
Enduring, Sexually Dimorphic Impact of In Utero Exposure to Elevated Levels of Glucocorticoids on Midbrain Dopaminergic Populations

Abstract
:1. Introduction
2. Midbrain Dopaminergic Systems
2.1. Function
2.2. Malfunction, Neurobiological Programming and Sex Dimorphisms
2.2.1. Malfunction
2.2.2. Neurobiological Programming
2.2.3. Sex Dimorphisms
3. Glucocorticoids and Programming of Midbrain Dopaminergic Systems
3.1. Antenatal GC Treatment (AGT)
3.2. Macrostructure
3.3. Microstructure
3.3.1. Neurons
3.3.2. Astrocytes
3.3.3. Behaviour
3.4. Why Should the Influences of AGT Be Sexually Dimorphic?
4. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
References
- McEwen, B.S.; Bowles, N.P.; Gray, J.D.; Hill, M.N.; Hunter, R.G.; Karatsoreos, I.N.; Nasca, C. Mechanisms of stress in the brain. Nat. Neurosci. 2015, 18, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Heim, C.; Binder, E.B. Current research trends in early life stress and depression: Review of human studies on sensitive periods, gene-environment interactions, and epigenetics. Exp. Neurol. 2012, 233, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Lupien, S.J.; McEwen, B.S.; Gunnar, M.R.; Heim, C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat. Rev. Neurosci. 2009, 10, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Cahill, L. Why sex matters for neuroscience. Nat. Rev. Neurosci. 2006, 7, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, K.P.; Mazure, C.M.; Staley, J.K. Evolving knowledge of sex differences in brain structure, function, and chemistry. Biol. Psychiatry 2007, 62, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J. The wellcome foundation lecture, 1994. The fetal origins of adult disease. Proc. Biol. Sci. 1995, 262, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Newman, L.; Judd, F.; Olsson, C.A.; Castle, D.; Bousman, C.; Sheehan, P.; Pantelis, C.; Craig, J.M.; Komiti, A.; Everall, I. Early origins of mental disorder—Risk factors in the perinatal and infant period. BMC Psychiatry 2016, 16, 270. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Hanson, M.A.; Mitchell, M.D. Developmental origins of health and disease: Reducing the burden of chronic disease in the next generation. Genome Med. 2010, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Charil, A.; Laplante, D.P.; Vaillancourt, C.; King, S. Prenatal stress and brain development. Brain Res. Rev. 2010, 65, 56–79. [Google Scholar] [CrossRef] [PubMed]
- French, N.P.; Hagan, R.; Evans, S.F.; Mullan, A.; Newnham, J.P. Repeated antenatal corticosteroids: Effects on cerebral palsy and childhood behavior. Am. J. Obstet. Gynecol. 2004, 190, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Baud, O.; Sola, A. Corticosteroids in perinatal medicine: How to improve outcomes without affecting the developing brain? Semin. Fetal. Neonatal. Med. 2007, 12, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Petropoulos, S.; Matthews, S.G. Fetal programming of hypothalamic-pituitary-adrenal (hpa) axis function and behavior by synthetic glucocorticoids. Brain Res. Rev. 2008, 57, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.V.; Goldman-Rakic, P.S. Modulation of memory fields by dopamine d1 receptors in prefrontal cortex. Nature 1995, 376, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Robbins, T.W.; Everitt, B.J. Neurobehavioural mechanisms of reward and motivation. Curr. Opin. Neurobiol. 1996, 6, 228–236. [Google Scholar] [CrossRef]
- Bjorklund, A.; Dunnett, S.B. Dopamine neuron systems in the brain: An update. Trends Neurosci. 2007, 30, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.Y.; Bloom, F.E. Central catecholamine neuron systems: Anatomy and physiology of the dopamine systems. Annu. Rev. Neurosci. 1978, 1, 129–169. [Google Scholar] [CrossRef] [PubMed]
- Everitt, B.J.; Belin, D.; Economidou, D.; Pelloux, Y.; Dalley, J.W.; Robbins, T.W. Review. Neural mechanisms underlying the vulnerability to develop compulsive drug-seeking habits and addiction. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 3125–3135. [Google Scholar] [CrossRef] [PubMed]
- Thierry, A.M.; Tassin, J.P.; Blanc, G.; Glowinski, J. Selective activation of mesocortical da system by stress. Nature 1976, 263, 242–244. [Google Scholar] [CrossRef] [PubMed]
- Roth, R.H.; Tam, S.Y.; Ida, Y.; Yang, J.X.; Deutch, A.Y. Stress and the mesocorticolimbic dopamine systems. Ann. N. Y. Acad. Sci. 1988, 537, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Abercrombie, E.D.; Keefe, K.A.; DiFrischia, D.S.; Zigmond, M.J. Differential effect of stress on in vivo dopamine release in striatum, nucleus accumbens, and medial frontal cortex. J. Neurochem. 1989, 52, 1655–1658. [Google Scholar] [CrossRef] [PubMed]
- Trainor, B.C. Stress responses and the mesolimbic dopamine system: Social contexts and sex differences. Horm. Behav. 2011, 60, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Guarraci, F.A.; Frohardt, R.J.; Falls, W.A.; Kapp, B.S. The effects of intra-amygdaloid infusions of a d2 dopamine receptor antagonist on pavlovian fear conditioning. Behav. Neurosci. 2000, 114, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Sapolsky, R. Any kind of mother in a storm. Nat. Neurosci. 2009, 12, 1355–1356. [Google Scholar] [CrossRef] [PubMed]
- Brischoux, F.; Chakraborty, S.; Brierley, D.I.; Ungless, M.A. Phasic excitation of dopamine neurons in ventral vta by noxious stimuli. Proc. Natl. Acad. Sci. USA 2009, 106, 4894–4899. [Google Scholar] [CrossRef] [PubMed]
- Bromberg-Martin, E.S.; Matsumoto, M.; Hikosaka, O. Dopamine in motivational control: Rewarding, aversive, and alerting. Neuron 2010, 68, 815–834. [Google Scholar] [CrossRef] [PubMed]
- Roeper, J. Dissecting the diversity of midbrain dopamine neurons. Trends Neurosci. 2013, 36, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Arnsten, A.F. Stress weakens prefrontal networks: Molecular insults to higher cognition. Nat. Neurosci. 2015, 18, 1376–1385. [Google Scholar] [CrossRef] [PubMed]
- De Kloet, E.R.; Joels, M.; Holsboer, F. Stress and the brain: From adaptation to disease. Nat. Rev. Neurosci. 2005, 6, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Han, M.H.; Graham, D.L.; Berton, O.; Renthal, W.; Russo, S.J.; Laplant, Q.; Graham, A.; Lutter, M.; Lagace, D.C.; et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 2007, 131, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Han, M.H.; Mazei-Robison, M.; Iniguez, S.D.; Ables, J.L.; Vialou, V.; Berton, O.; Ghose, S.; Covington, H.E., 3rd; Wiley, M.D.; et al. Akt signaling within the ventral tegmental area regulates cellular and behavioral responses to stressful stimuli. Biol. Psychiatry 2008, 64, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Levitt, P. Schizophrenia as a disorder of neurodevelopment. Annu. Rev. Neurosci. 2002, 25, 409–432. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, B.W.; Nemeroff, C.B. The role of dopamine in the pathophysiology of depression. Arch. Gen. Psychiatry 2007, 64, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Melichar, J.K.; Daglish, M.R.; Nutt, D.J. Addiction and withdrawal--current views. Curr. Opin. Pharmacol. 2001, 1, 84–90. [Google Scholar] [CrossRef]
- Goeders, N.E. The impact of stress on addiction. Eur. Neuropsychopharmacol. 2003, 13, 435–441. [Google Scholar] [CrossRef]
- Biederman, J.; Faraone, S.V.; Monuteaux, M.C. Differential effect of environmental adversity by gender: Rutter’s index of adversity in a group of boys and girls with and without adhd. Am. J. Psychiatry 2002, 159, 1556–1562. [Google Scholar] [CrossRef] [PubMed]
- Solanto, M.V. Dopamine dysfunction in ad/hd: Integrating clinical and basic neuroscience research. Behav. Brain Res. 2002, 130, 65–71. [Google Scholar] [CrossRef]
- Moore, H.; Rose, H.J.; Grace, A.A. Chronic cold stress reduces the spontaneous activity of ventral tegmental dopamine neurons. Neuropsychopharmacology 2001, 24, 410–419. [Google Scholar] [CrossRef]
- Mizoguchi, K.; Yuzurihara, M.; Ishige, A.; Sasaki, H.; Chui, D.H.; Tabira, T. Chronic stress induces impairment of spatial working memory because of prefrontal dopaminergic dysfunction. J. Neurosci. 2000, 20, 1568–1574. [Google Scholar] [PubMed]
- Mizoguchi, K.; Ikeda, R.; Shoji, H.; Tanaka, Y.; Tabira, T. Suppression of glucocorticoid secretion induces a behaviorally depressive state in rotarod performance in rat. Pharmacol. Biochem. Behav. 2008, 90, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Brake, W.G.; Sullivan, R.M.; Gratton, A. Perinatal distress leads to lateralized medial prefrontal cortical dopamine hypofunction in adult rats. J. Neurosci. 2000, 20, 5538–5543. [Google Scholar] [PubMed]
- Hemmerle, A.M.; Herman, J.P.; Seroogy, K.B. Stress, depression and parkinson’s disease. Exp. Neurol. 2012, 233, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Pienaar, I.S.; Kellaway, L.A.; Russell, V.A.; Smith, A.D.; Stein, D.J.; Zigmond, M.J.; Daniels, W.M. Maternal separation exaggerates the toxic effects of 6-hydroxydopamine in rats: Implications for neurodegenerative disorders. Stress 2008, 11, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Talge, N.M.; Neal, C.; Glover, V. Antenatal maternal stress and long-term effects on child neurodevelopment: How and why? J. Child. Psychol. Psychiatry 2007, 48, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Van den Bergh, B.R.; Mulder, E.J.; Mennes, M.; Glover, V. Antenatal maternal anxiety and stress and the neurobehavioural development of the fetus and child: Links and possible mechanisms. A review. Neurosci. Biobehav. Rev. 2005, 29, 237–258. [Google Scholar] [CrossRef] [PubMed]
- Sandman, C.A.; Davis, E.P.; Buss, C.; Glynn, L.M. Exposure to prenatal psychobiological stress exerts programming influences on the mother and her fetus. Neuroendocrinology 2012, 95, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Boksa, P. Animal models of obstetric complications in relation to schizophrenia. Brain Res. Brain Res. Rev. 2004, 45, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Susser, E.; Hoek, H.W.; Brown, A. Neurodevelopmental disorders after prenatal famine: The story of the dutch famine study. Am. J. Epidemiol. 1998, 147, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Khashan, A.S.; Abel, K.M.; McNamee, R.; Pedersen, M.G.; Webb, R.T.; Baker, P.N.; Kenny, L.C.; Mortensen, P.B. Higher risk of offspring schizophrenia following antenatal maternal exposure to severe adverse life events. Arch. Gen. Psychiatry 2008, 65, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Koenig, J.I.; Kirkpatrick, B.; Lee, P. Glucocorticoid hormones and early brain development in schizophrenia. Neuropsychopharmacology 2002, 27, 309–318. [Google Scholar] [PubMed]
- Meyer, U.; Feldon, J. Prenatal exposure to infection: A primary mechanism for abnormal dopaminergic development in schizophrenia. Psychopharmacology (Berl.) 2009, 206, 587–602. [Google Scholar] [CrossRef] [PubMed]
- Ben Amor, L.; Grizenko, N.; Schwartz, G.; Lageix, P.; Baron, C.; Ter-Stepanian, M.; Zappitelli, M.; Mbekou, V.; Joober, R. Perinatal complications in children with attention-deficit hyperactivity disorder and their unaffected siblings. J. Psychiatry Neurosci. 2005, 30, 120–126. [Google Scholar] [PubMed]
- Szpir, M. Tracing the origins of autism: A spectrum of new studies. Environ. Health Perspect. 2006, 114, A412–A418. [Google Scholar] [CrossRef] [PubMed]
