A Translational Study on Acute Traumatic Brain Injury: High Incidence of Epileptiform Activity on Human and Rat Electrocorticograms and Histological Correlates in Rats

 , , and
, , and 
Abstract
1. Introduction
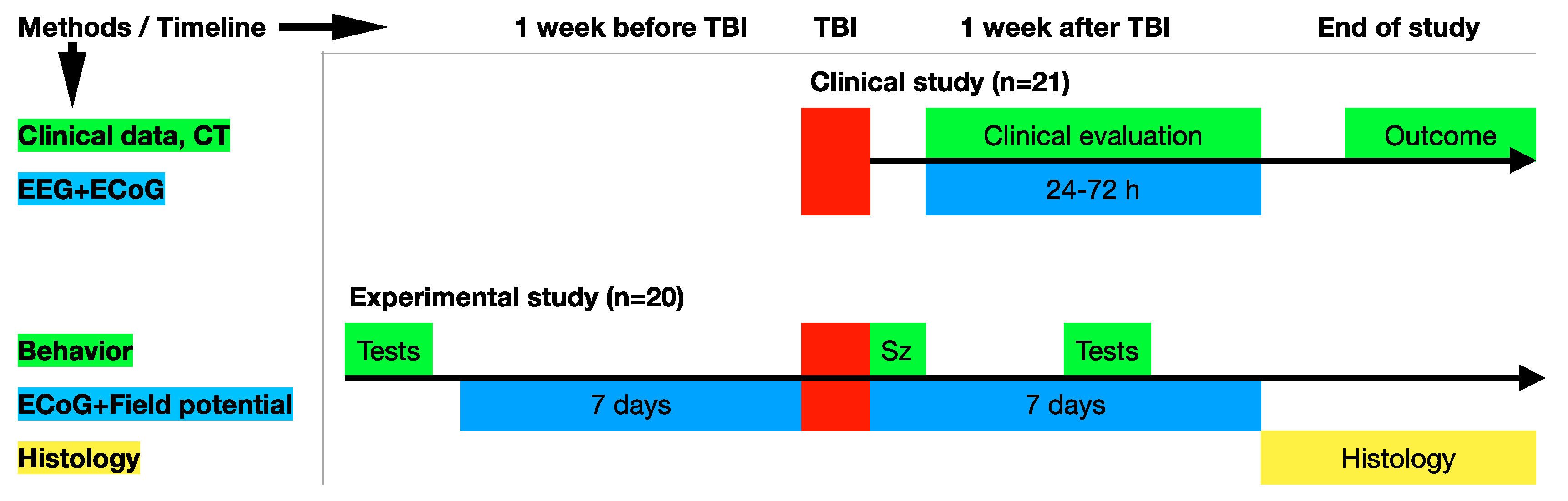
2. Materials and Methods
2.1. The Human Study
2.1.1. Group Description
2.1.2. Exclusion Criteria
2.1.3. Electroencephalograms (EEGs) and Electrocorticograms (ECoGs)
2.2. Animals
2.2.1. Electrode Implantation
