5 alpha-androstane-3 beta,17 beta-diol by rat testicular cells in vitro. J. Reprod. Fertil. 1979, 57, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Do Rego, J.L.; Seong, J.Y.; Burel, D.; Leprince, J.; Luu-The, V.; Tsutsui, K.; Tonon, M.C.; Pelletier, G.; Vaudry, H. Neurosteroid biosynthesis: Enzymatic pathways and neuroendocrine regulation by neurotransmitters and neuropeptides. Front. Neuroendocrinol. 2009, 30, 259–301. [Google Scholar] [CrossRef] [PubMed]
- Abdelgadir, S.E.; Roselli, C.E.; Choate, J.V.; Resko, J.A. Distribution of aromatase cytochrome P450 messenger ribonucleic acid in adult rhesus monkey brains. Biol. Reprod. 1997, 57, 772–777. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Biegon, A.; Kim, S.W.; Alexoff, D.L.; Jayne, M.; Carter, P.; Hubbard, B.; King, P.; Logan, J.; Muench, L.; Pareto, D.; et al. Unique distribution of aromatase in the human brain: In vivo studies with pet and [n-methyl-11c]vorozole. Synapse 2010, 64, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Boon, W.C.; Chow, J.D.; Simpson, E.R. The multiple roles of estrogens and the enzyme aromatase. Prog. Brain Res. 2010, 181, 209–232. [Google Scholar] [PubMed]
- Hojo, Y.; Higo, S.; Kawato, S.; Hatanaka, Y.; Ooishi, Y.; Murakami, G.; Ishii, H.; Komatsuzaki, Y.; Ogiue-Ikeda, M.; Mukai, H.; et al. Hippocampal synthesis of sex steroids and corticosteroids: Essential for modulation of synaptic plasticity. Front. Endocrinol. 2011, 2, 43. [Google Scholar] [CrossRef] [PubMed]
- Bakker, J.; Honda, S.; Harada, N.; Balthazart, J. The aromatase knock-out mouse provides new evidence that estradiol is required during development in the female for the expression of sociosexual behaviors in adulthood. J. Neurosci. 2002, 22, 9104–9112. [Google Scholar] [PubMed]
- Wu, M.V.; Manoli, D.S.; Fraser, E.J.; Coats, J.K.; Tollkuhn, J.; Honda, S.; Harada, N.; Shah, N.M. Estrogen masculinizes neural pathways and sex-specific behaviors. Cell 2009, 139, 61–72. [Google Scholar] [CrossRef] [PubMed]
- George, F.W.; Ojeda, S.R. Changes in aromatase activity in the rat brain during embryonic, neonatal, and infantile development. Endocrinology 1982, 111, 522–529. [Google Scholar] [CrossRef] [PubMed]
- George, F.W.; Tobleman, W.T.; Milewich, L.; Wilson, J.D. Aromatase activity in the developing rabbit brain. Endocrinology 1978, 102, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Auchus, R.J. The backdoor pathway to dihydrotestosterone. Trends Endocrinol. Metab. TEM 2004, 15, 432–438. [Google Scholar] [CrossRef]
- Fan, X.; Xu, H.; Warner, M.; Gustafsson, J.A. Erbeta in CNS: New roles in development and function. Prog. Brain Res. 2010, 181, 233–250. [Google Scholar] [PubMed]
- Lemmen, J.G.; Broekhof, J.L.; Kuiper, G.G.; Gustafsson, J.A.; van der Saag, P.T.; van der Burg, B. Expression of estrogen receptor alpha and beta during mouse embryogenesis. Mech. Dev. 1999, 81, 163–167. [Google Scholar] [CrossRef]
- Sugiyama, N.; Barros, R.P.; Warner, M.; Gustafsson, J.A. Erbeta: Recent understanding of estrogen signaling. Trends Endocrinol. Metab. TEM 2010, 21, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Walf, A.A.; Koonce, C.J.; Frye, C.A. Adult female wildtype, but not oestrogen receptor beta knockout, mice have decreased depression-like behaviour during pro-oestrus and following administration of oestradiol or diarylpropionitrile. J. Psychopharmacol. 2009, 23, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Andersson, S.; Warner, M.; Gustafsson, J.A. Estrogen receptor (er)beta knockout mice reveal a role for erbeta in migration of cortical neurons in the developing brain. Proc. Natl. Acad. Sci. USA 2003, 100, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Nah, H.Y.; Lee, Y.J.; Lee, J.W.; Park, J.H.; Kim, S.J.; Lee, J.B.; Yoon, H.S.; Kim, C.H. Expression of estrogen receptor-alpha and -beta, glucocorticoid receptor, and progesterone receptor genes in human embryonic stem cells and embryoid bodies. Mol. Cells 2004, 18, 320–325. [Google Scholar] [PubMed]