- Raikkonen, K.; Pesonen, A.K.; Heinonen, K.; Kajantie, E.; Hovi, P.; Jarvenpaa, A.L.; Eriksson, J.G.; Andersson, S. Depression in young adults with very low birth weight: The helsinki study of very low-birth-weight adults. Arch. Gen. Psychiatry 2008, 65, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Phillips, N.K.; Hammen, C.L.; Brennan, P.A.; Najman, J.M.; Bor, W. Early adversity and the prospective prediction of depressive and anxiety disorders in adolescents. J. Abnorm. Child. Psychol. 2005, 33, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Barlow, B.K.; Cory-Slechta, D.A.; Richfield, E.K.; Thiruchelvam, M. The gestational environment and parkinson’s disease: Evidence for neurodevelopmental origins of a neurodegenerative disorder. Reprod. Toxicol. 2007, 23, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.; Guegant, G.; Cador, M.; Arnauld, E.; Arsaut, J.; Le Moal, M.; Demotes-Mainard, J. Prenatal stress in rats facilitates amphetamine-induced sensitization and induces long-lasting changes in dopamine receptors in the nucleus accumbens. Brain Res. 1995, 685, 179–186. [Google Scholar] [CrossRef]
- Kippin, T.E.; Szumlinski, K.K.; Kapasova, Z.; Rezner, B.; See, R.E. Prenatal stress enhances responsiveness to cocaine. Neuropsychopharmacology 2008, 33, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Gatzke-Kopp, L.M. The canary in the coalmine: The sensitivity of mesolimbic dopamine to environmental adversity during development. Neurosci. Biobehav. Rev. 2010, 35, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.J.; Leao, P.; Carvalho, M.; Almeida, O.F.; Sousa, N. Potential programming of dopaminergic circuits by early life stress. Psychopharmacology (Berl.) 2011, 214, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Vuillermot, S.; Joodmardi, E.; Perlmann, T.; Ogren, S.O.; Feldon, J.; Meyer, U. Prenatal immune activation interacts with genetic nurr1 deficiency in the development of attentional impairments. J. Neurosci. 2012, 32, 436–451. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.S. Early adrenalectomy stimulates subsequent growth and development of the rat brain. Exp. Neurol. 1983, 82, 432–446. [Google Scholar] [CrossRef]
- Fowden, A.L.; Li, J.; Forhead, A.J. Glucocorticoids and the preparation for life after birth: Are there long-term consequences of the life insurance? Proc. Nutr. Soc. 1998, 57, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, R.M. Glucocorticoid excess and the developmental origins of disease: Two decades of testing the hypothesis--2012 curt richter award winner. Psychoneuroendocrinology 2013, 38, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.; Andrews, M.H.; Matthews, S.G. Maternal adversity, glucocorticoids and programming of neuroendocrine function and behaviour. Neurosci. Biobehav. Rev. 2005, 29, 209–226. [Google Scholar] [CrossRef] [PubMed]
- Seckl, J.R.; Holmes, M.C. Mechanisms of disease: Glucocorticoids, their placental metabolism and fetal ‘programming’ of adult pathophysiology. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.P.; Waffarn, F.; Sandman, C.A. Prenatal treatment with glucocorticoids sensitizes the hpa axis response to stress among full-term infants. Dev. Psychobiol. 2010, 53, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.P.; Waffarn, F.; Uy, C.; Hobel, C.J.; Glynn, L.M.; Sandman, C.A. Effect of prenatal glucocorticoid treatment on size at birth among infants born at term gestation. J. Perinatol. 2009, 29, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Sandman, C.A.; Davis, E.P. Neurobehavioral risk is associated with gestational exposure to stress hormones. Expert Rev. Endocrinol. Metab. 2012, 7, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Barbazanges, A.; Piazza, P.V.; Le Moal, M.; Maccari, S. Maternal glucocorticoid secretion mediates long-term effects of prenatal stress. J. Neurosci. 1996, 16, 3943–3949. [Google Scholar] [PubMed]
- Welberg, L.A.; Seckl, J.R. Prenatal stress, glucocorticoids and the programming of the brain. J. Neuroendocrinol. 2001, 13, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, M. The long-term behavioural consequences of prenatal stress. Neurosci. Biobehav. Rev. 2008, 32, 1073–1086. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, R.M.; Jacobsen, G.H.; Drake, A.J. What is the evidence in humans that DNA methylation changes link events in utero and later life disease? Clin. Endocrinol. (Oxf.) 2013, 78, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Gillies, G.E.; Pienaar, I.S.; Vohra, S.; Qamhawi, Z. Sex differences in Parkinson’s disease. Front. Neuroendocrinol. 2014, 35, 370–384. [Google Scholar] [CrossRef] [PubMed]
- Gillies, G.E.; Virdee, K.; McArthur, S.; Dalley, J.W. Sex-dependent diversity in ventral tegmental dopaminergic neurons and developmental programing: A molecular, cellular and behavioral analysis. Neuroscience 2014, 282C, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Pruessner, J.C.; Champagne, F.; Meaney, M.J.; Dagher, A. Dopamine release in response to a psychological stress in humans and its relationship to early life maternal care: A positron emission tomography study using [11c]raclopride. J. Neurosci. 2004, 24, 2825–2831. [Google Scholar] [CrossRef] [PubMed]
- Buss, C.; Davis, E.P.; Class, Q.A.; Gierczak, M.; Pattillo, C.; Glynn, L.M.; Sandman, C.A. Maturation of the human fetal startle response: Evidence for sex-specific maturation of the human fetus. Early Hum. Dev. 2009, 85, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Buss, C.; Lord, C.; Wadiwalla, M.; Hellhammer, D.H.; Lupien, S.J.; Meaney, M.J.; Pruessner, J.C. Maternal care modulates the relationship between prenatal risk and hippocampal volume in women but not in men. J. Neurosci. 2007, 27, 2592–2595. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, R.M.; Hii, H.L.; Pennell, C.E.; McKeague, I.W.; Kloet, E.R.; Lye, S.; Stanley, F.J.; Mattes, E.; Foster, J.K. Analysis of baseline hypothalamic-pituitary-adrenal activity in late adolescence reveals gender specific sensitivity of the stress axis. Psychoneuroendocrinology 2013, 38, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Luine, V.N.; Beck, K.D.; Bowman, R.E.; Frankfurt, M.; Maclusky, N.J. Chronic stress and neural function: Accounting for sex and age. J. Neuroendocrinol. 2007, 19, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Gabory, A.; Attig, L.; Junien, C. Sexual dimorphism in environmental epigenetic programming. Mol. Cell Endocrinol. 2009, 304, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Brummelte, S.; Lieblich, S.E.; Galea, L.A. Gestational and postpartum corticosterone exposure to the dam affects behavioral and endocrine outcome of the offspring in a sexually-dimorphic manner. Neuropharmacology 2012, 62, 406–418. [Google Scholar] [CrossRef] [PubMed]
- Kreider, M.L.; Levin, E.D.; Seidler, F.J.; Slotkin, T.A. Gestational dexamethasone treatment elicits sex-dependent alterations in locomotor activity, reward-based memory and hippocampal cholinergic function in adolescent and adult rats. Neuropsychopharmacology 2005, 30, 1617–1623. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, M. Sex-dependent changes induced by prenatal stress in cortical and hippocampal morphology and behaviour in rats: An update. Stress 2011, 14, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.B.; Hu, M.; Lee, T.M.; Bhatnagar, S.; Becker, J.B. Sex-specific susceptibility to cocaine in rats with a history of prenatal stress. Physiol. Behav. 2009, 97, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Gillies, G.E.; McArthur, S. Estrogen actions in the brain and the basis for differential action in men and women: A case for sex-specific medicines. Pharmacol. Rev. 2010, 62, 155–198. [Google Scholar] [CrossRef] [PubMed]
- Baron-Cohen, S.; Knickmeyer, R.C.; Belmonte, M.K. Sex differences in the brain: Implications for explaining autism. Science 2005, 310, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Aleman, A.; Kahn, R.S.; Selten, J.P. Sex differences in the risk of schizophrenia: Evidence from meta-analysis. Arch. Gen. Psychiatry 2003, 60, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.E.; Lynch, W.J.; Roth, M.E.; Morgan, A.D.; Cosgrove, K.P. Sex and estrogen influence drug abuse. Trends Pharmacol. Sci. 2004, 25, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Lynch, W.J. Sex differences in vulnerability to drug self-administration. Exp. Clin. Psychopharmacol. 2006, 14, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.B.; Hu, M. Sex differences in drug abuse. Front. Neuroendocrinol. 2008, 29, 36–47. [Google Scholar] [CrossRef] [PubMed]
- McArthur, S.; McHale, E.; Gillies, G.E. The size and distribution of midbrain dopaminergic populations are permanently altered by perinatal glucocorticoid exposure in a sex- region- and time-specific manner. Neuropsychopharmacology 2007, 32, 1462–1476. [Google Scholar] [CrossRef] [PubMed]
- Welberg, L.A.; Seckl, J.R.; Holmes, M.C. Prenatal glucocorticoid programming of brain corticosteroid receptors and corticotrophin-releasing hormone: Possible implications for behaviour. Neuroscience 2001, 104, 71–79. [Google Scholar] [CrossRef]
- Oliveira, M.; Bessa, J.M.; Mesquita, A.; Tavares, H.; Carvalho, A.; Silva, R.; Pego, J.M.; Cerqueira, J.J.; Palha, J.A.; Almeida, O.F.; et al. Induction of a hyperanxious state by antenatal dexamethasone: A case for less detrimental natural corticosteroids. Biol. Psychiatry 2006, 59, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Hauser, J.; Feldon, J.; Pryce, C.R. Direct and dam-mediated effects of prenatal dexamethasone on emotionality, cognition and hpa axis in adult wistar rats. Horm. Behav. 2009, 56, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Borges, S.; Coimbra, B.; Soares-Cunha, C.; Miguel Pego, J.; Sousa, N.; Joao Rodrigues, A. Dopaminergic modulation of affective and social deficits induced by prenatal glucocorticoid exposure. Neuropsychopharmacology 2013, 38, 2068–2079. [Google Scholar] [CrossRef] [PubMed]
- Meaney, M.J.; Brake, W.; Gratton, A. Environmental regulation of the development of mesolimbic dopamine systems: A neurobiological mechanism for vulnerability to drug abuse? Psychoneuroendocrinology 2002, 27, 127–138. [Google Scholar] [CrossRef]
- Theogaraj, E.; John, C.D.; Christian, H.C.; Morris, J.F.; Smith, S.F.; Buckingham, J.C. Perinatal glucocorticoid treatment produces molecular, functional, and morphological changes in the anterior pituitary gland of the adult male rat. Endocrinology 2005, 146, 4804–4813. [Google Scholar] [CrossRef] [PubMed]
- McArthur, S.; Siddique, Z.L.; Christian, H.C.; Capone, G.; Theogaraj, E.; John, C.D.; Smith, S.F.; Morris, J.F.; Buckingham, J.C.; Gillies, G.E. Perinatal glucocorticoid treatment disrupts the hypothalamo-lactotroph axis in adult female, but not male, rats. Endocrinology 2006, 147, 1904–1915. [Google Scholar] [CrossRef] [PubMed]
- McArthur, S.; Murray, H.E.; Dhankot, A.; Dexter, D.T.; Gillies, G.E. Striatal susceptibility to a dopaminergic neurotoxin is independent of sex hormone effects on cell survival and dat expression but is exacerbated by central aromatase inhibition. J. Neurochem. 2007, 100, 678–692. [Google Scholar] [CrossRef] [PubMed]
- McArthur, S.; McHale, E.; Dalley, J.W.; Buckingham, J.C.; Gillies, G.E. Altered mesencephalic dopaminergic populations in adulthood as a consequence of brief perinatal glucocorticoid exposure. J. Neuroendocrinol. 2005, 17, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, Y.; Okazaki, M.; Kitamura, Y.; Taniguchi, T. Developmental expression of p-glycoprotein (multidrug resistance gene product) in the rat brain. J. Neurobiol. 1999, 39, 383–392. [Google Scholar] [CrossRef]
- Meaney, M.J.; Aitken, D.H. [3h]dexamethasone binding in rat frontal cortex. Brain Res. 1985, 328, 176–180. [Google Scholar] [CrossRef]