2.2.2. TBI Induction
2.2.3. In Vivo Electrophysiology
2.2.4. Behavioral Testing
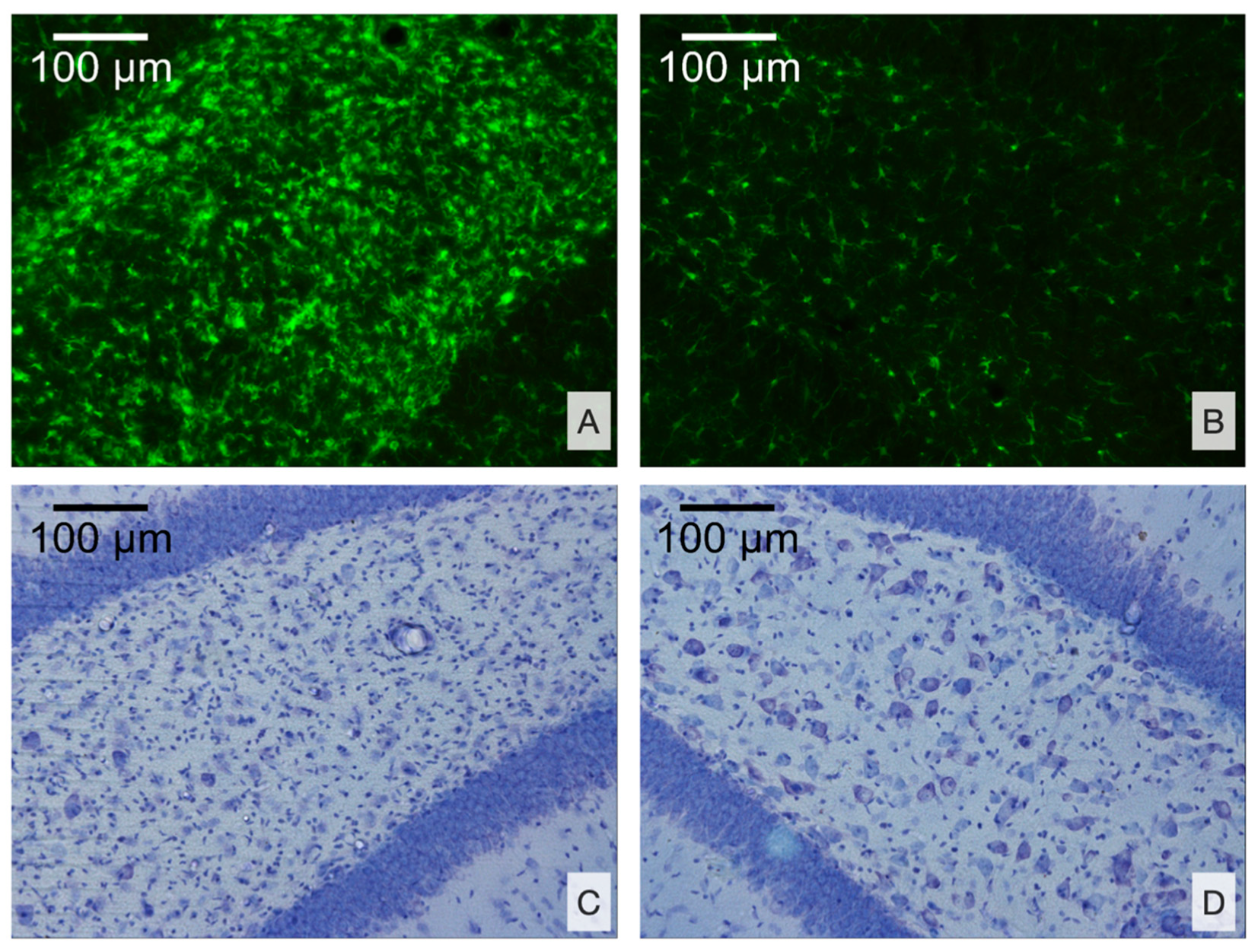
2.2.5. Histology and Immunohistochemistry
2.2.6. Morphometry
2.3. Statistical Analysis
3. Results
3.1. Clinical Study
3.2. Experimental Study
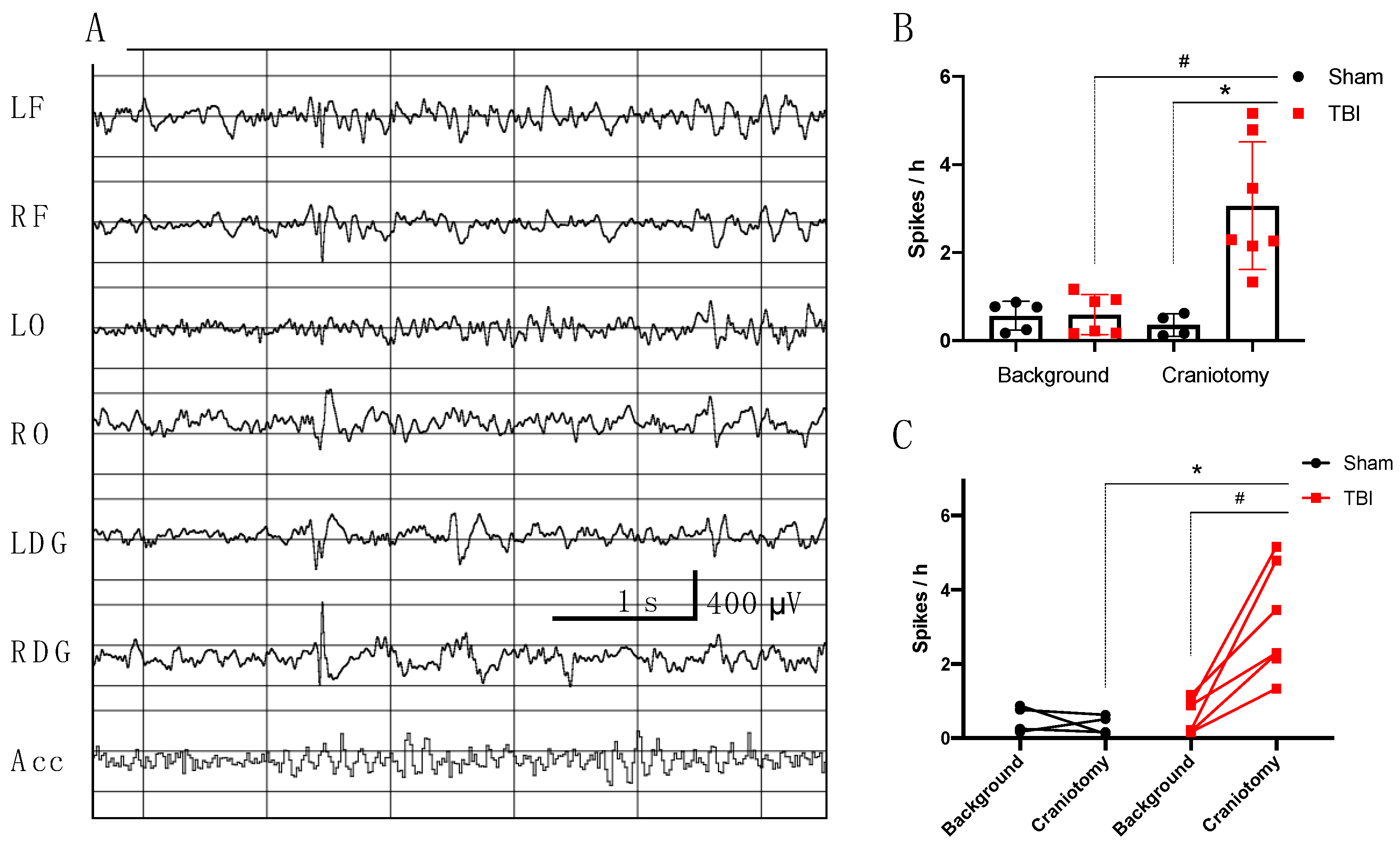
3.2.1. Electrophysiological Analysis in Rats
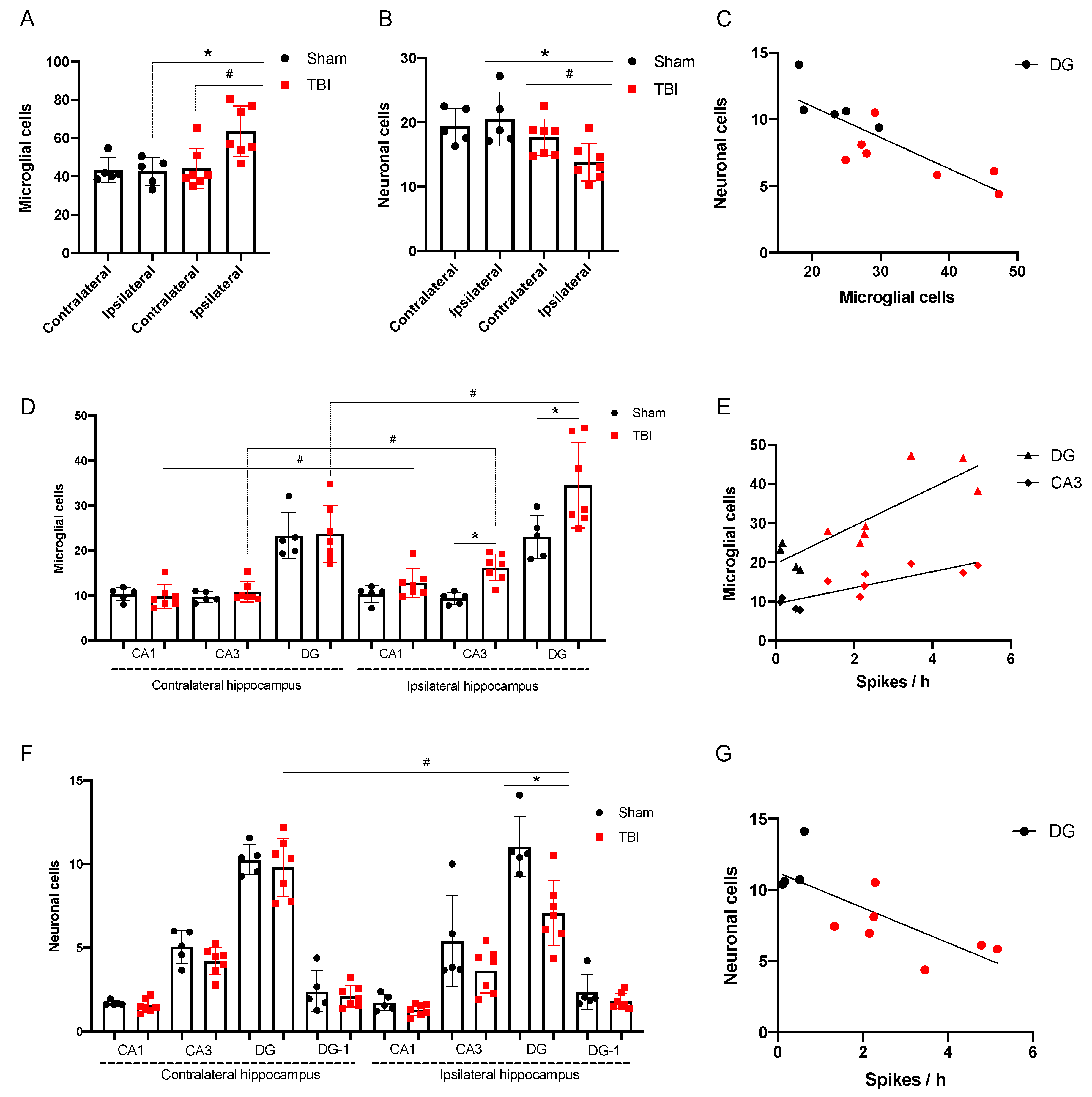
3.2.2. Morphological Analysis