- Liu, Y.; Duong, W.; Krawczyk, C.; Bretschneider, N.; Borbely, G.; Varshney, M.; Zinser, C.; Schar, P.; Ruegg, J. Oestrogen receptor beta regulates epigenetic patterns at specific genomic loci through interaction with thymine DNA glycosylase. Epigenet. Chromatin 2016, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Cerdeno, V.; Noctor, S.C.; Kriegstein, A.R. Estradiol stimulates progenitor cell division in the ventricular and subventricular zones of the embryonic neocortex. Eur. J. Neurosci. 2006, 24, 3475–3488. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Lubahn, D.B.; Korach, K.S.; Pfaff, D.W. Behavioral effects of estrogen receptor gene disruption in male mice. Proc. Natl. Acad. Sci. USA 1997, 94, 1476–1481. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Gieske, M.C.; Trudgen, K.L.; Hudgins-Spivey, S.; Kim, B.G.; Krust, A.; Chambon, P.; Jeong, J.W.; Blalock, E.; Ko, C. Identification of estradiol/eralpha-regulated genes in the mouse pituitary. J. Endocrinol. 2011, 210, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Durbak, L.; Chan, J.; Smithies, O.; Gustafsson, J.A.; Korach, K.S.; Pfaff, D.W.; Ogawa, S. Genotype/age interactions on aggressive behavior in gonadally intact estrogen receptor beta knockout (betaerko) male mice. Horm. Behav. 2002, 41, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Chan, J.; Chester, A.E.; Gustafsson, J.A.; Korach, K.S.; Pfaff, D.W. Survival of reproductive behaviors in estrogen receptor beta gene-deficient (betaerko) male and female mice. Proc. Natl. Acad. Sci. USA 1999, 96, 12887–12892. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Day, M.; Muniz, L.C.; Bitran, D.; Arias, R.; Revilla-Sanchez, R.; Grauer, S.; Zhang, G.; Kelley, C.; Pulito, V.; et al. Activation of estrogen receptor-beta regulates hippocampal synaptic plasticity and improves memory. Nat. Neurosci. 2008, 11, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Rissman, E.F.; Heck, A.L.; Leonard, J.E.; Shupnik, M.A.; Gustafsson, J.A. Disruption of estrogen receptor beta gene impairs spatial learning in female mice. Proc. Natl. Acad. Sci. USA 2002, 99, 3996–4001. [Google Scholar] [CrossRef] [PubMed]
- Imwalle, D.B.; Gustafsson, J.A.; Rissman, E.F. Lack of functional estrogen receptor beta influences anxiety behavior and serotonin content in female mice. Physiol. Behav. 2005, 84, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, G.G.; Lemmen, J.G.; Carlsson, B.; Corton, J.C.; Safe, S.H.; van der Saag, P.T.; van der Burg, B.; Gustafsson, J.A. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 1998, 139, 4252–4263. [Google Scholar] [PubMed]
- Donner, N.; Handa, R.J. Estrogen receptor beta regulates the expression of tryptophan-hydroxylase 2 mrna within serotonergic neurons of the rat dorsal raphe nuclei. Neuroscience 2009, 163, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Gundlah, C.; Alves, S.E.; Clark, J.A.; Pai, L.Y.; Schaeffer, J.M.; Rohrer, S.P. Estrogen receptor-beta regulates tryptophan hydroxylase-1 expression in the murine midbrain raphe. Biol. Psychiatry 2005, 57, 938–942. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Barros, R.P.; Sugiyama, N.; Krishnan, V.; Yaden, B.C.; Kim, H.J.; Warner, M.; Gustafsson, J.A. Involvement of estrogen receptor beta in maintenance of serotonergic neurons of the dorsal raphe. Mol. Psychiatry 2013, 18, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Shughrue, P.J.; Merchenthaler, I. Distribution of estrogen receptor beta immunoreactivity in the rat central nervous system. J. Comp. Neurol. 2001, 436, 64–81. [Google Scholar] [CrossRef] [PubMed]