- Diaz, R.; Brown, R.W.; Seckl, J.R. Distinct ontogeny of glucocorticoid and mineralocorticoid receptor and 11β-hydroxysteroid dehydrogenase types I and II mRNAs in the fetal rat brain suggest a complex control of glucocorticoid actions. J. Neurosci. 1998, 18, 2570–2580. [Google Scholar] [PubMed]
- Samtani, M.N.; Pyszczynski, N.A.; Dubois, D.C.; Almon, R.R.; Jusko, W.J. Modeling glucocorticoid-mediated fetal lung maturation: II. Temporal patterns of gene expression in fetal rat lung. J. Pharmacol. Exp. Ther. 2006, 317, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Samtani, M.N.; Pyszczynski, N.A.; Dubois, D.C.; Almon, R.R.; Jusko, W.J. Modeling glucocorticoid-mediated fetal lung maturation: I. Temporal patterns of corticosteroids in rat pregnancy. J. Pharmacol. Exp. Ther. 2006, 317, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Liggins, G.C. The role of cortisol in preparing the fetus for birth. Reprod. Fertil. Dev. 1994, 6, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Jobe, A.H.; Soll, R.F. Choice and dose of corticosteroid for antenatal treatments. Am. J. Obstet. Gynecol. 2004, 190, 878–881. [Google Scholar] [CrossRef] [PubMed]
- Ballard, P.L.; Ballard, R.A. Scientific basis and therapeutic regimens for use of antenatal glucocorticoids. Am. J. Obstet. Gynecol. 1995, 173, 254–262. [Google Scholar] [CrossRef]
- McArthur, S.; Pienaar, I.S.; Siddiqi, S.M.; Gillies, G.E. Sex-specific disruption of murine midbrain astrocytic and dopaminergic developmental trajectories following antenatal gc treatment. Brain Struct. Funct. 2015, 221, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Geuze, E.; Vermetten, E.; Bremner, J.D. MR-based in vivo hippocampal volumetrics: 2. Findings in neuropsychiatric disorders. Mol. Psychiatry 2005, 10, 160–184. [Google Scholar] [CrossRef] [PubMed]
- Buss, C.; Davis, E.P.; Muftuler, L.T.; Head, K.; Sandman, C.A. High pregnancy anxiety during mid-gestation is associated with decreased gray matter density in 6–9-year-old children. Psychoneuroendocrinology 2010, 35, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Lupien, S.J.; Evans, A.; Lord, C.; Miles, J.; Pruessner, M.; Pike, B.; Pruessner, J.C. Hippocampal volume is as variable in young as in older adults: Implications for the notion of hippocampal atrophy in humans. Neuroimage 2007, 34, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Rao, U.; Chen, L.A.; Bidesi, A.S.; Shad, M.U.; Thomas, M.A.; Hammen, C.L. Hippocampal changes associated with early-life adversity and vulnerability to depression. Biol. Psychiatry 2010, 67, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Uno, H.; Eisele, S.; Sakai, A.; Shelton, S.; Baker, E.; DeJesus, O.; Holden, J. Neurotoxicity of glucocorticoids in the primate brain. Horm. Behav. 1994, 28, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Buss, C.; Davis, E.P.; Shahbaba, B.; Pruessner, J.C.; Head, K.; Sandman, C.A. Maternal cortisol over the course of pregnancy and subsequent child amygdala and hippocampus volumes and affective problems. Proc. Natl. Acad. Sci. USA 2012, 109, E1312–E1319. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Smith, D.; Hof, P.R.; Foerster, B.; Hamilton, S.; Blackband, S.J.; Yu, M.; Benveniste, H. In vivo 3D digital atlas database of the adult C57BL/6J mouse brain by magnetic resonance microscopy. Front. Neuroanat. 2008, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Menke, R.A.; Jbabdi, S.; Miller, K.L.; Matthews, P.M.; Zarei, M. Connectivity-based segmentation of the substantia nigra in human and its implications in Parkinson’s disease. Neuroimage 2010, 52, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Murray, H.E.; Pillai, A.V.; McArthur, S.R.; Razvi, N.; Datla, K.P.; Dexter, D.T.; Gillies, G.E. Dose- and sex-dependent effects of the neurotoxin 6-hydroxydopamine on the nigrostriatal dopaminergic pathway of adult rats: Differential actions of estrogen in males and females. Neuroscience 2003, 116, 213–222. [Google Scholar] [CrossRef]
- Virdee, K.; McArthur, S.; Brischoux, F.; Caprioli, D.; Ungless, M.A.; Robbins, T.W.; Dalley, J.W.; Gillies, G.E. Antenatal glucocorticoid treatment induces adaptations in adult midbrain dopamine neurons, which underpin sexually dimorphic behavioral resilience. Neuropsychopharmacology 2014, 39, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Carman, L.S.; Gage, F.H.; Shults, C.W. Partial lesion of the substantia nigra: Relation between extent of lesion and rotational behavior. Brain Res. 1991, 553, 275–283. [Google Scholar] [CrossRef]
- Forger, N.G. Control of cell number in the sexually dimorphic brain and spinal cord. J. Neuroendocrinol. 2009, 21, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Shende, V.H.; McArthur, S.; Gillies, G.E.; Opacka-Juffry, J. Astroglial plasticity is implicated in hippocampal remodelling in adult rats exposed to antenatal dexamethasone. Neural. Plast. 2015, 2015, 694347. [Google Scholar] [CrossRef] [PubMed]
- Menke, R.A.; Scholz, J.; Miller, K.L.; Deoni, S.; Jbabdi, S.; Matthews, P.M.; Zarei, M. Mri characteristics of the substantia nigra in parkinson’s disease: A combined quantitative t1 and dti study. Neuroimage 2009, 47, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Smidt, M.P.; Burbach, J.P. How to make a mesodiencephalic dopaminergic neuron. Nat. Rev. Neurosci. 2007, 8, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Tassin, J.P.; Simon, H.; Herve, D.; Blanc, G.; Le Moal, M.; Glowinski, J.; Bockaert, J. Non-dopaminergic fibres may regulate dopamine-sensitive adenylate cyclase in the prefrontal cortex and nucleus accumbens. Nature 1982, 295, 696–698. [Google Scholar] [CrossRef] [PubMed]
- Brodski, C.; Weisenhorn, D.M.; Signore, M.; Sillaber, I.; Oesterheld, M.; Broccoli, V.; Acampora, D.; Simeone, A.; Wurst, W. Location and size of dopaminergic and serotonergic cell populations are controlled by the position of the midbrain-hindbrain organizer. J. Neurosci. 2003, 23, 4199–4207. [Google Scholar] [PubMed]
- Dewing, P.; Chiang, C.W.; Sinchak, K.; Sim, H.; Fernagut, P.O.; Kelly, S.; Chesselet, M.F.; Micevych, P.E.; Albrecht, K.H.; Harley, V.R.; et al. Direct regulation of adult brain function by the male-specific factor SRY. Curr. Biol. 2006, 16, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Sillitoe, R.V.; Vogel, M.W. Desire, disease, and the origins of the dopaminergic system. Schizophr. Bull. 2008, 34, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Bayer, S.A.; Wills, K.V.; Triarhou, L.C.; Ghetti, B. Time of neuron origin and gradients of neurogenesis in midbrain dopaminergic neurons in the mouse. Exp. Brain Res. 1995, 105, 191–199. [Google Scholar] [CrossRef]
- Lieb, K.; Andersen, C.; Lazarov, N.; Zienecker, R.; Urban, I.; Reisert, I.; Pilgrim, C. Pre- and postnatal development of dopaminergic neuron numbers in the male and female mouse midbrain. Brain Res. Dev. Brain Res. 1996, 94, 37–43. [Google Scholar] [CrossRef]
- Kawano, H.; Ohyama, K.; Kawamura, K.; Nagatsu, I. Migration of dopaminergic neurons in the embryonic mesencephalon of mice. Brain Res. Dev. Brain Res. 1995, 86, 101–113. [Google Scholar] [CrossRef]
- Kholodilov, N.; Yarygina, O.; Oo, T.F.; Zhang, H.; Sulzer, D.; Dauer, W.; Burke, R.E. Regulation of the development of mesencephalic dopaminergic systems by the selective expression of glial cell line-derived neurotrophic factor in their targets. J. Neurosci. 2004, 24, 3136–3146. [Google Scholar] [CrossRef] [PubMed]
- Jackson-Lewis, V.; Vila, M.; Djaldetti, R.; Guegan, C.; Liberatore, G.; Liu, J.; O’Malley, K.L.; Burke, R.E.; Przedborski, S. Developmental cell death in dopaminergic neurons of the substantia nigra of mice. J. Comp. Neurol. 2000, 424, 476–488. [Google Scholar] [CrossRef]
- Hagerty, T.; Morgan, W.W.; Elango, N.; Strong, R. Identification of a glucocorticoid-responsive element in the promoter region of the mouse tyrosine hydroxylase gene. J. Neurochem. 2001, 76, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Oo, T.F.; Kholodilov, N.; Burke, R.E. Regulation of natural cell death in dopaminergic neurons of the substantia nigra by striatal glial cell line-derived neurotrophic factor in vivo. J. Neurosci. 2003, 23, 5141–5148. [Google Scholar] [PubMed]
- Burke, R.E. Ontogenic cell death in the nigrostriatal system. Cell Tissue Res. 2004, 318, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Vitalis, T.; Cases, O.; Parnavelas, J.G. Development of the dopaminergic neurons in the rodent brainstem. Exp. Neurol. 2005, 191, S104–S112. [Google Scholar] [CrossRef]
- Gould, E.; Tanapat, P.; Cameron, H.A. Adrenal steroids suppress granule cell death in the developing dentate gyrus through an nmda receptor-dependent mechanism. Brain Res. Dev. Brain Res. 1997, 103, 91–93. [Google Scholar] [CrossRef]
- Abraham, I.M.; Harkany, T.; Horvath, K.M.; Luiten, P.G. Action of glucocorticoids on survival of nerve cells: Promoting neurodegeneration or neuroprotection? J. Neuroendocrinol. 2001, 13, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Fleckenstein, A.E.; Volz, T.J.; Riddle, E.L.; Gibb, J.W.; Hanson, G.R. New insights into the mechanism of action of amphetamines. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 681–698. [Google Scholar] [CrossRef] [PubMed]
- DeLong, M.R.; Wichmann, T. Basal ganglia circuits as targets for neuromodulation in parkinson disease. JAMA Neurol. 2015, 72, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Chiodo, L.A.; Bannon, M.J.; Grace, A.A.; Roth, R.H.; Bunney, B.S. Evidence for the absence of impulse-regulating somatodendritic and synthesis-modulating nerve terminal autoreceptors on subpopulations of mesocortical dopamine neurons. Neuroscience 1984, 12, 1–16. [Google Scholar] [CrossRef]
- Benes, F.M. The development of the prefrontal cortex: The maturation of neurotransmitter systems and their interactions. In Developmental Psychopathology, 2nd ed.; Cicchetti, D.J.C., Ed.; Wiley: Hoboken, NJ, USA, 2006. [Google Scholar]
- Doherty, M.D.; Gratton, A. Effects of medial prefrontal cortical injections of gaba receptor agonists and antagonists on the local and nucleus accumbens dopamine responses to stress. Synapse 1999, 32, 288–300. [Google Scholar] [CrossRef]
- Grace, A.A.; Floresco, S.B.; Goto, Y.; Lodge, D.J. Regulation of firing of dopaminergic neurons and control of goal-directed behaviors. Trends Neurosci. 2007, 30, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Brog, J.S.; Salyapongse, A.; Deutch, A.Y.; Zahm, D.S. The patterns of afferent innervation of the core and shell in the “accumbens” part of the rat ventral striatum: Immunohistochemical detection of retrogradely transported fluoro-gold. J. Comp. Neurol. 1993, 338, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.R.; Robinson, E.S.; Theobald, D.E.; Dalley, J.W.; Robbins, T.W. Contrasting effects of selective lesions of nucleus accumbens core or shell on inhibitory control and amphetamine-induced impulsive behaviour. Eur. J. Neurosci. 2008, 28, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Schultz, W. Multiple dopamine functions at different time courses. Annu. Rev. Neurosci. 2007, 30, 259–288. [Google Scholar] [CrossRef] [PubMed]
- Barres, B.A. The mystery and magic of glia: A perspective on their roles in health and disease. Neuron 2008, 60, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Halassa, M.M.; Haydon, P.G. Integrated brain circuits: Astrocytic networks modulate neuronal activity and behavior. Annu. Rev. Physiol. 2010, 72, 335–355. [Google Scholar] [CrossRef] [PubMed]
- Rappold, P.M.; Tieu, K. Astrocytes and therapeutics for parkinson’s disease. Neurotherapeutics 2010, 7, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Gillies, G.E.; McArthur, S.; Imperial College London, London, UK. Unpublished work. 2014.