3.2.3. Spike Occurrence and Hippocampal Damage
3.2.4. Behavior
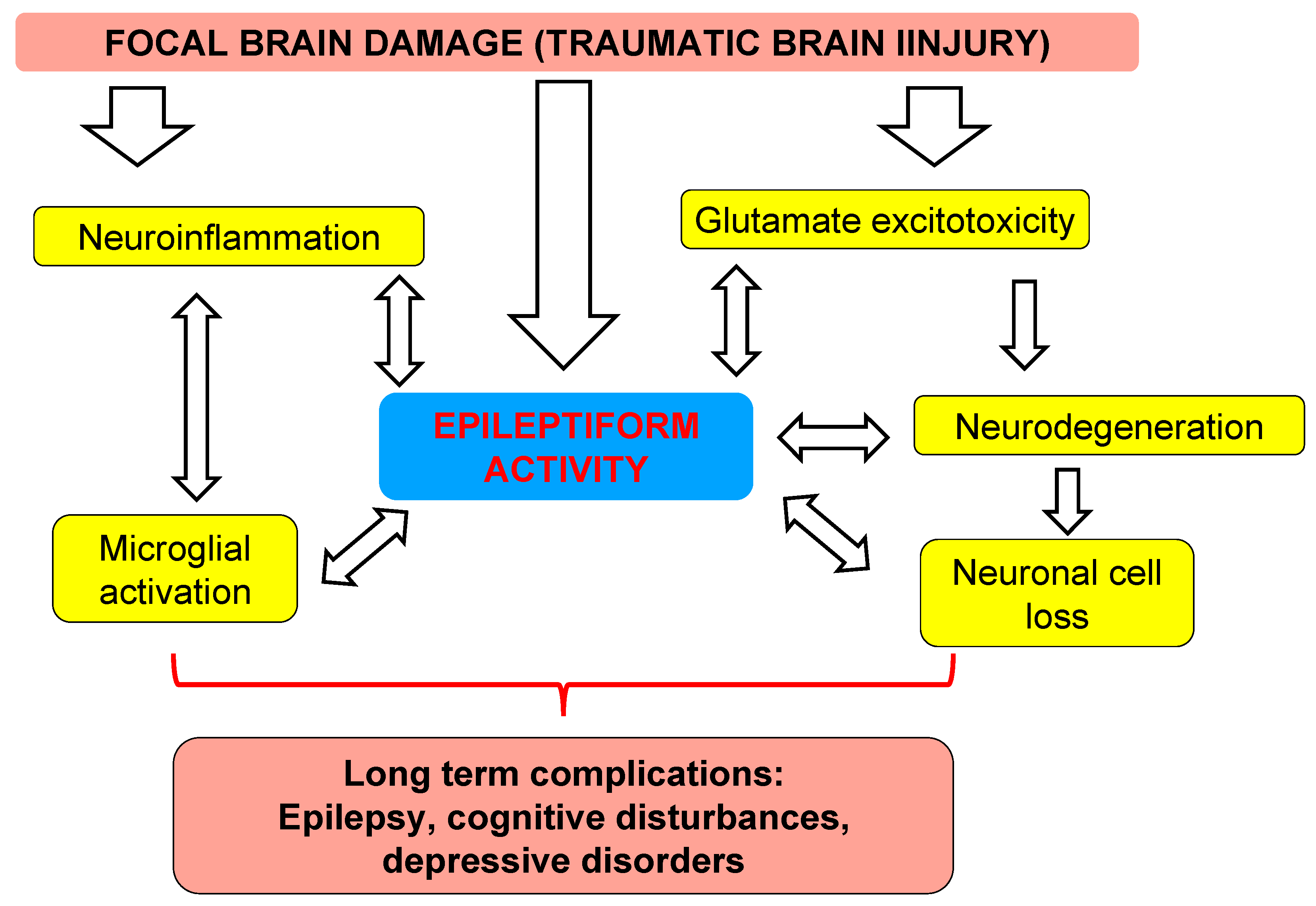
4. Discussion
4.1. Electrophysiological Characteristics of Acute TBI in Humans
4.2. Translational Links between Clinical Study and Animal Model
4.3. Distant Damage to the Hippocampus and Epileptiform Activity in Rats
4.4. Clinical Implication
5. Conclusions
- ECoG is more sensitive for epileptiform activity and periodic patterns as compared with scalp EEG.