- Dluzen, D.; Horstink, M. Estrogen as neuroprotectant of nigrostriatal dopaminergic system: Laboratory and clinical studies. Endocrine 2003, 21, 67–75. [Google Scholar] [CrossRef]
- Jourdain, S.; Morissette, M.; Morin, N.; Di Paolo, T. Oestrogens prevent loss of dopamine transporter (DAT) and vesicular monoamine transporter (VMAT2) in substantia nigra of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mice. J. Neuroendocrinol. 2005, 17, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Sawada, H.; Ibi, M.; Kihara, T.; Honda, K.; Nakamizo, T.; Kanki, R.; Nakanishi, M.; Sakka, N.; Akaike, A.; Shimohama, S. Estradiol protects dopaminergic neurons in a MPP+Parkinson’s disease model. Neuropharmacology 2002, 42, 1056–1064. [Google Scholar] [CrossRef]
- Shughrue, P.J. Estrogen attenuates the MPTP-induced loss of dopamine neurons from the mouse snc despite a lack of estrogen receptors (eralpha and erbeta). Exp. Neurol. 2004, 190, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Kruijver, F.P.; Balesar, R.; Espila, A.M.; Unmehopa, U.A.; Swaab, D.F. Estrogen receptor-alpha distribution in the human hypothalamus in relation to sex and endocrine status. J. Comp. Neurol. 2002, 454, 115–139. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.F.; Tan, X.J.; Dai, Y.B.; Krishnan, V.; Warner, M.; Gustafsson, J.A. Targeting estrogen receptor beta in microglia and T cells to treat experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2013, 110, 3543–3548. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Dent, S.Y. Chromatin modifiers and remodellers: Regulators of cellular differentiation. Nat. Rev. Genet. 2014, 15, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Martens, J.H.; Rao, N.A.; Stunnenberg, H.G. Genome-wide interplay of nuclear receptors with the epigenome. Biochim. Biophys. Acta 2011, 1812, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Patra, S.K.; Patra, A.; Rizzi, F.; Ghosh, T.C.; Bettuzzi, S. Demethylation of (cytosine-5-c-methyl) DNA and regulation of transcription in the epigenetic pathways of cancer development. Cancer Metastasis Rev. 2008, 27, 315–334. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Su, Y.; Zhong, C.; Ming, G.L.; Song, H. Emerging roles of tet proteins and 5-hydroxymethylcytosines in active DNA demethylation and beyond. Cell Cycle 2011, 10, 2662–2668. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, A.L.; Schar, P. DNA glycosylases: In DNA repair and beyond. Chromosoma 2012, 121, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Maiti, A.; Drohat, A.C. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: Potential implications for active demethylation of cpg sites. J. Biol. Chem. 2011, 286, 35334–35338. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Zhang, Y. 5-hydroxymethylcytosine: Generation, fate, and genomic distribution. Curr. Opin. Cell Biol. 2013, 25, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Xia, Y.; Wang, J.; Luo, H.; Gao, Z.; Han, X.; Zhang, J.; Huang, X.; Yao, Y.; Lu, H.; et al. Integrated detection of both 5-mC and 5-hmC by high-throughput tag sequencing technology highlights methylation reprogramming of bivalent genes during cellular differentiation. Epigenetics Off. J. DNA Methylation Soc. 2013, 8, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Coskun, V.; Tsoa, R.; Sun, Y.E. Epigenetic regulation of stem cells differentiating along the neural lineage. Curr. Opin. Neurobiol. 