- Vardimon, L.; Ben-Dror, I.; Avisar, N.; Oren, A.; Shiftan, L. Glucocorticoid control of glial gene expression. J. Neurobiol. 1999, 40, 513–527. [Google Scholar] [CrossRef]
- Freeman, M.R. Specification and morphogenesis of astrocytes. Science 2010, 330, 774–778. [Google Scholar] [CrossRef]
- Bandeira, F.; Lent, R.; Herculano-Houzel, S. Changing numbers of neuronal and non-neuronal cells underlie postnatal brain growth in the rat. Proc. Natl. Acad. Sci. USA 2009, 106, 14108–14113. [Google Scholar] [CrossRef] [PubMed]
- Zschocke, J.; Bayatti, N.; Clement, A.M.; Witan, H.; Figiel, M.; Engele, J.; Behl, C. Differential promotion of glutamate transporter expression and function by glucocorticoids in astrocytes from various brain regions. J. Biol. Chem. 2005, 280, 34924–34932. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.J.; Leao, P.; Pego, J.M.; Cardona, D.; Carvalho, M.M.; Oliveira, M.; Costa, B.M.; Carvalho, A.F.; Morgado, P.; Araujo, D.; et al. Mechanisms of initiation and reversal of drug-seeking behavior induced by prenatal exposure to glucocorticoids. Mol. Psychiatry 2012, 17, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Vuillermot, S.; Weber, L.; Feldon, J.; Meyer, U. A longitudinal examination of the neurodevelopmental impact of prenatal immune activation in mice reveals primary defects in dopaminergic development relevant to schizophrenia. J. Neurosci. 2010, 30, 1270–1287. [Google Scholar] [CrossRef] [PubMed]
- Meyer, U.; Yee, B.K.; Feldon, J. The neurodevelopmental impact of prenatal infections at different times of pregnancy: The earlier the worse? Neuroscientist 2007, 13, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.H.; Robbins, T.W. Differential effects of mesocortical, mesolimbic, and mesostriatal dopamine depletion on spontaneous, conditioned, and drug-induced locomotor activity. Pharmacol. Biochem. Behav. 1992, 43, 887–895. [Google Scholar] [CrossRef]
- Humby, T.; Wilkinson, L.S.; Robbins, T.W.; Geyer, M.A. Prepulses inhibit startle-induced reductions of extracellular dopamine in the nucleus accumbens of rat. J. Neurosci. 1996, 16, 2149–2156. [Google Scholar] [PubMed]
- Campbell, A.; Villavicencio, A.T.; Yeghiayan, S.K.; Balikian, R.; Baldessarini, R.J. Mapping of locomotor behavioral arousal induced by microinjections of dopamine within nucleus accumbens septi of rat forebrain. Brain Res. 1997, 771, 55–62. [Google Scholar] [CrossRef]
- Roberts, D.C.; Corcoran, M.E.; Fibiger, H.C. On the role of ascending catecholaminergic systems in intravenous self-administration of cocaine. Pharmacol. Biochem. Behav. 1977, 6, 615–620. [Google Scholar] [CrossRef]
- Parkinson, J.A.; Dalley, J.W.; Cardinal, R.N.; Bamford, A.; Fehnert, B.; Lachenal, G.; Rudarakanchana, N.; Halkerston, K.M.; Robbins, T.W.; Everitt, B.J. Nucleus accumbens dopamine depletion impairs both acquisition and performance of appetitive pavlovian approach behaviour: Implications for mesoaccumbens dopamine function. Behav. Brain Res. 2002, 137, 149–163. [Google Scholar] [CrossRef]
- Dalley, J.W.; Laane, K.; Theobald, D.E.; Armstrong, H.C.; Corlett, P.R.; Chudasama, Y.; Robbins, T.W. Time-limited modulation of appetitive pavlovian memory by d1 and nmda receptors in the nucleus accumbens. Proc. Natl. Acad. Sci. USA 2005, 102, 6189–6194. [Google Scholar] [CrossRef] [PubMed]
- Virdee, K.; Kentrop, J.; Jupp, B.; Venus, B.; Hensman, D.; McArthur, S.; Wilkinson, J.; Robbins, T.W.; Gillies, G.; Dalley, J.W. Counteractive effects of antenatal glucocorticoid treatment on d1 receptor modulation of spatial working memory. Psychopharmacology (Berl.) 2016, 233, 3751–3761. [Google Scholar] [CrossRef] [PubMed]
- Van den Heuvel, D.M.; Pasterkamp, R.J. Getting connected in the dopamine system. Prog. Neurobiol. 2008, 85, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Pryce, C.R.; Aubert, Y.; Maier, C.; Pearce, P.C.; Fuchs, E. The developmental impact of prenatal stress, prenatal dexamethasone and postnatal social stress on physiology, behaviour and neuroanatomy of primate offspring: Studies in rhesus macaque and common marmoset. Psychopharmacology (Berl.) 2009, 214, 33–53. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, R.M.; Labad, J.; Buss, C.; Ghaemmaghami, P.; Raikkonen, K. Transmitting biological effects of stress in utero: Implications for mother and offspring. Psychoneuroendocrinology 2013, 38, 1843–1849. [Google Scholar] [CrossRef] [PubMed]
- Boksa, P.; El-Khodor, B.F. Birth insult interacts with stress at adulthood to alter dopaminergic function in animal models: Possible implications for schizophrenia and other disorders. Neurosci. Biobehav. Rev. 2003, 27, 91–101. [Google Scholar] [CrossRef]
- McEwen, B.S. Stress, adaptation, and disease. Allostasis and allostatic load. Ann. N. Y. Acad. Sci. 1998, 840, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Beauchaine, T.P.; Neuhaus, E.; Zalewski, M.; Crowell, S.E.; Potapova, N. The effects of allostatic load on neural systems subserving motivation, mood regulation, and social affiliation. Dev. Psychopathol. 2011, 23, 975–999. [Google Scholar] [CrossRef] [PubMed]
- Arnold, A.P.; Gorski, R.A. Gonadal steroid induction of structural sex differences in the central nervous system. Annu. Rev. Neurosci. 1984, 7, 413–442. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.A.; Davies, D.C. The control of sexual differentiation of the reproductive system and brain. Reproduction 2007, 133, 331–359. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.M. Estradiol and the developing brain. Physiol. Rev. 2008, 88, 91–124. [Google Scholar] [CrossRef] [PubMed]
- Arnold, A.P. The organizational-activational hypothesis as the foundation for a unified theory of sexual differentiation of all mammalian tissues. Horm. Behav. 2009, 55, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.L.; Ward, O.B.; Affuso, J.D.; Long, W.D., 3rd; French, J.A.; Hendricks, S.E. Fetal testosterone surge: Specific modulations induced in male rats by maternal stress and/or alcohol consumption. Horm. Behav. 2003, 43, 531–539. [Google Scholar] [CrossRef]
- Weisz, J.; Ward, I.L. Plasma testosterone and progesterone titers of pregnant rats, their male and female fetuses, and neonatal offspring. Endocrinology 1980, 106, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Abramovich, D.R. Human sexual differentiation--in utero influences. J. Obstet. Gynaecol. Br. Commonw. 1974, 81, 448–453. [Google Scholar] [CrossRef]
- Luine, V.N.; Renner, K.J.; McEwen, B.S. Sex-dependent differences in estrogen regulation of choline acetyltransferase are altered by neonatal treatments. Endocrinology 1986, 119, 874–878. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S.; Alves, S.E. Estrogen actions in the central nervous system. Endocr. Rev. 1999, 20, 279–307. [Google Scholar] [CrossRef] [PubMed]