- The incidence of epileptiform abnormalities is very high in patients subjected to surgical treatment after TBI.
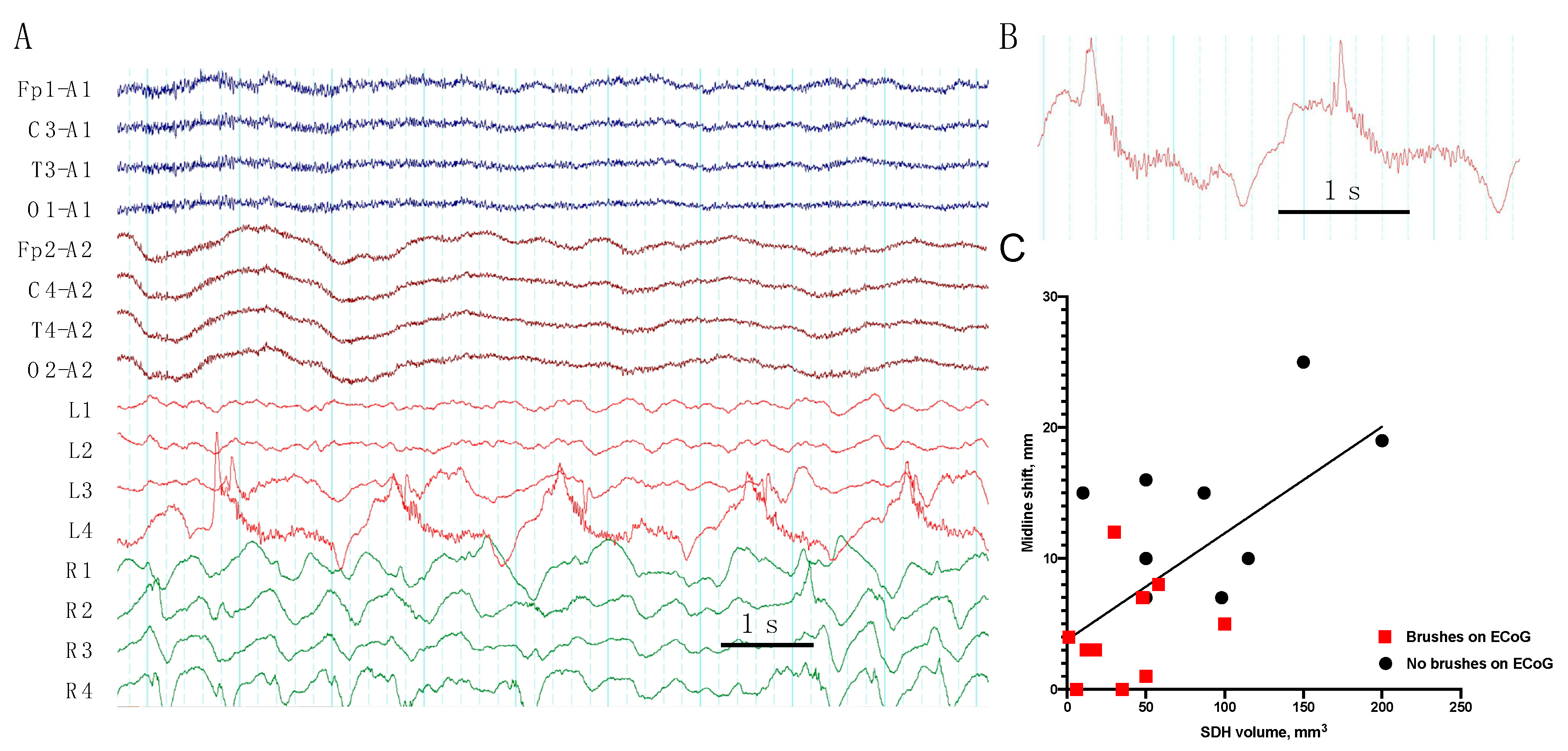
- We describe, for the first time, a “brush”-like pattern on RDA and PD on ECoG, associated with epileptiform activity, low grade midline shift, and subdural hematoma volume.
- Lateral fluid percussion brain injury in rats is valid for studying early mechanisms of post-traumatic pathology.
- Incidence of high amplitude epileptiform spikes is elevated seven days after FPI in rats with maximum amplitude in the ipsilateral hippocampus.
- Microglial cell density is elevated in the CA3 field and DG of the ipsilateral hippocampus and is associated with neuronal loss in the polymorph layer of the ipsilateral DG.
- The occurrence of epileptiform spikes correlates with microglial cell density and neuronal loss in the ipsilateral hippocampus of rat.