2012, 22, 762–767. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Saitou, M.; Kagiwada, S.; Kurimoto, K. Epigenetic reprogramming in mouse pre-implantation development and primordial germ cells. Development 2012, 139, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, A.; Ivanek, R.; Murr, R.; Gaidatzis, D.; Burger, L.; Schubeler, D. Transcription factor occupancy can mediate active turnover of DNA methylation at regulatory regions. PLoS Genet. 2013, 9, e1003994. [Google Scholar] [CrossRef] [PubMed]
- Lienert, F.; Wirbelauer, C.; Som, I.; Dean, A.; Mohn, F.; Schubeler, D. Identification of genetic elements that autonomously determine DNA methylation states. Nat. Genet. 2011, 43, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Stadler, M.B.; Murr, R.; Burger, L.; Ivanek, R.; Lienert, F.; Scholer, A.; van Nimwegen, E.; Wirbelauer, C.; Oakeley, E.J.; Gaidatzis, D.; et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011, 480, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Kangaspeska, S.; Stride, B.; Metivier, R.; Polycarpou-Schwarz, M.; Ibberson, D.; Carmouche, R.P.; Benes, V.; Gannon, F.; Reid, G. Transient cyclical methylation of promoter DNA. Nature 2008, 452, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Kondo, T.; Takada, I.; Youn, M.Y.; Yamamoto, Y.; Takahashi, S.; Matsumoto, T.; Fujiyama, S.; Shirode, Y.; Yamaoka, I.; et al. DNA demethylation in hormone-induced transcriptional derepression. Nature 2009, 461, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.; Laflamme, L.; Gaudreau, L. Estrogen receptor alpha can selectively repress dioxin receptor-mediated gene expression by targeting DNA methylation. Nucleic Acids Res. 2013, 41, 8094–8106. [Google Scholar] [CrossRef] [PubMed]
- Metivier, R.; Gallais, R.; Tiffoche, C.; Le Peron, C.; Jurkowska, R.Z.; Carmouche, R.P.; Ibberson, D.; Barath, P.; Demay, F.; Reid, G.; et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature 2008, 452, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Ruegg, J.; Cai, W.; Karimi, M.; Kiss, N.B.; Swedenborg, E.; Larsson, C.; Ekstrom, T.J.; Pongratz, I. Epigenetic regulation of glucose transporter 4 by estrogen receptor beta. Mol. Endocrinol. 2011, 25, 2017–2028. [Google Scholar] [CrossRef] [PubMed]
- Thomassin, H.; Flavin, M.; Espinas, M.L.; Grange, T. Glucocorticoid-induced DNA demethylation and gene memory during development. EMBO J. 2001, 20, 1974–1983. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Lucey, M.J.; Phoenix, F.; Lopez-Garcia, J.; Hart, S.M.; Losson, R.; Buluwela, L.; Coombes, R.C.; Chambon, P.; Schar, P.; et al. T:G mismatch-specific thymine-DNA glycosylase potentiates transcription of estrogen-regulated genes through direct interaction with estrogen receptor alpha. J. Biol. Chem. 2003, 278, 38586–38592. [Google Scholar] [CrossRef] [PubMed]
- Dey, P.; Barros, R.P.; Warner, M.; Strom, A.; Gustafsson, J.A. Insight into the mechanisms of action of estrogen receptor beta in the breast, prostate, colon, and CNS. J. Mol. Endocrinol. 2013, 51, T61–T74. [Google Scholar] [CrossRef] [PubMed]