- Mitsushima, D.; Takase, K.; Takahashi, T.; Kimura, F. Activational and organisational effects of gonadal steroids on sex-specific acetylcholine release in the dorsal hippocampus. J. Neuroendocrinol. 2009, 21, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.T.; Breedlove, S.M.; Jordan, C.L. Androgen receptors mediate masculinization of astrocytes in the rat posterodorsal medial amygdala during puberty. J. Comp. Neurol. 2013, 521, 2298–2309. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.L.; Weisz, J. Maternal stress alters plasma testosterone in fetal males. Science 1980, 207, 328–329. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.L.; Ward, O.B.; Winn, R.J.; Bielawski, D. Male and female sexual behavior potential of male rats prenatally exposed to the influence of alcohol, stress, or both factors. Behav. Neurosci. 1994, 108, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.; Leao, P.; Rodrigues, A.J.; Pego, J.M.; Cerqueira, J.J.; Sousa, N. Programming effects of antenatal corticosteroids exposure in male sexual behavior. J. Sex Med. 2011, 8, 1965–1974. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.B.; Arnold, A.P.; Berkley, K.J.; Blaustein, J.D.; Eckel, L.A.; Hampson, E.; Herman, J.P.; Marts, S.; Sadee, W.; Steiner, M.; et al. Strategies and methods for research on sex differences in brain and behavior. Endocrinology 2005, 146, 1650–1673. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.B. Gender differences in dopaminergic function in striatum and nucleus accumbens. Pharmacol. Biochem. Behav. 1999, 64, 803–812. [Google Scholar] [CrossRef]
- Dluzen, D.E.; Mickley, K.R. Gender differences in modulatory effects of tamoxifen upon the nigrostriatal dopaminergic system. Pharmacol. Biochem. Behav. 2005, 80, 27–33. [Google Scholar] [CrossRef]
- McDermott, J.L.; Liu, B.; Dluzen, D.E. Sex differences and effects of estrogen on dopamine and dopac release from the striatum of male and female cd-1 mice. Exp. Neurol. 1994, 125, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Serova, L.I.; Maharjan, S.; Huang, A.; Sun, D.; Kaley, G.; Sabban, E.L. Response of tyrosine hydroxylase and gtp cyclohydrolase i gene expression to estrogen in brain catecholaminergic regions varies with mode of administration. Brain Res. 2004, 1015, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Moroz, I.A.; Rajabi, H.; Rodaros, D.; Stewart, J. Effects of sex and hormonal status on astrocytic basic fibroblast growth factor-2 and tyrosine hydroxylase immunoreactivity after medial forebrain bundle 6-hydroxydopamine lesions of the midbrain dopamine neurons. Neuroscience 2003, 118, 463–476. [Google Scholar] [CrossRef]
- Ferraz, A.C.; Matheussi, F.; Szawka, R.E.; Rizelio, V.; Delattre, A.M.; Rigon, P.; Hermel Edo, E.; Xavier, L.L.; Achaval, M.; Anselmo-Franci, J.A. Evaluation of estrogen neuroprotective effect on nigrostriatal dopaminergic neurons following 6-hydroxydopamine injection into the substantia nigra pars compacta or the medial forebrain bundle. Neurochem. Res. 2008, 33, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Sibug, R.; Kuppers, E.; Beyer, C.; Maxson, S.C.; Pilgrim, C.; Reisert, I. Genotype-dependent sex differentiation of dopaminergic neurons in primary cultures of embryonic mouse brain. Brain Res. Dev. Brain Res. 1996, 93, 136–142. [Google Scholar] [CrossRef]
- Arnold, A.P.; Burgoyne, P.S. Are xx and xy brain cells intrinsically different? Trends Endocrinol. Metab. 2004, 15, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Ngun, T.C.; Ghahramani, N.; Sanchez, F.J.; Bocklandt, S.; Vilain, E. The genetics of sex differences in brain and behavior. Front. Neuroendocrinol. 2011, 32, 227–246. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.; Mosler, G.; Just, W.; Pilgrim, C.; Reisert, I. Developmental profile of sry transcripts in mouse brain. Neurogenetics 2000, 3, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Czech, D.P.; Lee, J.; Sim, H.; Parish, C.L.; Vilain, E.; Harley, V.R. The human testis-determining factor sry localizes in midbrain dopamine neurons and regulates multiple components of catecholamine synthesis and metabolism. J. Neurochem. 2012, 122, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Czech, D.P.; Lee, J.; Correia, J.; Loke, H.; Moller, E.K.; Harley, V.R. Transient neuroprotection by sry upregulation in dopamine cells following injury in males. Endocrinology 2014, 155, 2602–2612. [Google Scholar] [CrossRef] [PubMed]
- Kreider, M.L.; Tate, C.A.; Cousins, M.M.; Oliver, C.A.; Seidler, F.J.; Slotkin, T.A. Lasting effects of developmental dexamethasone treatment on neural cell number and size, synaptic activity, and cell signaling: Critical periods of vulnerability, dose-effect relationships, regional targets, and sex selectivity. Neuropsychopharmacology 2006, 31, 12–35. [Google Scholar] [CrossRef] [PubMed]
- Baud, O. Antenatal corticosteroid therapy: Benefits and risks. Acta Paediatr. Suppl. 2004, 93, 6–10. [Google Scholar] [CrossRef]
- Asztalos, E. The need to go beyond: Evaluating antenatal corticosteroid trials with long-term outcomes. J. Obstet. Gynaecol. Can. 2007, 29, 429–432. [Google Scholar] [CrossRef]
- Asztalos, E. Antenatal corticosteroids: A risk factor for the development of chronic disease. J. Nutr. Metab. 2012, 2012, 930591. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, R.L.; Cohen, W.R. Antenatal steroid therapy: Have we undervalued the risks? J. Matern. Fetal. Neonatal. Med. 2006, 19, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.P.; Townsend, E.L.; Gunnar, M.R.; Guiang, S.F.; Lussky, R.C.; Cifuentes, R.F.; Georgieff, M.K. Antenatal betamethasone treatment has a persisting influence on infant hpa axis regulation. J. Perinatol. 2006, 26, 147–153. [Google Scholar] [CrossRef]
- Davis, E.P.; Sandman, C.A. The timing of prenatal exposure to maternal cortisol and psychosocial stress is associated with human infant cognitive development. Child. Dev. 2010, 81, 131–148. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.P.; Glynn, L.M.; Waffarn, F.; Sandman, C.A. Prenatal maternal stress programs infant stress regulation. J. Child. Psychol. Psychiatry 2010, 52, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Bale, T.L. Stress sensitivity and the development of affective disorders. Horm. Behav. 2006, 50, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Kessler, R.C. Epidemiology of women and depression. J. Affect. Disord. 2003, 74, 5–13. [Google Scholar] [CrossRef]
- Kumari, V.; Antonova, E.; Geyer, M.A. Prepulse inhibition and “psychosis-proneness” in healthy individuals: An fmri study. Eur. Psychiatry 2008, 23, 274–280. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.M.; Auger, A.P.; Bale, T.L.; De Vries, G.J.; Dunn, G.A.; Forger, N.G.; Murray, E.K.; Nugent, B.M.; Schwarz, J.M.; Wilson, M.E. The epigenetics of sex differences in the brain. J. Neurosci. 2009, 29, 12815–12823. [Google Scholar] [CrossRef]
- Schwarz, J.M.; McCarthy, M.M. Cellular mechanisms of estradiol-mediated masculinization of the brain. J. Steroid. Biochem. Mol. Biol. 2008, 109, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Crudo, A.; Suderman, M.; Moisiadis, V.G.; Petropoulos, S.; Kostaki, A.; Hallett, M.; Szyf, M.; Matthews, S.G. Glucocorticoid programming of the fetal male hippocampal epigenome. Endocrinology 2013, 154, 1168–1180. [Google Scholar] [CrossRef] [PubMed]
- Bale, T.L.; Baram, T.Z.; Brown, A.S.; Goldstein, J.M.; Insel, T.R.; McCarthy, M.M.; Nemeroff, C.B.; Reyes, T.M.; Simerly, R.B.; Susser, E.S.; et al. Early life programming and neurodevelopmental disorders. Biol. Psychiatry 2010, 68, 314–319. [Google Scholar] [CrossRef] [PubMed]
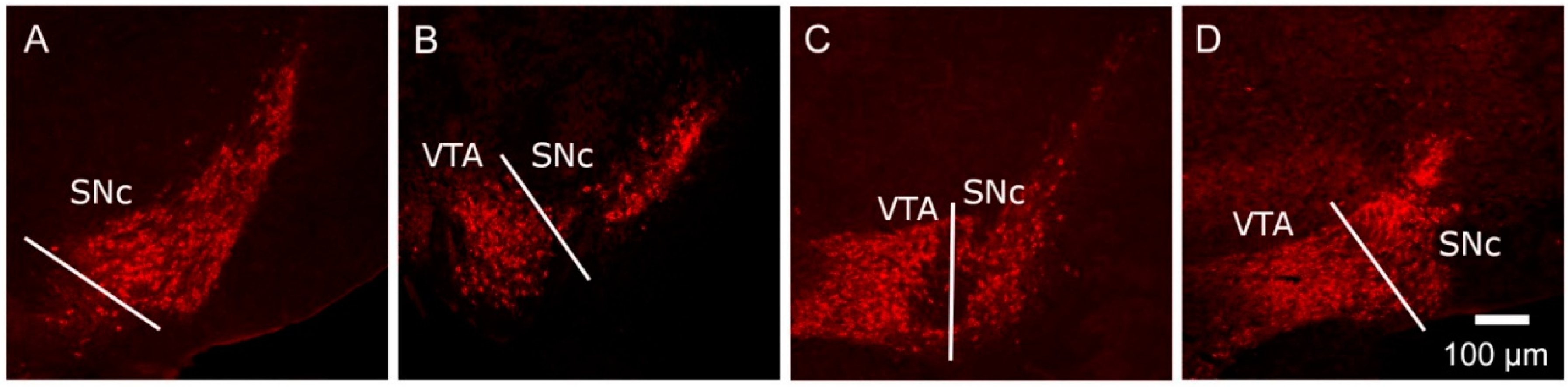
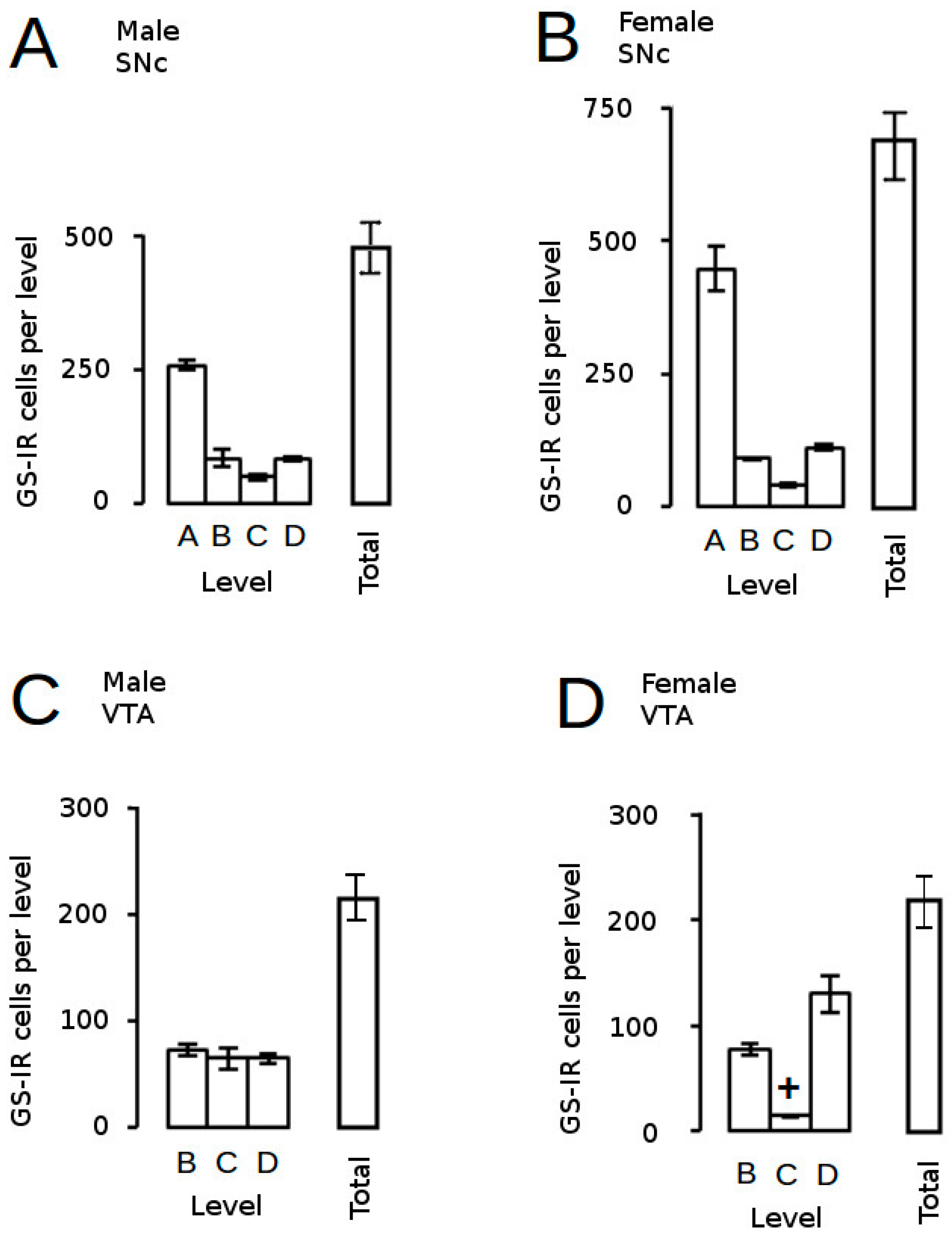
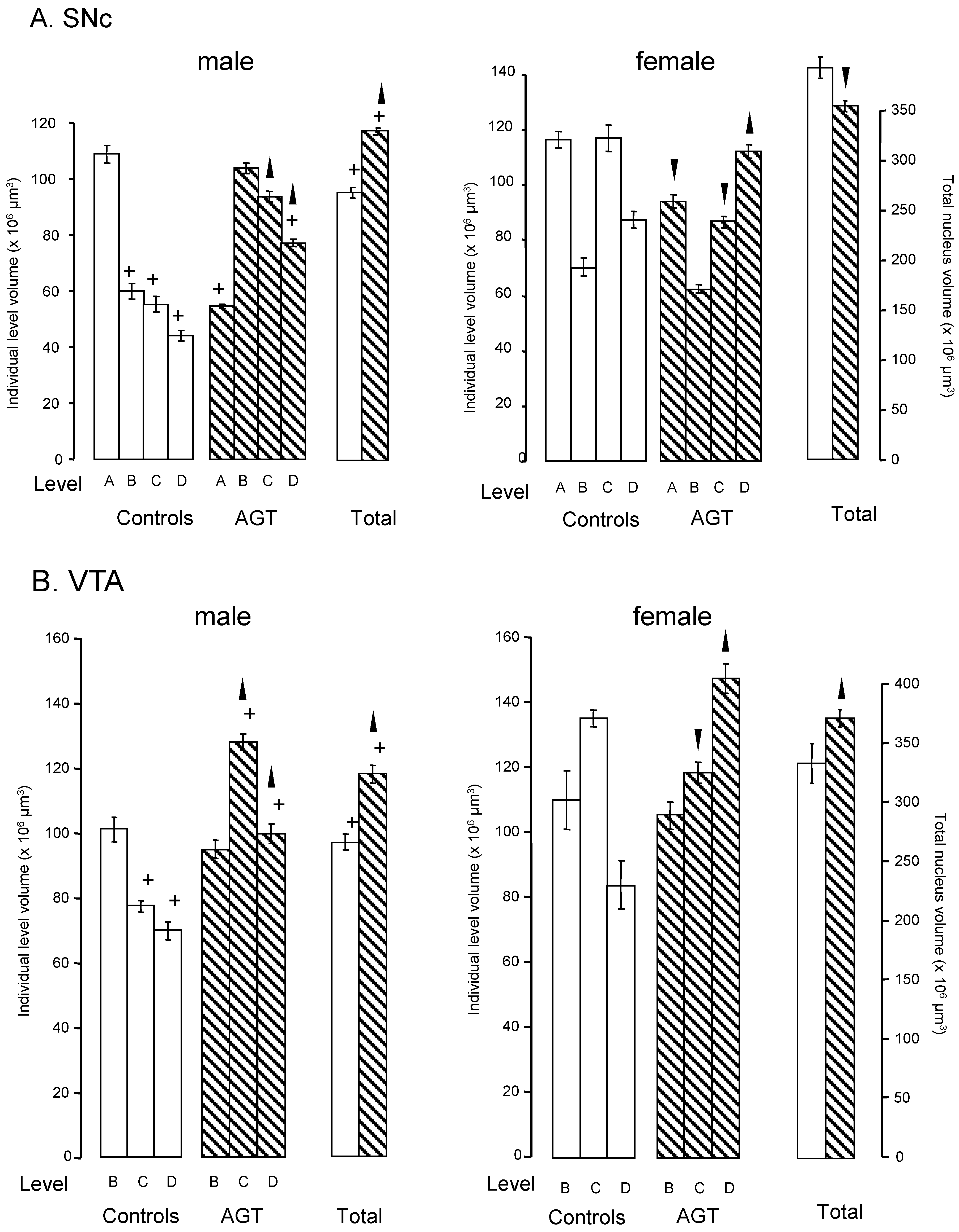
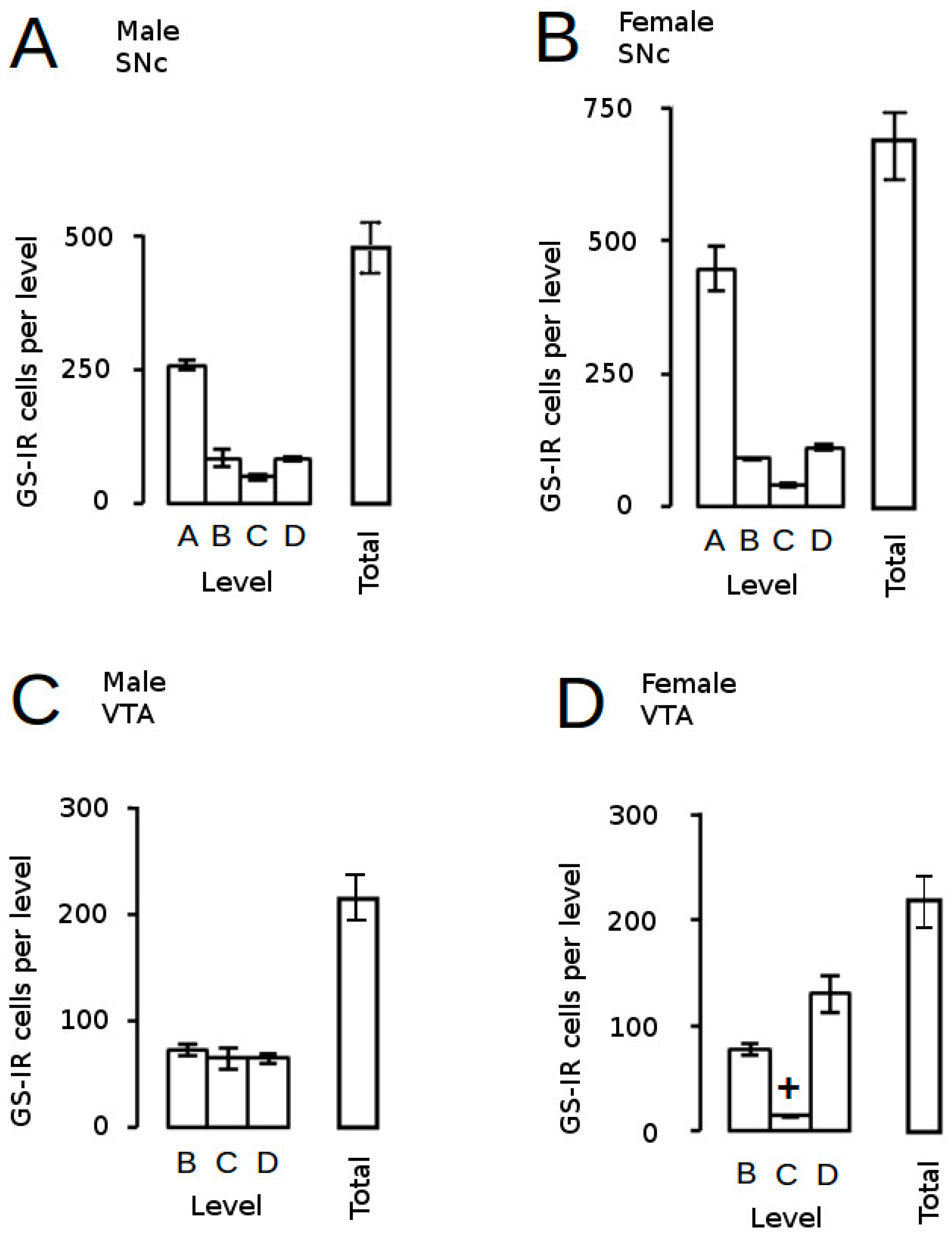
 ) and volumes of the SNc (A) and VTA (B) in adulthood were compared to the control offspring of dams receiving normal drinking water (
) and volumes of the SNc (A) and VTA (B) in adulthood were compared to the control offspring of dams receiving normal drinking water (  ). Data are means ± s.e.m., n = 8 animals per treatment group.