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Reilly, P. The impact of neurotrauma on society: An international perspective. In Progress in Brain Research; Elsevier: Amsterdam, The Netherlands, 2007; Volume 161, pp. 3–9. ISBN 9780444530172. [Google Scholar]
- Bramlett, H.M.; Dietrich, W.D. Long-Term Consequences of Traumatic Brain Injury: Current Status of Potential Mechanisms of Injury and Neurological Outcomes. J. Neurotrauma 2015, 32, 1834–1848. [Google Scholar] [CrossRef]
- Trinka, E.; Cock, H.; Hesdorffer, D.; Rossetti, A.O.; Scheffer, I.E.; Shinnar, S.; Shorvon, S.; Lowenstein, D.H. A definition and classification of status epilepticus–Report of the ILAE Task Force on Classification of Status Epilepticus. Epilepsia 2015, 56, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.X.; Ray, A.; Hawes-Ebersole, S.; Ebersole, J.S. Intracranial EEG Substrates of Scalp EEG Interictal Spikes. Epilepsia 2005, 46, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Pitkänen, A.; Kyyriäinen, J.; Andrade, P.; Pasanen, L.; Ndode-Ekane, X.E. Epilepsy After Traumatic Brain Injury. In Models of Seizures and Epilepsy; Elsevier: Amsterdam, The Netherlands, 2017; pp. 661–681. ISBN 9780128040669. [Google Scholar]
- Hirsch, L.J.; LaRoche, S.M.; Gaspard, N.; Gerard, E.; Svoronos, A.; Herman, S.T.; Mani, R.; Arif, H.; Jette, N.; Minazad, Y.; et al. American Clinical Neurophysiology Society’s Standardized Critical Care EEG Terminology. J. Clin. Neurophysiol. 2013, 30, 1–27. [Google Scholar] [CrossRef]
- Hashiguchi, K.; Morioka, T.; Yoshida, F.; Miyagi, Y.; Nagata, S.; Sakata, A.; Sasaki, T. Correlation between scalp-recorded electroencephalographic and electrocorticographic activities during ictal period. Seizure 2007, 16, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Ronne-Engstrom, E.; Winkler, T. Continuous EEG monitoring in patients with traumatic brain injury reveals a high incidence of epileptiform activity. Acta Neurol. Scand. 2006, 114, 47–53. [Google Scholar] [CrossRef]
- Perucca, P.; Smith, G.; Santana-Gomez, C.; Bragin, A.; Staba, R. Electrophysiological biomarkers of epileptogenicity after traumatic brain injury. Neurobiol. Dis. 2019, 123, 69–74. [Google Scholar] [CrossRef]
- Jennett, B.; Sande, J. EEG Prediction of Post-Traumatic Epilepsy. Epilepsia 1975, 16, 251–256. [Google Scholar] [CrossRef]
- Frey, L.C. Epidemiology of posttraumatic epilepsy: A critical review. Epilepsia 2003, 44, 11–17. [Google Scholar] [CrossRef]
- Kim, J.A.; Boyle, E.J.; Wu, A.C.; Cole, A.J.; Staley, K.J.; Zafar, S.; Cash, S.S.; Westover, M.B. Epileptiform activity in traumatic brain injury predicts post-traumatic epilepsy. Ann. Neurol. 2018, 83, 858–862. [Google Scholar] [CrossRef]
- Veciana, M.; Becerra, J.L.; Fossas, P.; Muriana, D.; Sansa, G.; Santamarina, E.; Gaig, C.; Carreño, M.; Molins, A.; Escofet, C.; et al. EEG extreme delta brush: An ictal pattern in patients with anti-NMDA receptor encephalitis. Epilepsy Behav. 2015, 49, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Baykan, B.; Gungor Tuncer, O.; Vanli-Yavuz, E.N.; Baysal Kirac, L.; Gundogdu, G.; Bebek, N.; Gurses, C.; Altindag, E.; Tuzun, E. Delta Brush Pattern Is Not Unique to NMDAR Encephalitis: Evaluation of Two Independent Long-Term EEG Cohorts. Clin. EEG Neurosci. 2018, 49, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Perucca, P.; Dubeau, F.; Gotman, J. Intracranial electroencephalographic seizure-onset patterns: Effect of underlying pathology. Brain 2014, 137, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Lagarde, S.; Buzori, S.; Trebuchon, A.; Carron, R.; Scavarda, D.; Milh, M.; McGonigal, A.; Bartolomei, F. The repertoire of seizure onset patterns in human focal epilepsies: Determinants and prognostic values. Epilepsia 2019, 60, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Ferrari-Marinho, T.; Perucca, P.; Dubeau, F.; Gotman, J. Intracranial EEG seizure onset-patterns correlate with high-frequency oscillations in patients with drug-resistant epilepsy. Epilepsy Res. 2016, 127, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Ruiz, A.; Vlachy, J.; Lee, J.W.; Gilmore, E.J.; Ayer, T.; Haider, H.A.; Gaspard, N.; Ehrenberg, J.A.; Tolchin, B.; Fantaneanu, T.A.; et al. Association of Periodic and Rhythmic Electroencephalographic Patterns With Seizures in Critically Ill Patients. JAMA Neurol. 2017, 74, 181. [Google Scholar] [CrossRef]