- Calafat, A.M.; Ye, X.; Wong, L.Y.; Reidy, J.A.; Needham, L.L. Exposure of the U.S. Population to bisphenol a and 4-tertiary-octylphenol: 2003–2004. Environ. Health Perspect. 2008, 116, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Dodson, R.E.; Nishioka, M.; Standley, L.J.; Perovich, L.J.; Brody, J.G.; Rudel, R.A. Endocrine disruptors and asthma-associated chemicals in consumer products. Environ. Health Perspect. 2012, 120, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Bergman, A.; Heindel, J.J.; Kasten, T.; Kidd, K.A.; Jobling, S.; Neira, M.; Zoeller, R.T.; Becher, G.; Bjerregaard, P.; Bornman, R.; et al. The impact of endocrine disruption: A consensus statement on the state of the science. Environ. Health Perspect. 2013, 121, A104–A106. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Genuis, S.J.; Frye, R.E. Environmental toxicants and autism spectrum disorders: A systematic review. Transl. Psychiatry 2014, 4, e360. [Google Scholar] [CrossRef] [PubMed]
- Chevrier, J.; Gunier, R.B.; Bradman, A.; Holland, N.T.; Calafat, A.M.; Eskenazi, B.; Harley, K.G. Maternal urinary bisphenol a during pregnancy and maternal and neonatal thyroid function in the chamacos study. Environ. Health Perspect. 2013, 121, 138–144. [Google Scholar] [PubMed]
- Xu, X.B.; He, Y.; Song, C.; Ke, X.; Fan, S.J.; Peng, W.J.; Tan, R.; Kawata, M.; Matsuda, K.; Pan, B.X.; et al. Bisphenol a regulates the estrogen receptor alpha signaling in developing hippocampus of male rats through estrogen receptor. Hippocampus 2014, 24, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Furr, J.R.; Lambright, C.S.; Wilson, V.S.; Foster, P.M.; Gray, L.E., Jr. A short-term in vivo screen using fetal testosterone production, a key event in the phthalate adverse outcome pathway, to predict disruption of sexual differentiation. Toxicol. Sci. Off. J. Soc. Toxicol. 2014, 140, 403–424. [Google Scholar] [CrossRef] [PubMed]
- Betz, A.; Jayatilaka, S.; Joshi, J.; Ramanan, S.; Debartolo, D.; Pylypiw, H.; Franke, E. Chronic exposure to benzyl butyl phthalate (BBP) alters social interaction and fear conditioning in male adult rats: Alterations in amygdalar MeCP2, ERK1/2 and eralpha. Neuro Endocrinol. Lett. 2013, 34, 347–358. [Google Scholar] [PubMed]
- Ji, K.; Hong, S.; Kho, Y.; Choi, K. Effects of bisphenol s exposure on endocrine functions and reproduction of zebrafish. Environ. Sci. Technol. 2013, 47, 8793–8800. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Jiang, L.; Chen, L.; Chen, H.S.; Li, X. Neurotoxicity of dibutyl phthalate in brain development following perinatal exposure: A study in rats. Environ. Toxicol. Pharmacol. 2013, 36, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Masutomi, N.; Shibutani, M.; Takagi, H.; Uneyama, C.; Takahashi, N.; Hirose, M. Impact of dietary exposure to methoxychlor, genistein, or diisononyl phthalate during the perinatal period on the development of the rat endocrine/reproductive systems in later life. Toxicology 2003, 192, 149–170. [Google Scholar] [CrossRef]
- Patisaul, H.B.; Polston, E.K. Influence of endocrine active compounds on the developing rodent brain. Brain Res. Rev. 2008, 57, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, L.N.; Colborn, T.; Hayes, T.B.; Heindel, J.J.; Jacobs, D.R., Jr.; Lee, D.H.; Shioda, T.; Soto, A.M.; vom Saal, F.S.; Welshons, W.V.; et al. Hormones and endocrine-disrupting chemicals: Low-dose effects and nonmonotonic dose responses. Endocr. Rev. 2012, 33, 378–455. [Google Scholar] [CrossRef] [PubMed]
- Ejaredar, M.; Lee, Y.; Roberts, D.J.; Sauve, R.; Dewey, D. Bisphenol a exposure and children’s behavior: A systematic review. J. Expo. Sci. Environ. Epidemiol. 2017, 27, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Joyner, L.; Mickens, J.A.; Leyrer, S.M.; Patisaul, H.B. Sex-specific ESR2 mRNA expression in the rat hypothalamus and amygdala is altered by neonatal bisphenol a exposure. Reproduction 2014, 147, 537–554. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Rebuli, M.E.; Rogers, J.; Todd, K.L.; Leyrer, S.M.; Ferguson, S.A.; Patisaul, H.B. Prenatal bisphenol a exposure alters sex-specific estrogen receptor expression in the neonatal rat hypothalamus and amygdala. Toxicol. Sci. Off. J. Soc. Toxicol. 2013, 133, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhou, L.; Bai, Y.; Zhou, R.; Chen, L. Sex differences in the adult HPA axis and affective behaviors are altered by perinatal exposure to a low dose of bisphenol A. Brain Res. 2014, 1571, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Gioiosa, L.; Parmigiani, S.; Vom Saal, F.S.; Palanza, P. The effects of bisphenol a on emotional behavior depend upon the timing of exposure, age and gender in mice. Horm. Behav. 2013, 63, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Jasarevic, E.; Williams, S.A.; Vandas, G.M.; Ellersieck, M.R.; Liao, C.; Kannan, K.; Roberts, R.M.; Geary, D.C.; Rosenfeld, C.S. Sex and dose-dependent effects of developmental exposure to bisphenol a on anxiety and spatial learning in deer mice (peromyscus maniculatus bairdii) offspring. Horm. Behav. 2013, 63, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, J.T.; Rissman, E.F.; Connelly, J.J. The role of bisphenol a in shaping the brain, epigenome and behavior. Horm. Behav. 2011, 59, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Dong, F.; Yang, Y.; Wang, Y.; Wang, R.; Shen, X. Sex-specific effects of long-term exposure to bisphenol-A on anxiety- and depression-like behaviors in adult mice. Chemosphere 2015, 120, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.M.; Kalkbrenner, A.E.; Calafat, A.M.; Yolton, K.; Ye, X.; Dietrich, K.N.; Lanphear, B.P. Impact of early-life bisphenol a exposure on behavior and executive function in children. Pediatrics 2011, 128, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.F.; Kobrosly, R.W.; Barrett, E.S.; Thurston, S.W.; Calafat, A.M.; Weiss, B.; Stahlhut, R.; Yolton, K.; Swan, S.H. Prenatal bisphenol a exposure and maternally reported behavior in boys and girls. Neurotoxicology 2014, 45, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Harley, K.G.; Gunier, R.B.; Kogut, K.; Johnson, C.; Bradman, A.; Calafat, A.M.; Eskenazi, B. Prenatal and early childhood bisphenol a concentrations and behavior in school-aged children. Environ. Res. 2013, 126, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Kitraki, E.; Nalvarte, I.; Alavian-Ghavanini, A.; Ruegg, J. Developmental exposure to bisphenol A alters expression and DNA methylation of Fkbp5, an important regulator of the stress response. Mol. Cell. Endocrinol. 2015, 417, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Peterson, R.L.; Pennington, B.F. Developmental dyslexia. Lancet 2012, 379, 1997–2007. [Google Scholar] [CrossRef]
- Schulte-Korne, G. The prevention, diagnosis, and treatment of dyslexia. Deutsch. Arzteblatt Int. 2010, 107, 718–726. [Google Scholar]
- Lambe, E.K. Dyslexia, gender, and brain imaging. Neuropsychologia 1999, 37, 521–536. [Google Scholar] [CrossRef]
- Fisher, S.E.; DeFries, J.C. Developmental dyslexia: Genetic dissection of a complex cognitive trait. Nat. Rev. Neurosci. 2002, 3, 767–780. [Google Scholar] [CrossRef] [PubMed]
- McGrath, L.M.; Smith, S.D.; Pennington, B.F. Breakthroughs in the search for dyslexia candidate genes. Trends Mol. Med. 2006, 12, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Anthoni, H.; Sucheston, L.E.; Lewis, B.A.; Tapia-Paez, I.; Fan, X.; Zucchelli, M.; Taipale, M.; Stein, C.M.; Hokkanen, M.E.; Castren, E.; et al. The aromatase gene CYP19A1: Several genetic and functional lines of evidence supporting a role in reading, speech and language. Behav. Genet. 2012, 42, 509–527. [Google Scholar] [CrossRef] [PubMed]