). Data are means ± s.e.m., n = 8 animals per treatment group.  ,
,  indicates significant effect of treatment, p < 0.05, increased or decreased respectively, for dexamethasone treated vs. control animals; + indicates significant sex difference p < 0.05 vs. females in the same treatment group.
) and volumes of the SNc (A) and VTA (B) in adulthood were compared to the control offspring of dams receiving normal drinking water ( ). Data are means ± s.e.m., n = 8 animals per treatment group. , indicates significant effect of treatment, p < 0.05, increased or decreased respectively, for dexamethasone treated vs. control animals; + indicates significant sex difference p < 0.05 vs. females in the same treatment group.
indicates significant effect of treatment, p < 0.05, increased or decreased respectively, for dexamethasone treated vs. control animals; + indicates significant sex difference p < 0.05 vs. females in the same treatment group.
) and volumes of the SNc (A) and VTA (B) in adulthood were compared to the control offspring of dams receiving normal drinking water ( ). Data are means ± s.e.m., n = 8 animals per treatment group. , indicates significant effect of treatment, p < 0.05, increased or decreased respectively, for dexamethasone treated vs. control animals; + indicates significant sex difference p < 0.05 vs. females in the same treatment group.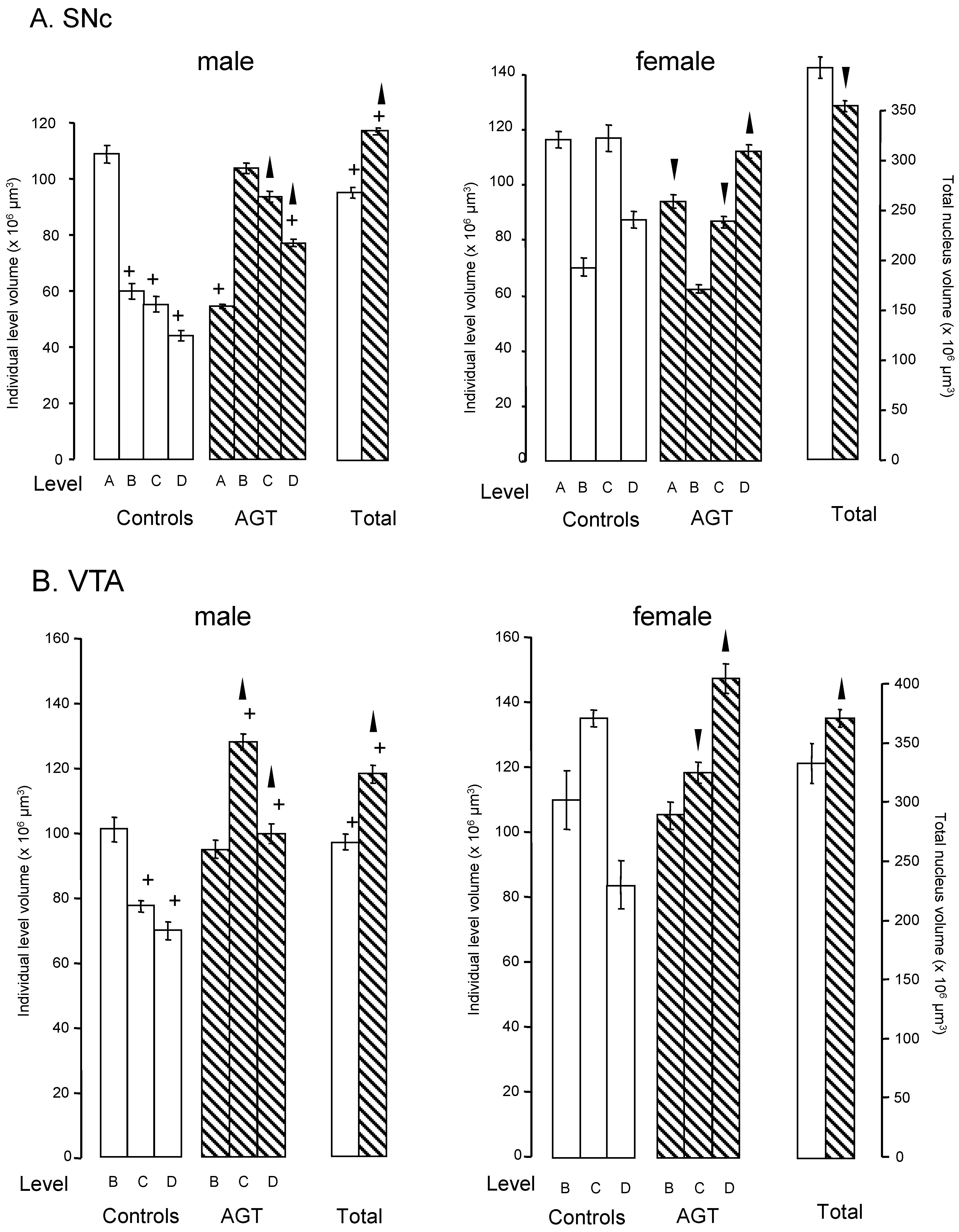
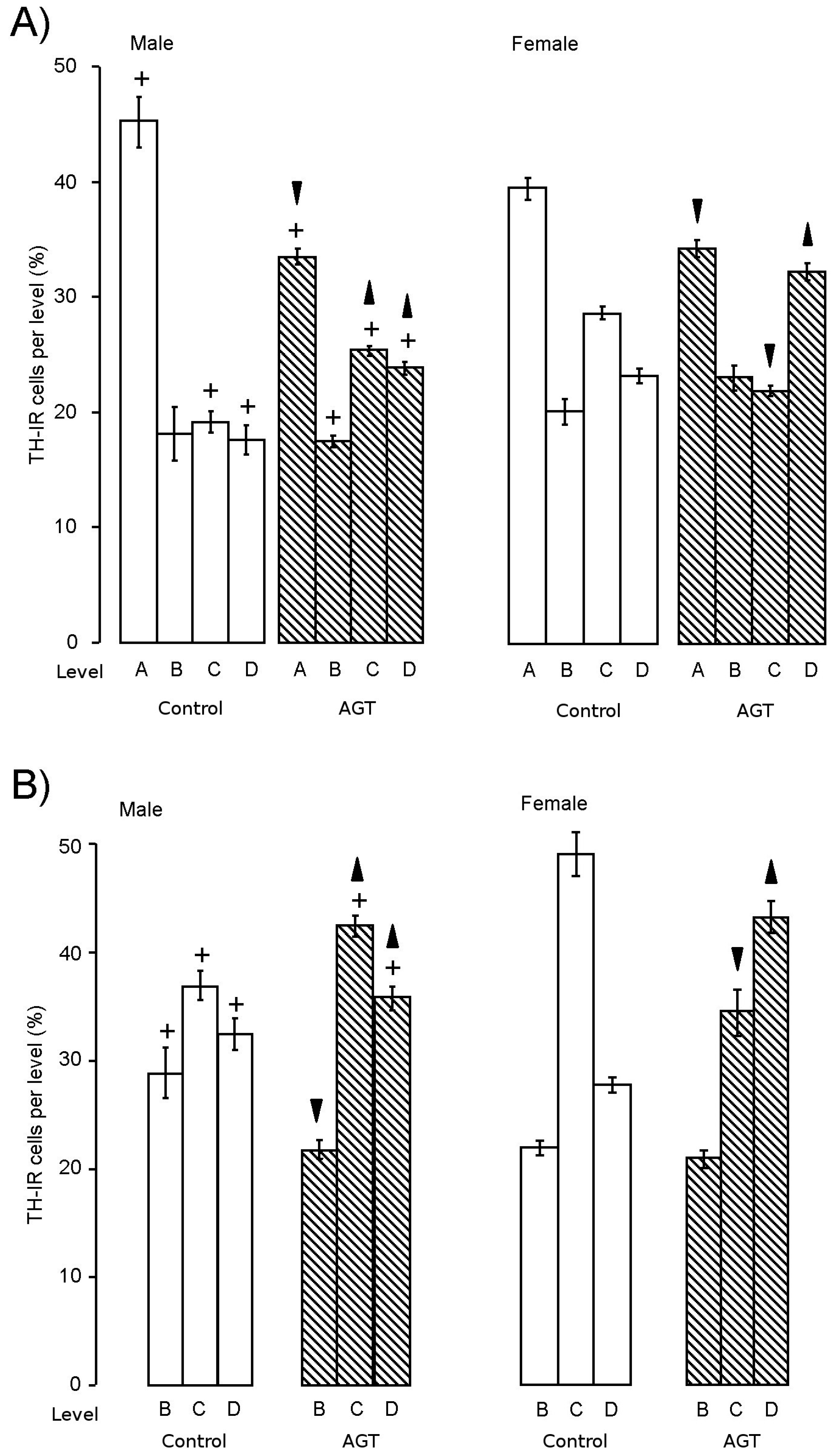
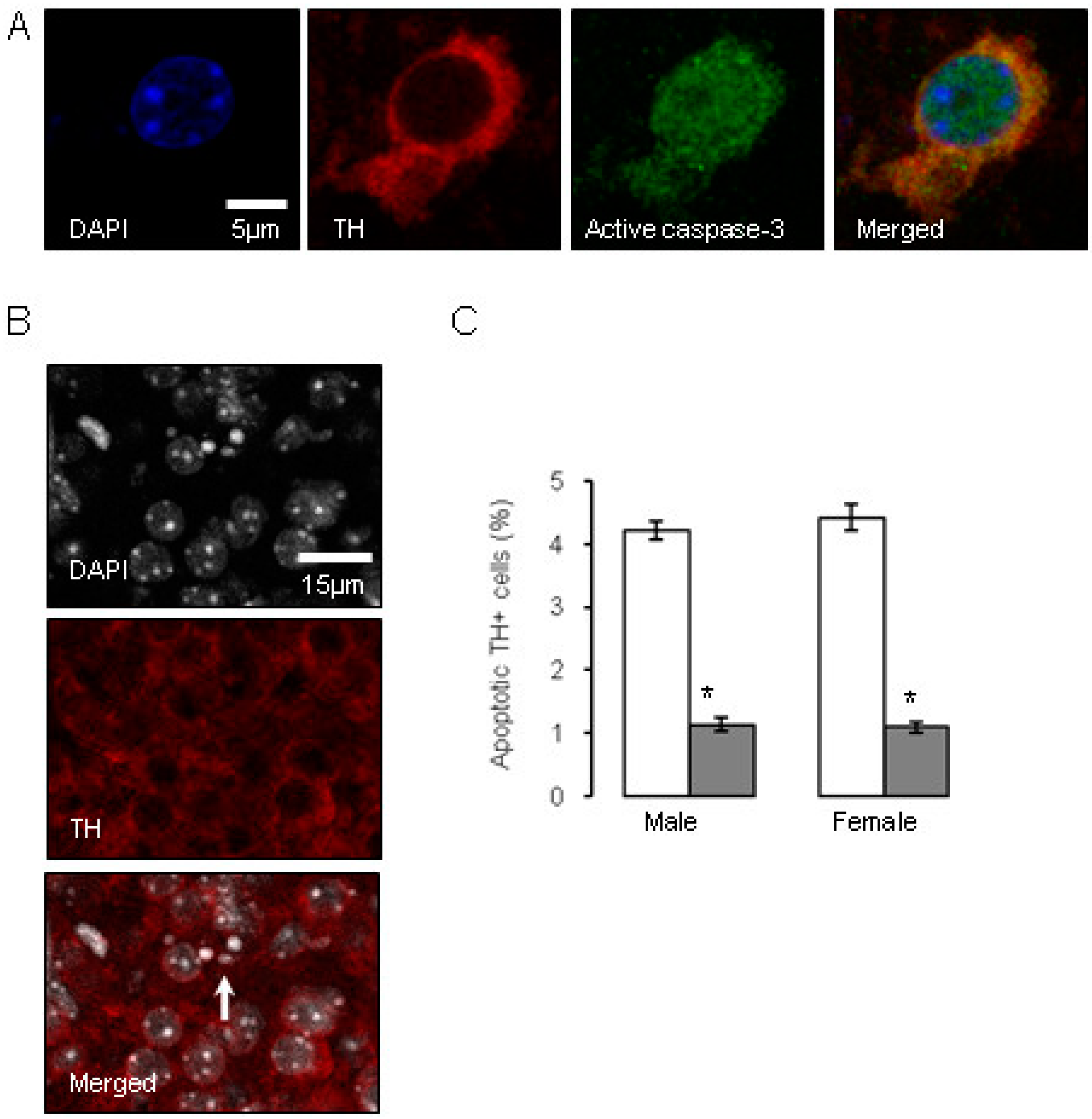
 ) compared to the control offspring of dams receiving normal drinking water ( ). Data are means ± s.e.m., n = 8 animals per treatment group. , indicates significant effect of treatment, p < 0.05 increased or decreased, respectively, for dexamethasone treated vs. control animals; + indicates significant sex difference p < 0.05 vs. females in the same treatment group.