- Bragin, A.; Li, L.; Almajano, J.; Alvarado-Rojas, C.; Reid, A.Y.; Staba, R.J.; Engel, J. Pathologic electrographic changes after experimental traumatic brain injury. Epilepsia 2016, 57, 735–745. [Google Scholar] [CrossRef]
- Dumoulin, S.O.; Fracasso, A.; van der Zwaag, W.; Siero, J.C.W.; Petridou, N. Ultra-high field MRI: Advancing systems neuroscience towards mesoscopic human brain function. Neuroimage 2018, 168, 345–357. [Google Scholar] [CrossRef]
- Ovsepian, S.V.; Olefir, I.; Ntziachristos, V. Advances in Optoacoustic Neurotomography of Animal Models. Trends Biotechnol. 2019, 37, 1315–1326. [Google Scholar] [CrossRef]
- Ovsepian, S.V.; Olefir, I.; Westmeyer, G.; Razansky, D.; Ntziachristos, V. Pushing the Boundaries of Neuroimaging with Optoacoustics. Neuron 2017, 96, 966–988. [Google Scholar] [CrossRef]
- Buzsáki, G.; Anastassiou, C.A.; Koch, C. The origin of extracellular fields and currents—EEG, ECoG, FPI and spikes. Nat. Rev. Neurosci. 2012, 13, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.; Fujioka, W.; Lifshitz, J.; Crockett, D.P.; Thakker-Varia, S. Lateral Fluid Percussion: Model of Traumatic Brain Injury in Mice. JoVE 2011, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kabadi, S.V.; Hilton, G.D.; Stoica, B.A.; Zapple, D.N.; Faden, A.I. Fluid-percussion-induced traumatic brain injury model in rats. Nat. Protoc. 2010, 5, 1552–1563. [Google Scholar] [CrossRef] [PubMed]
- Pitkänen, A.; Immonen, R. Epilepsy Related to Traumatic Brain Injury. Neurotherapeutics 2014, 11, 286–296. [Google Scholar] [CrossRef]
- D’Ambrosio, R.; Fender, J.S.; Fairbanks, J.P.; Simon, E.A.; Born, D.E.; Doyle, D.L.; Miller, J.W. Progression from frontal-parietal to mesial-temporal epilepsy after fluid percussion injury in the rat. Brain 2005, 128, 174–188. [Google Scholar] [CrossRef]
- Santana-Gomez, C.; Andrade, P.; Hudson, M.R.; Paananen, T.; Ciszek, R.; Smith, G.; Ali, I.; Rundle, B.K.; Ndode-Ekane, X.E.; Casillas-Espinosa, P.M.; et al. Harmonization of pipeline for detection of HFOs in a rat model of post-traumatic epilepsy in preclinical multicenter study on post-traumatic epileptogenesis. Epilepsy Res. 2019, 156, 106110. [Google Scholar] [CrossRef]
- Komoltsev, I.G.; Frankevich, S.O.; Shirobokova, N.I.; Volkova, A.A.; Levshina, I.P.; Novikova, M.R.; Manolova, A.O.; Gulyaeva, N.V. Differential early effects of traumatic brain injury on spike-wave discharges in Sprague-Dawley rats. Neurosci. Res. 2020. [Google Scholar] [CrossRef]
- Aronica, E.; Mühlebner, A.; van Vliet, E.A.; Gorter, J.A. Characterization of Pathology. In Models of Seizures and Epilepsy, 2nd ed.; Academic Press: Cambridge, MA, USA, 2017; ISBN 9780128040669. [Google Scholar]
- Gulyaeva, N.V. Aberrant neurogenesis in adult epileptic brain: Compensatory or pathologic. Neurochem. J. 2010, 4, 84–89. [Google Scholar] [CrossRef]
- Hester, M.S.; Danzer, S.C. Accumulation of Abnormal Adult-Generated Hippocampal Granule Cells Predicts Seizure Frequency and Severity. J. Neurosci. 2013, 33, 8926–8936. [Google Scholar] [CrossRef]
- Lowenstein, D.H.; Thomas, M.J.; Smith, D.H.; McIntosh, T.K. Selective vulnerability of dentate hilar neurons following traumatic brain injury: A potential mechanistic link between head trauma and disorders of the hippocampus. J. Neurosci. 1992, 12, 4846–4853. [Google Scholar] [CrossRef]
- Grady, M.S.; Charleston, J.S.; Maris, D.; Witgen, B.M.; Lifshitz, J. Neuronal and glial cell number in the hippocampus after experimental traumatic brain injury: Analysis by stereological estimation. J. Neurotrauma 2003, 20, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Huusko, N.; Römer, C.; Ndode-Ekane, X.E.; Lukasiuk, K.; Pitkänen, A. Loss of hippocampal interneurons and epileptogenesis: A comparison of two animal models of acquired epilepsy. Brain Struct. Funct. 2013, 220, 153–191. [Google Scholar] [CrossRef] [PubMed]
- Ruth, R.E. Kainic-acid lesions of hippocampus produced iontophoretically: The problem of distant damage. Exp. Neurol. 1982, 76, 508–527. [Google Scholar] [CrossRef]
- Bagetta, G.; Iannone, M.; Palma, E.; Nisticò, G.; Dolly, J.O. N-methyl-d-aspartate and non-N-methyl-d-aspartate receptors mediate seizures and CA1 hippocampal damage induced by dendrotoxin-K in rats. Neuroscience 1996, 71, 613–624. [Google Scholar] [CrossRef]