- Ylisaukko-Oja, T.; Peyrard-Janvid, M.; Lindgren, C.M.; Rehnstrom, K.; Vanhala, R.; Peltonen, L.; Jarvela, I.; Kere, J. Family-based association study of DYX1C1 variants in autism. Eur. J. Hum. Genet. EJHG 2005, 13, 127–130. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Geschwind, N.; Galaburda, A.M. Cerebral lateralization. Biological mechanisms, associations, and pathology: I. A hypothesis and a program for research. Arch. Neurol. 1985, 42, 428–459. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.A.; Jordan, C.L.; Breedlove, S.M. Sexual differentiation of the vertebrate nervous system. Nat. Neurosci. 2004, 7, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
- Waddell, J.; McCarthy, M.M. Sexual differentiation of the brain and ADHD: What is a sex difference in prevalence telling us? Curr. Top. Behav. Neurosci. 2012, 9, 341–360. [Google Scholar] [PubMed]
- Seidman, L.J.; Valera, E.M.; Makris, N. Structural brain imaging of attention-deficit/hyperactivity disorder. Biol. Psychiatry 2005, 57, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Thiebaut de Schotten, M.; Altarelli, I.; Dubois, J.; Ramus, F. Altered hemispheric lateralization of white matter pathways in developmental dyslexia: Evidence from spherical deconvolution tractography. Cortex J. Devoted Study Nerv. Syst. Behav. 2016, 76, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.S.; Katzir, T.; Liu, T.; Corriveau, K.; Barzillai, M.; Apse, K.A.; Bodell, A.; Hackney, D.; Alsop, D.; Wong, S.T.; et al. A structural basis for reading fluency: White matter defects in a genetic brain malformation. Neurology 2007, 69, 2146–2154. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.S.; Ly, J.; Appignani, B.; Bodell, A.; Apse, K.A.; Ravenscroft, R.S.; Sheen, V.L.; Doherty, M.J.; Hackney, D.B.; O’Connor, M.; et al. Reading impairment in the neuronal migration disorder of periventricular nodular heterotopia. Neurology 2005, 64, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Galaburda, A.M.; Sherman, G.F.; Rosen, G.D.; Aboitiz, F.; Geschwind, N. Developmental dyslexia: Four consecutive patients with cortical anomalies. Ann. Neurol. 1985, 18, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, P.; Kaufmann, W.E.; Galaburda, A.M. Developmental dyslexia in women: Neuropathological findings in three patients. Ann. Neurol. 1990, 28, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Paramasivam, M.; Thomas, A.; Bai, J.; Kaminen-Ahola, N.; Kere, J.; Voskuil, J.; Rosen, G.D.; Galaburda, A.M.; Loturco, J.J. Dyx1c1 functions in neuronal migration in developing neocortex. Neuroscience 2006, 143, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Paez, I.; Tammimies, K.; Massinen, S.; Roy, A.L.; Kere, J. The complex of TFII-I, PARP1, and SFPQ proteins regulates the DYX1C1 gene implicated in neuronal migration and dyslexia. FASEB J. 2008, 22, 3001–3009. [Google Scholar] [CrossRef] [PubMed]
- Rosen, G.D.; Bai, J.; Wang, Y.; Fiondella, C.G.; Threlkeld, S.W.; LoTurco, J.J.; Galaburda, A.M. Disruption of neuronal migration by rnai of DYX1C1 results in neocortical and hippocampal malformations. Cereb. Cortex 2007, 17, 2562–2572. [Google Scholar] [CrossRef] [PubMed]
- Threlkeld, S.W.; McClure, M.M.; Bai, J.; Wang, Y.; LoTurco, J.J.; Rosen, G.D.; Fitch, R.H. Developmental disruptions and behavioral impairments in rats following in utero rnai of DYX1C1. Brain Res. Bull. 2007, 71, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Tarkar, A.; Loges, N.T.; Slagle, C.E.; Francis, R.; Dougherty, G.W.; Tamayo, J.V.; Shook, B.; Cantino, M.; Schwartz, D.; Jahnke, C.; et al. DYX1C1 is required for axonemal dynein assembly and ciliary motility. Nat. Genet. 2013, 45, 995–1003. [Google Scholar] [CrossRef] [PubMed]