) compared to the control offspring of dams receiving normal drinking water ( ). Data are means ± s.e.m., n = 8 animals per treatment group. , indicates significant effect of treatment, p < 0.05 increased or decreased, respectively, for dexamethasone treated vs. control animals; + indicates significant sex difference p < 0.05 vs. females in the same treatment group.
) compared to the control offspring of dams receiving normal drinking water ( ). Data are means ± s.e.m., n = 8 animals per treatment group. , indicates significant effect of treatment, p < 0.05 increased or decreased, respectively, for dexamethasone treated vs. control animals; + indicates significant sex difference p < 0.05 vs. females in the same treatment group.
) compared to the control offspring of dams receiving normal drinking water ( ). Data are means ± s.e.m., n = 8 animals per treatment group. , indicates significant effect of treatment, p < 0.05 increased or decreased, respectively, for dexamethasone treated vs. control animals; + indicates significant sex difference p < 0.05 vs. females in the same treatment group.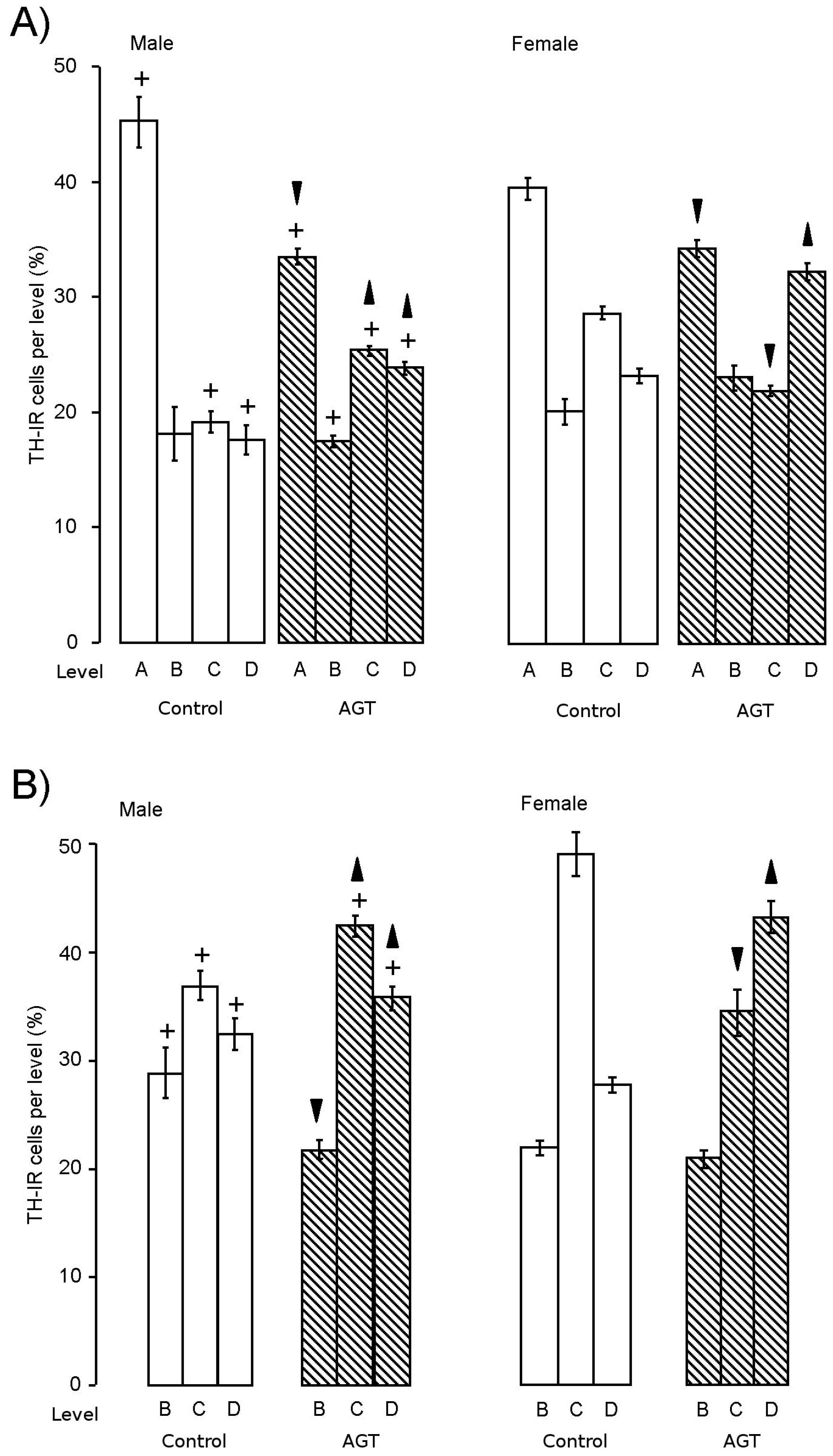
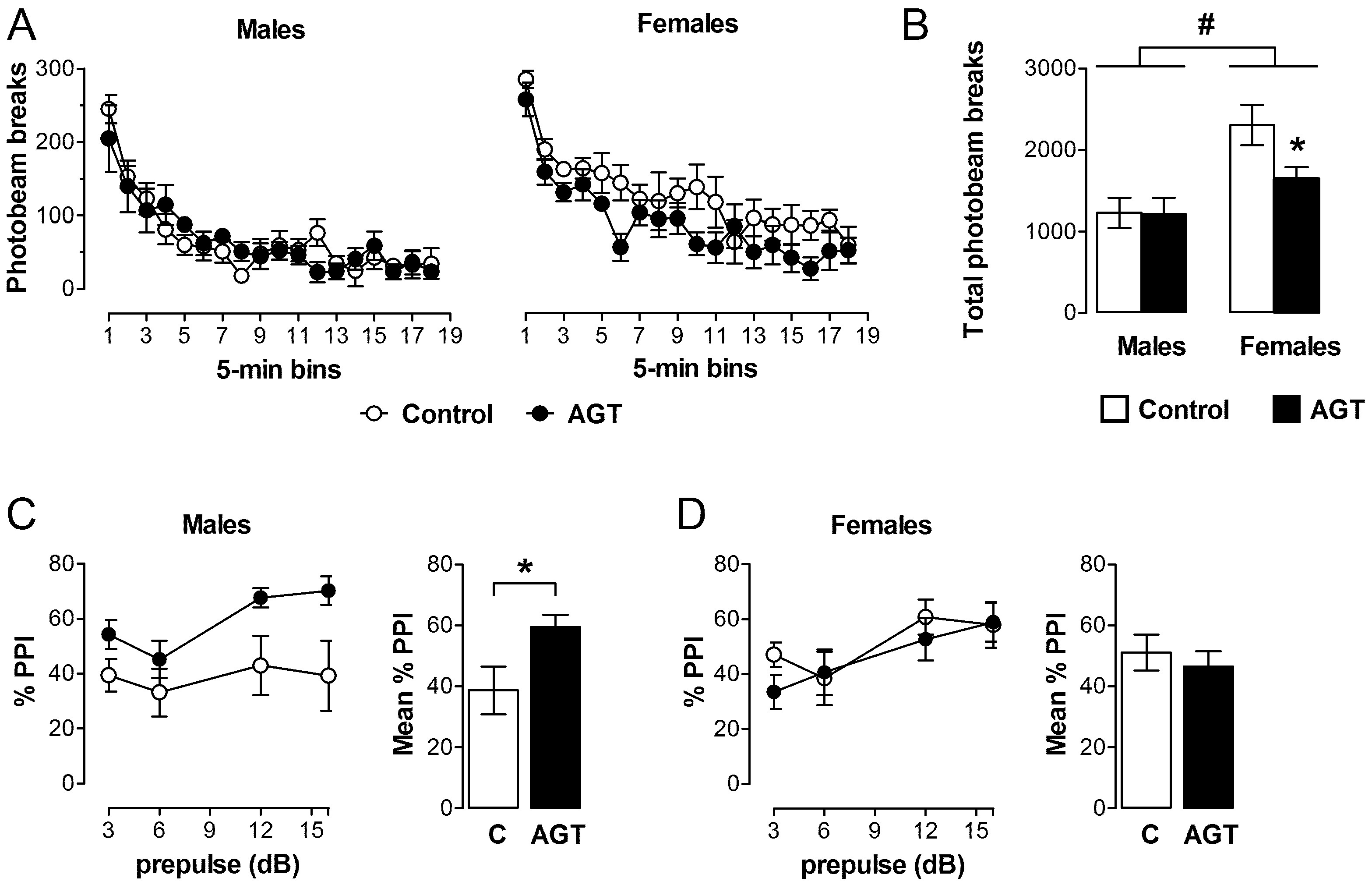
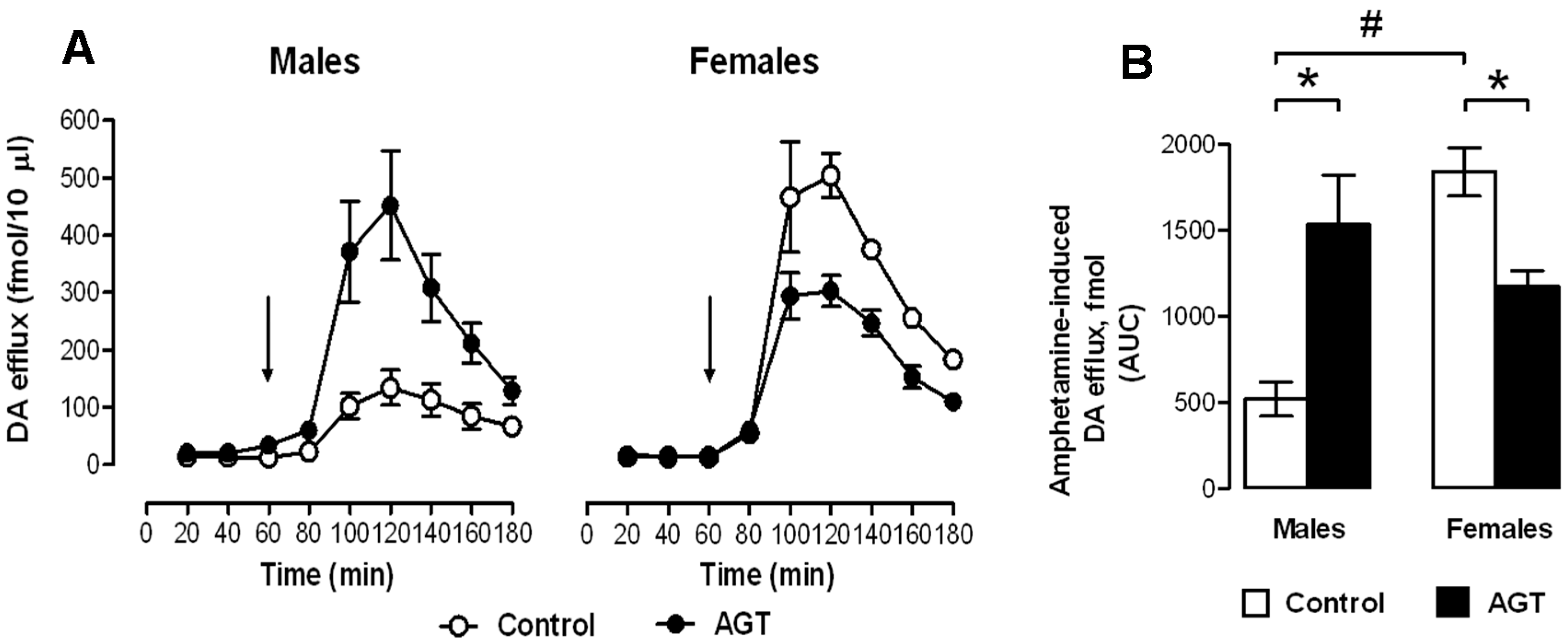
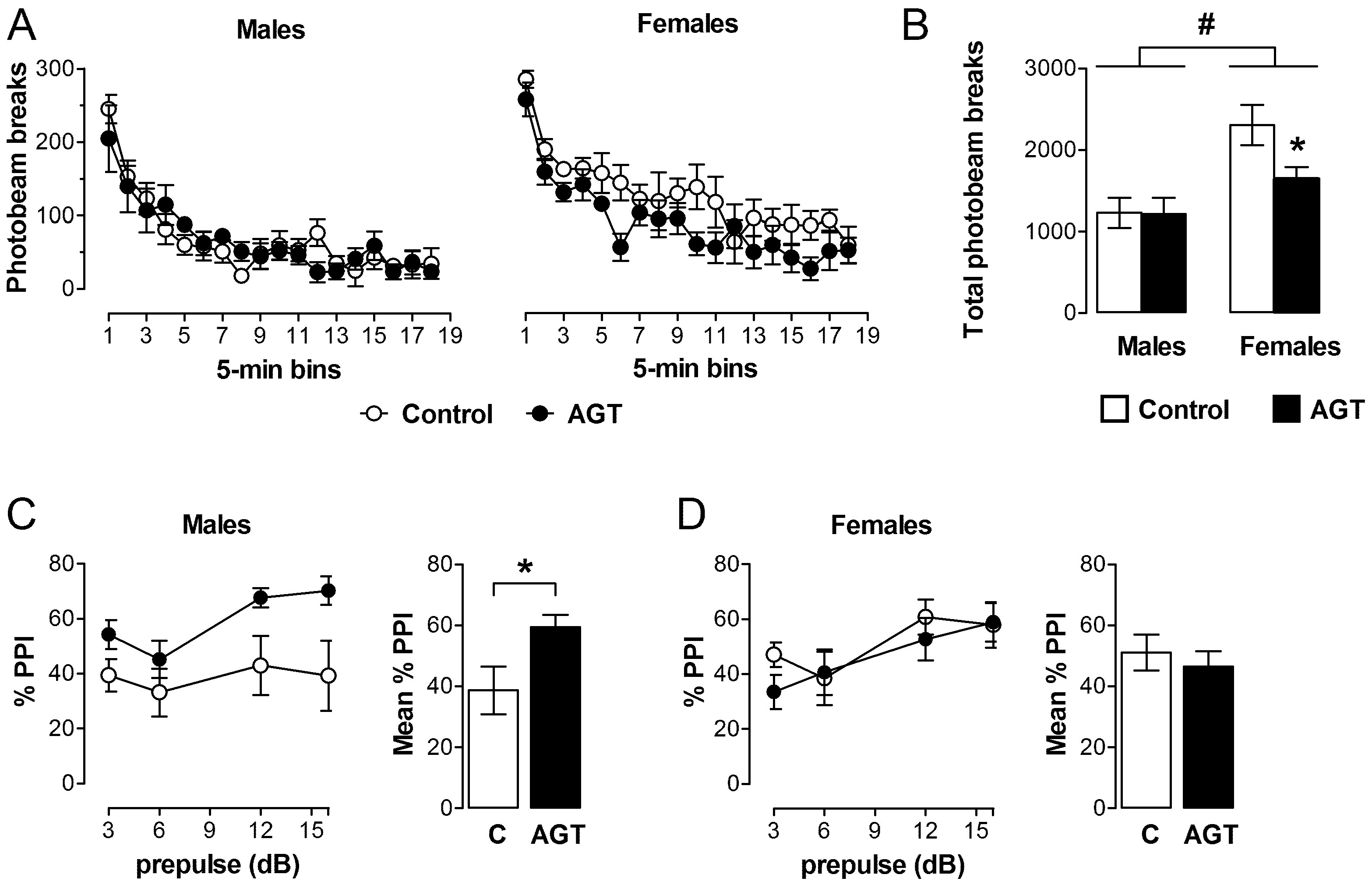
 0.5 µg/mL), and compared to the control offspring of dams receiving normal drinking water (
0.5 µg/mL), and compared to the control offspring of dams receiving normal drinking water (  ). Data are means ± SEM, n = 6 animals per treatment group. * Indicates a significant effect of treatment, p < 0.05 for dexamethasone treated vs. control animals.