- Becker, A.; Tiedge, A.; Grecksch, G.A. Diazepam—Its effects on the development of pentylenetetrazol kindling, related learning impairments, and neuronal cell loss. Pharmacol. Res. 1997, 35, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Aniol, V.A.; Stepanichev, M.; Lazareva, N.A.; Gulyaeva, N.V. An early decrease in cell proliferation after pentylenetetrazole-induced seizures. Epilepsy Behav. 2011, 22, 433–441. [Google Scholar] [CrossRef]
- Thompson, H.J.; Lifshitz, J.; Marklund, N.; Grady, M.S.; Graham, D.I.; Hovda, D.A.; McIntosh, T.K. Lateral fluid percussion brain injury: A 15-year review and evaluation. J. Neurotrauma 2005, 22, 42–75. [Google Scholar] [CrossRef]
- Hayes, R.L.; Jenkins, L.W.; Lyeth, B.G. Neurotransmitter-mediated mechanisms of traumatic brain injury: Acetylcholine and excitatory amino acids. J. Neurotrauma 1992, 9, S173–S187. [Google Scholar]
- Dubreuil, C.I.; Marklund, N.; Deschamps, K.; McIntosh, T.K.; McKerracher, L. Activation of Rho after traumatic brain injury and seizure in rats. Exp. Neurol. 2006, 198, 361–369. [Google Scholar] [CrossRef]
- Rink, A.; Fung, K.; Trojanowski, J.Q.; Lee, V.M.; Neugebauer, E.; Mcintosh, T.K. Evidence of Apoptotic Cell Death after Experimental Traumatic Brain Injury in the Rat. Am. J. Pathol. 1995, 63, 305–309. [Google Scholar] [CrossRef]
- Raghupathi, R. Cell death mechanisms following traumatic brain injury. In Brain Pathology; International Society of Neuropathology: Zürich, Switzerland, 2004; Volume 14, pp. 215–222. [Google Scholar] [CrossRef]
- Lerner-Natoli, M.; Rondouin, G.; Belaidi, M.; Baldy-Moulinier, M.; Kamenka, J.M. N-[1-(2-Thienyl)cyclohexyl]-piperidine (TCP) does not block kainic acid-induced status epilepticus but reduces secondary hippocampal damage. Neurosci. Lett. 1991, 122, 174–178. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Tremblay, E.; Ottersen, O.P.; Meldrum, B.S. The role of epileptic activity in hippocampal and “remote” cerebral lesions induced by kainic acid. Brain Res. 1980, 191, 79–97. [Google Scholar] [CrossRef]
- Aniol, V.A.; Ivanova-Dyatlova, A.Y.; Keren, O.; Guekht, A.B.; Sarne, Y.; Gulyaeva, N.V. A single pentylenetetrazole-induced clonic-tonic seizure episode is accompanied by a slowly developing cognitive decline in rats. Epilepsy Behav. 2013, 26, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Piccenna, L.; Shears, G.; O’Brien, T.J. Management of post-traumatic epilepsy: An evidence review over the last 5 years and future directions. Epilepsia Open 2017, 2, 123–144. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J. The Epidemiology of Posttraumatic Epilepsy. Semin. Neurol. 2015, 35, 218–222. [Google Scholar] [CrossRef]
- Chiu, C.; Liao, Y.; Yang, L.; Wang, J.; Tweedie, D. Neuroinflammation in animal models of traumatic brain injury. J. Neurosci. Methods 2016, 272, 38–49. [Google Scholar] [CrossRef]
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef]
- Gulyaeva, N.V. Functional Neurochemistry of the Ventral and Dorsal Hippocampus: Stress, Depression, Dementia and Remote Hippocampal Damage. Neurochem. Res. 2019, 44, 1306–1322. [Google Scholar] [CrossRef]
- Gulyaeva, N.V. Ventral hippocampus, Stress and phychopathology: Translational implications. Neurochem. J. 2015, 9, 85–94. [Google Scholar] [CrossRef]
- Vezzani, A.; Auvin, S.; Ravizza, T.; Aronica, E. Glia-neuronal interactions in ictogenesis and epileptogenesis: Role of inflammatory mediators. In Jasper’s Basic Mechanisms of the Epilepsies [Internet], 4th ed.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012; pp. 1–21. [Google Scholar]
- Gurkoff, G.G.; Gahan, J.D.; Ghiasvand, R.T.; Hunsaker, M.R.; Van, K.; Feng, J.-F.; Shahlaie, K.; Berman, R.F.; Lyeth, B.G.; Folkerts, M.M. Evaluation of Metric, Topological, and Temporal Ordering Memory Tasks after Lateral Fluid Percussion Injury. J. Neurotrauma 2013, 30, 292–300. [Google Scholar] [CrossRef]