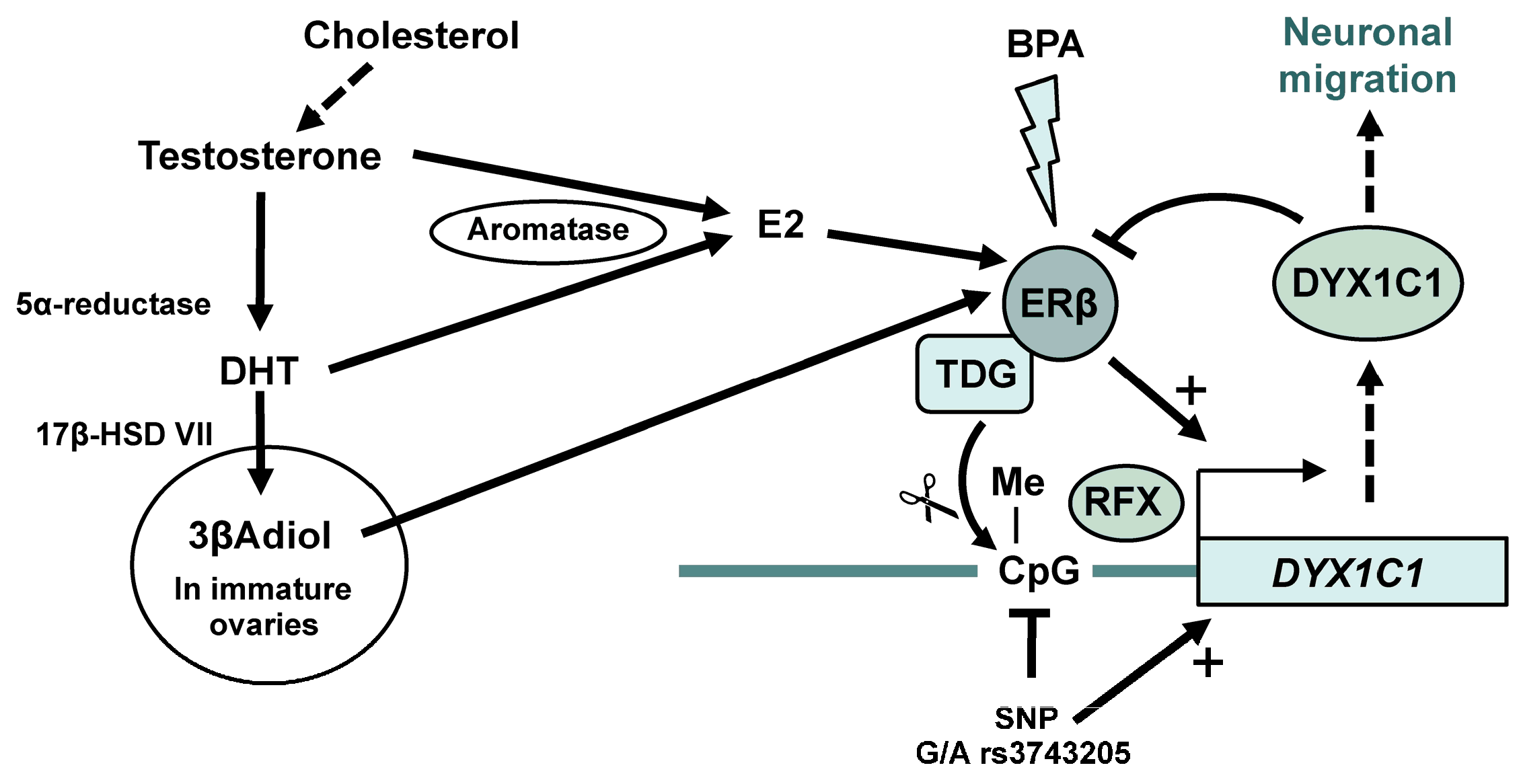
- Tammimies, K.; Tapia-Paez, I.; Ruegg, J.; Rosin, G.; Kere, J.; Gustafsson, J.A.; Nalvarte, I. The rs3743205 snp is important for the regulation of the dyslexia candidate gene DYX1C1 by estrogen receptor beta and DNA methylation. Mol. Endocrinol. 2012, 26, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Massinen, S.; Tammimies, K.; Tapia-Paez, I.; Matsson, H.; Hokkanen, M.E.; Soderberg, O.; Landegren, U.; Castren, E.; Gustafsson, J.A.; Treuter, E.; et al. Functional interaction of DYX1C1 with estrogen receptors suggests involvement of hormonal pathways in dyslexia. Hum. Mol. Genet. 2009, 18, 2802–2812. [Google Scholar] [CrossRef] [PubMed]
- Tammimies, K.; Bieder, A.; Lauter, G.; Sugiaman-Trapman, D.; Torchet, R.; Hokkanen, M.E.; Burghoorn, J.; Castren, E.; Kere, J.; Tapia-Paez, I.; et al. Ciliary dyslexia candidate genes DYX1C1 and DCDC2 are regulated by regulatory factor (RFX) transcription factors through x-box promoter motifs. FASEB J. 2016, 30, 3578–3587. [Google Scholar] [CrossRef] [PubMed]
- Choksi, S.P.; Lauter, G.; Swoboda, P.; Roy, S. Switching on cilia: Transcriptional networks regulating ciliogenesis. Development 2014, 141, 1427–1441. [Google Scholar] [CrossRef] [PubMed]
- Spruijt, C.G.; Gnerlich, F.; Smits, A.H.; Pfaffeneder, T.; Jansen, P.W.; Bauer, C.; Munzel, M.; Wagner, M.; Muller, M.; Khan, F.; et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 2013, 152, 1146–1159. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Sun, Y.; Gao, F.; Wu, X.; Tang, J.; Yin, H.; Luo, Y.; Richardson, B.; Lu, Q. Epigenetics and sle: Rfx1 downregulation causes cd11a and cd70 overexpression by altering epigenetic modifications in lupus CD4+ T cells. J. Autoimmun. 2010, 35, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Bakker, S.C.; van der Meulen, E.M.; Buitelaar, J.K.; Sandkuijl, L.A.; Pauls, D.L.; Monsuur, A.J.; van ‘t Slot, R.; Minderaa, R.B.; Gunning, W.B.; Pearson, P.L.; et al. A whole-genome scan in 164 dutch sib pairs with attention-deficit/hyperactivity disorder: Suggestive evidence for linkage on chromosomes 7p and 15q. Am. J. Hum. Genet. 2003, 72, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Jesmin, S.; Togashi, H.; Sakuma, I.; Mowa, C.N.; Ueno, K.; Yamaguchi, T.; Yoshioka, M.; Kitabatake, A. Gonadal hormones and frontocortical expression of vascular endothelial growth factor in male stroke-prone, spontaneously hypertensive rats, a model for attention-deficit/hyperactivity disorder. Endocrinology 2004, 145, 4330–4343. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varshney, M.; Nalvarte, I. Genes, Gender, Environment, and Novel Functions of Estrogen Receptor Beta in the Susceptibility to Neurodevelopmental Disorders. Brain Sci. 2017, 7, 24. https://doi.org/10.3390/brainsci7030024
Varshney M, Nalvarte I. Genes, Gender, Environment, and Novel Functions of Estrogen Receptor Beta in the Susceptibility to Neurodevelopmental Disorders. Brain Sciences. 2017; 7(3):24. https://doi.org/10.3390/brainsci7030024
Chicago/Turabian StyleVarshney, Mukesh, and Ivan Nalvarte. 2017. "Genes, Gender, Environment, and Novel Functions of Estrogen Receptor Beta in the Susceptibility to Neurodevelopmental Disorders" Brain Sciences 7, no. 3: 24. https://doi.org/10.3390/brainsci7030024
APA StyleVarshney, M., & Nalvarte, I. (2017). Genes, Gender, Environment, and Novel Functions of Estrogen Receptor Beta in the Susceptibility to Neurodevelopmental Disorders. Brain Sciences, 7(3), 24. https://doi.org/10.3390/brainsci7030024