0.5 µg/mL), and compared to the control offspring of dams receiving normal drinking water ( ). Data are means ± SEM, n = 6 animals per treatment group. * Indicates a significant effect of treatment, p < 0.05 for dexamethasone treated vs. control animals.
). Data are means ± SEM, n = 6 animals per treatment group. * Indicates a significant effect of treatment, p < 0.05 for dexamethasone treated vs. control animals.
0.5 µg/mL), and compared to the control offspring of dams receiving normal drinking water ( ). Data are means ± SEM, n = 6 animals per treatment group. * Indicates a significant effect of treatment, p < 0.05 for dexamethasone treated vs. control animals.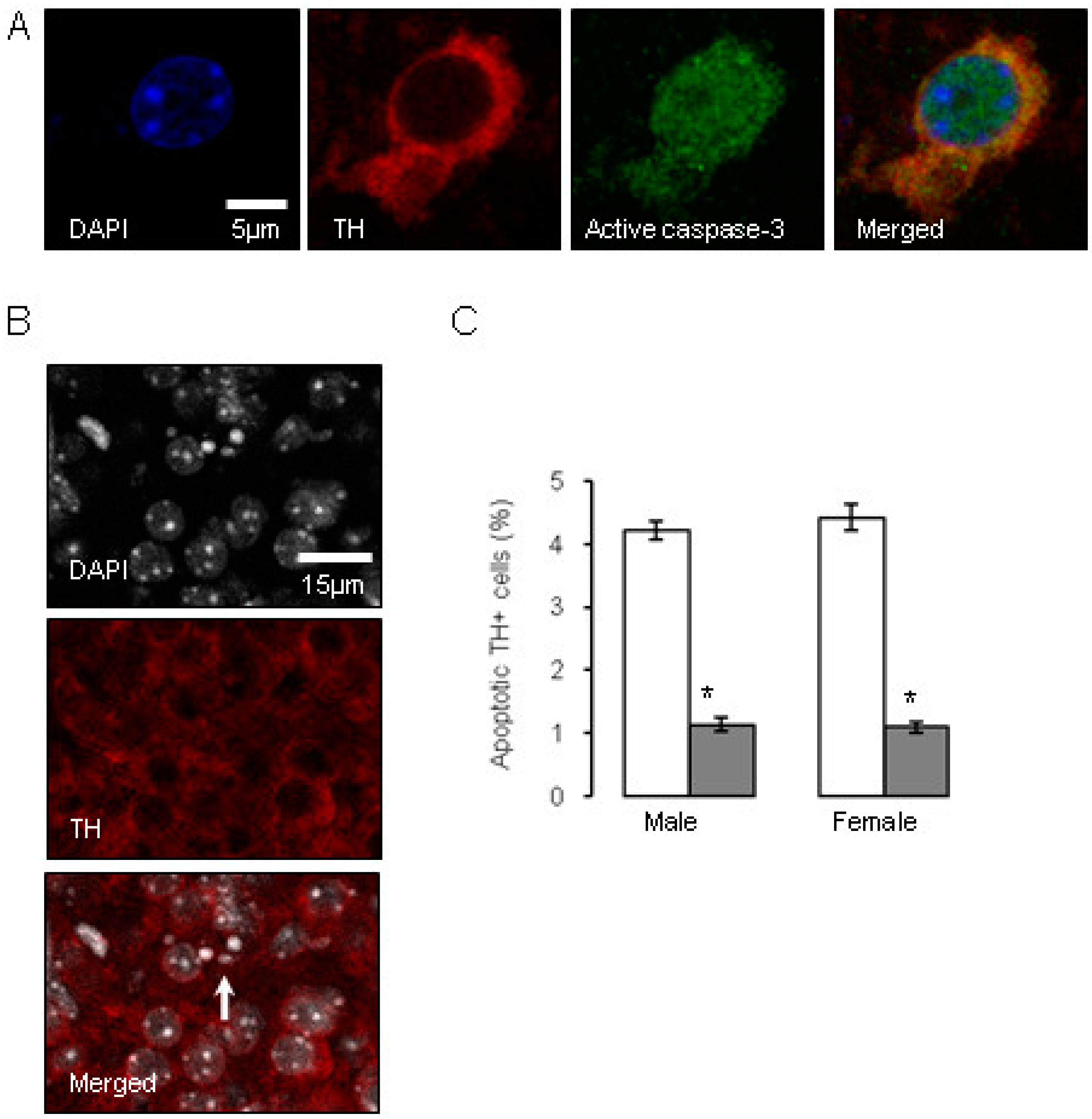
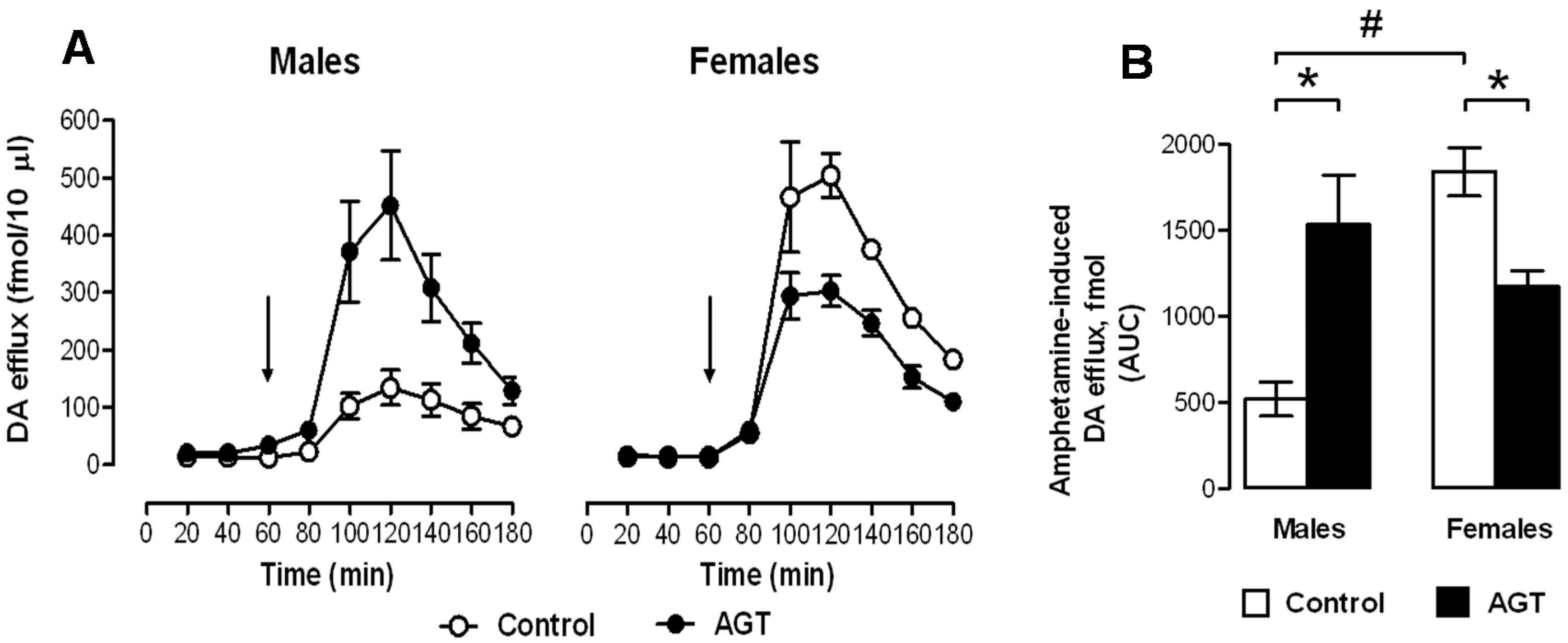

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Densities | Controls Males vs. Females | AGT Males (m) vs. Same Sex Controls | AGT Females (f) vs. Same Sex Controls | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CPu | NAc | CPu | Nac Core | Nac Shell | ILC | PLC | CPu | Nac Core | Nac Shell | ILC | PLC | |
| D1 | m > f | ns | ↓ | ns | ↓ | ns | ↓ | ↑ | ↑ | ns | ↑ | ↑ |
| D2 | ns | ns | ↑ | ↑ | ↑ | ns | ns | ↑ | ↑ | ↑ | ns | ns |
| DAT | ns | ns | ↑ | ↑ | ns | ns | ns | ↓ | ↓ | ↓ | ↓ | ↓ |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gillies, G.E.; Virdee, K.; Pienaar, I.; Al-Zaid, F.; Dalley, J.W. Enduring, Sexually Dimorphic Impact of In Utero Exposure to Elevated Levels of Glucocorticoids on Midbrain Dopaminergic Populations. Brain Sci. 2017, 7, 5. https://doi.org/10.3390/brainsci7010005
Gillies GE, Virdee K, Pienaar I, Al-Zaid F, Dalley JW. Enduring, Sexually Dimorphic Impact of In Utero Exposure to Elevated Levels of Glucocorticoids on Midbrain Dopaminergic Populations. Brain Sciences. 2017; 7(1):5. https://doi.org/10.3390/brainsci7010005
Chicago/Turabian StyleGillies, Glenda E., Kanwar Virdee, Ilse Pienaar, Felwah Al-Zaid, and Jeffrey W. Dalley. 2017. "Enduring, Sexually Dimorphic Impact of In Utero Exposure to Elevated Levels of Glucocorticoids on Midbrain Dopaminergic Populations" Brain Sciences 7, no. 1: 5. https://doi.org/10.3390/brainsci7010005
APA StyleGillies, G. E., Virdee, K., Pienaar, I., Al-Zaid, F., & Dalley, J. W. (2017). Enduring, Sexually Dimorphic Impact of In Utero Exposure to Elevated Levels of Glucocorticoids on Midbrain Dopaminergic Populations. Brain Sciences, 7(1), 5. https://doi.org/10.3390/brainsci7010005