- Dixon, C.E.; Kochanek, P.M.; Yan, H.Q.; Schiding, J.K.; Griffith, R.G.; Baum, E.; Marion, D.W.; DeKosky, S.T. One-Year Study of Spatial Memory Performance, Brain Morphology, and Cholinergic Markers After Moderate Controlled Cortical Impact in Rats. J. Neurotrauma 1999, 16, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.C.; Cardamone, L.; Williams, J.P.; Salzberg, M.R.; Myers, D.; O’Brien, T.J. Experimental traumatic brain injury induces a pervasive hyperanxious phenotype in rats. J. Neurotrauma 2008, 25, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, B.S. Prolonged Epileptic Seizures in Primates. Arch. Neurol. 1973, 28, 10. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, D.H.; Bleck, T.; Macdonald, R.L. It’s Time to Revise the Definition of Status Epilepticus. Epilepsia 1999, 40, 120–122. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | ED | PD | RDA | ES | NCSE | Injury (CT) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| sEEG | ECoG | sEEG | ECoG | sEEG | ECoG | sEEG | ECoG | |||
| 1 | frequent SW | PD + brushes | RDA | EDH, SDH (Multiple) | ||||||
| 2 | RDA + brushes | EDH (Frontal), SDH (Temporal) | ||||||||
| 3 | Contusion (Frontal), SDH (Multiple), SAH | |||||||||
| 4 | PD + brushes | RDA + brushes | Contusion (Frontal), SDH (Multiple), SAH | |||||||
| 5 | abundant ShW | Contusion (Temporal), EDH, SDH (Multiple), SAH | ||||||||
| 6 | occasion SW | RDA | cycl. Sz | cycl. Sz | Def | Contusion, EDH, SDH (Multiple), SAH | ||||
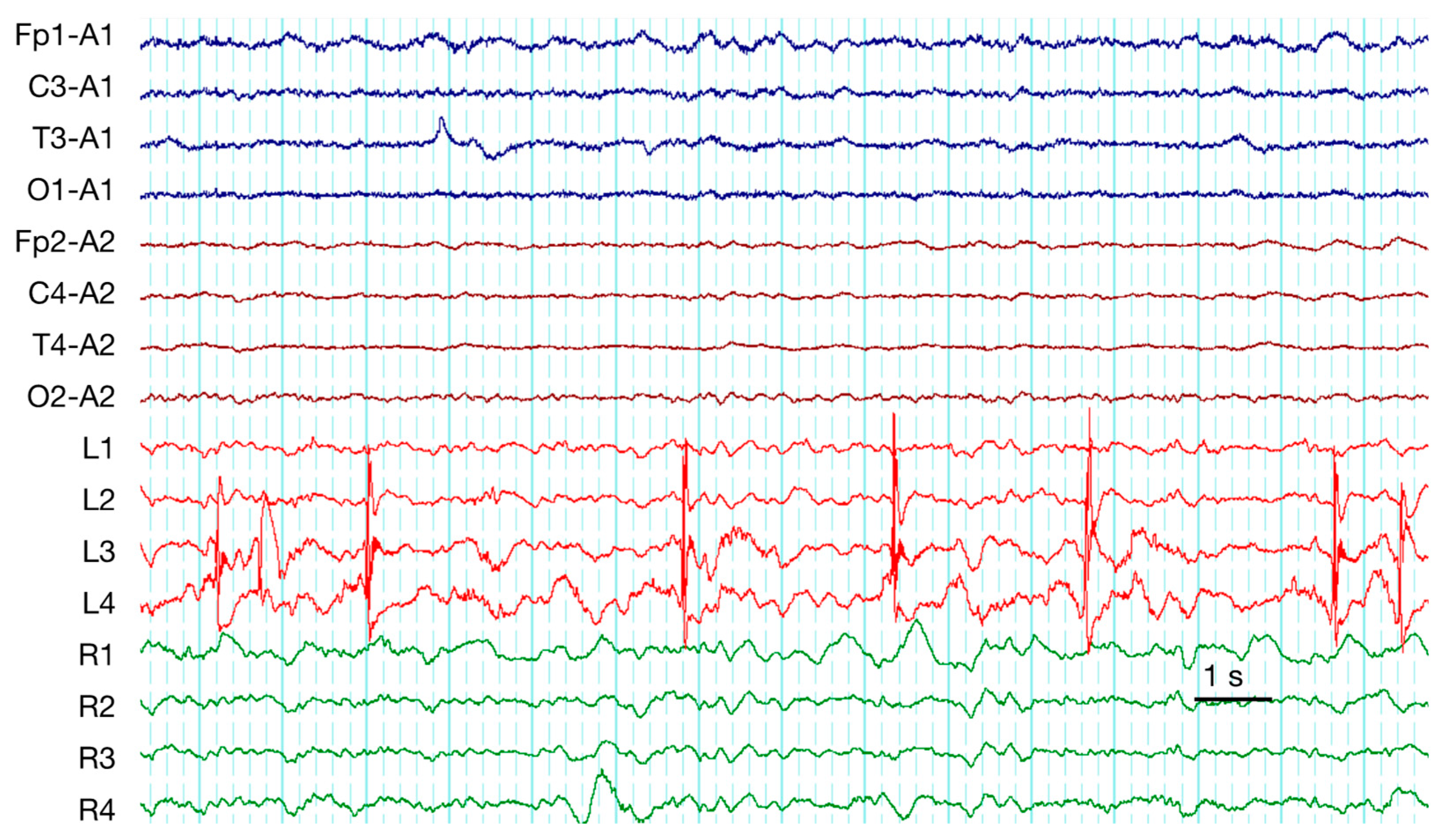
| 7 | occasion SW (Figure 2) | PD + brushes | RDA + F | RDA + brushes | Pos | Contusion, EDH (Multiple), SAH | ||||
| 8 | rare SW | rare S | PD | RDA | Contusion (Multiple), SAH | |||||
| 9 | occasion SW | PD + brushes (Figure 3) | RDA | RDA + brushes | cycl. Sz | cycl. Sz | Def | Contusion, SDH (Multiple), SAH | ||
| 10 | rare Sh | RDA+S | RDA + brushes | Pos | Contusion (Frontal), SDH (Multiple), SAH | |||||
| 11 | occasion SW | occasion S | RDA | Sz | SDH (Multiple), SAH | |||||
| 12 | rare S | rare SW | PD + brushes | Contusion, EDH, SDH (Multiple) | ||||||
| 13 | frequent SW | PD | PD + brushes | SDH (Multiple), SAH | ||||||
| 14 | frequent runs SW | RDA | RDA + brushes | Sz | Sz | SDH (Multiple), SAH | ||||
| 15 | rare SW | frequent runs S | PD | PD | SDH (Multiple) | |||||
| 16 | rare SW | Sz | Def | DAI, SDH (Multiple), Ventricular hemorrage | ||||||
| 17 | abundant SW | cycl. Sz | Contusion (Temporal), SDH (Parietal), SAH | |||||||
| 18 | rare SW | frequent runs S | SDH (Parietal), SAH | |||||||
| 19 | frequent SW | PD + brushes | RDA | Contusion, DAI, SDH (Multiple), SAH | ||||||
| 20 | rare runs S | RDA + F | RDA + brushes | Contusion, DAI, SDH (Multiple), SAH | ||||||
| 21 | n/a | rare S | n/a | PD | n/a | n/a | Sz | Contusion, SDH (Multiple) | ||
| Proportion * | 7/20 vs. 16/21 (p = 0.012) | 2/20 vs. 10/21 (p = 0.015) | 7/20 vs. 10/21 (p = 0.53) | 4/20 vs. 6/21 (p = 0.48) | ||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komoltsev, I.G.; Sinkin, M.V.; Volkova, A.A.; Smirnova, E.A.; Novikova, M.R.; Kordonskaya, O.O.; Talypov, A.E.; Guekht, A.B.; Krylov, V.V.; Gulyaeva, N.V. A Translational Study on Acute Traumatic Brain Injury: High Incidence of Epileptiform Activity on Human and Rat Electrocorticograms and Histological Correlates in Rats. Brain Sci. 2020, 10, 570. https://doi.org/10.3390/brainsci10090570
Komoltsev IG, Sinkin MV, Volkova AA, Smirnova EA, Novikova MR, Kordonskaya OO, Talypov AE, Guekht AB, Krylov VV, Gulyaeva NV. A Translational Study on Acute Traumatic Brain Injury: High Incidence of Epileptiform Activity on Human and Rat Electrocorticograms and Histological Correlates in Rats. Brain Sciences. 2020; 10(9):570. https://doi.org/10.3390/brainsci10090570
Chicago/Turabian StyleKomoltsev, Ilia G., Mikhail V. Sinkin, Aleksandra A. Volkova, Elizaveta A. Smirnova, Margarita R. Novikova, Olga O. Kordonskaya, Alexander E. Talypov, Alla B. Guekht, Vladimir V. Krylov, and Natalia V. Gulyaeva. 2020. "A Translational Study on Acute Traumatic Brain Injury: High Incidence of Epileptiform Activity on Human and Rat Electrocorticograms and Histological Correlates in Rats" Brain Sciences 10, no. 9: 570. https://doi.org/10.3390/brainsci10090570
APA StyleKomoltsev, I. G., Sinkin, M. V., Volkova, A. A., Smirnova, E. A., Novikova, M. R., Kordonskaya, O. O., Talypov, A. E., Guekht, A. B., Krylov, V. V., & Gulyaeva, N. V. (2020). A Translational Study on Acute Traumatic Brain Injury: High Incidence of Epileptiform Activity on Human and Rat Electrocorticograms and Histological Correlates in Rats. Brain Sciences, 10(9), 570. https://doi.org/10.3390/brainsci10090570