Hyperpolarised 1H–13C Benchtop NMR Spectroscopy


Abstract
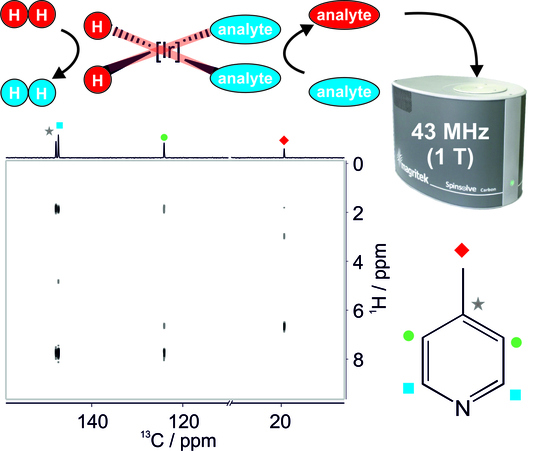
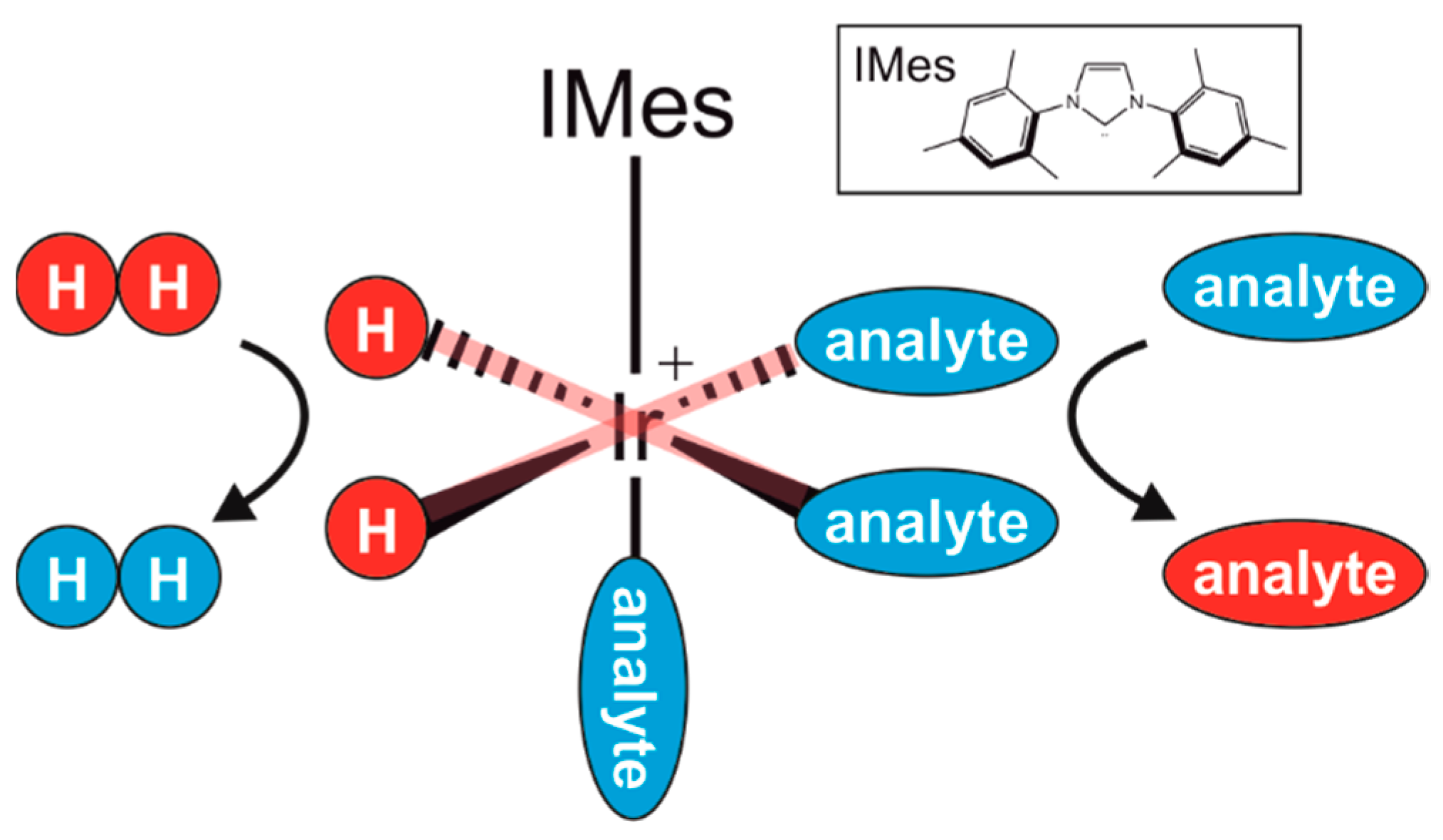
:
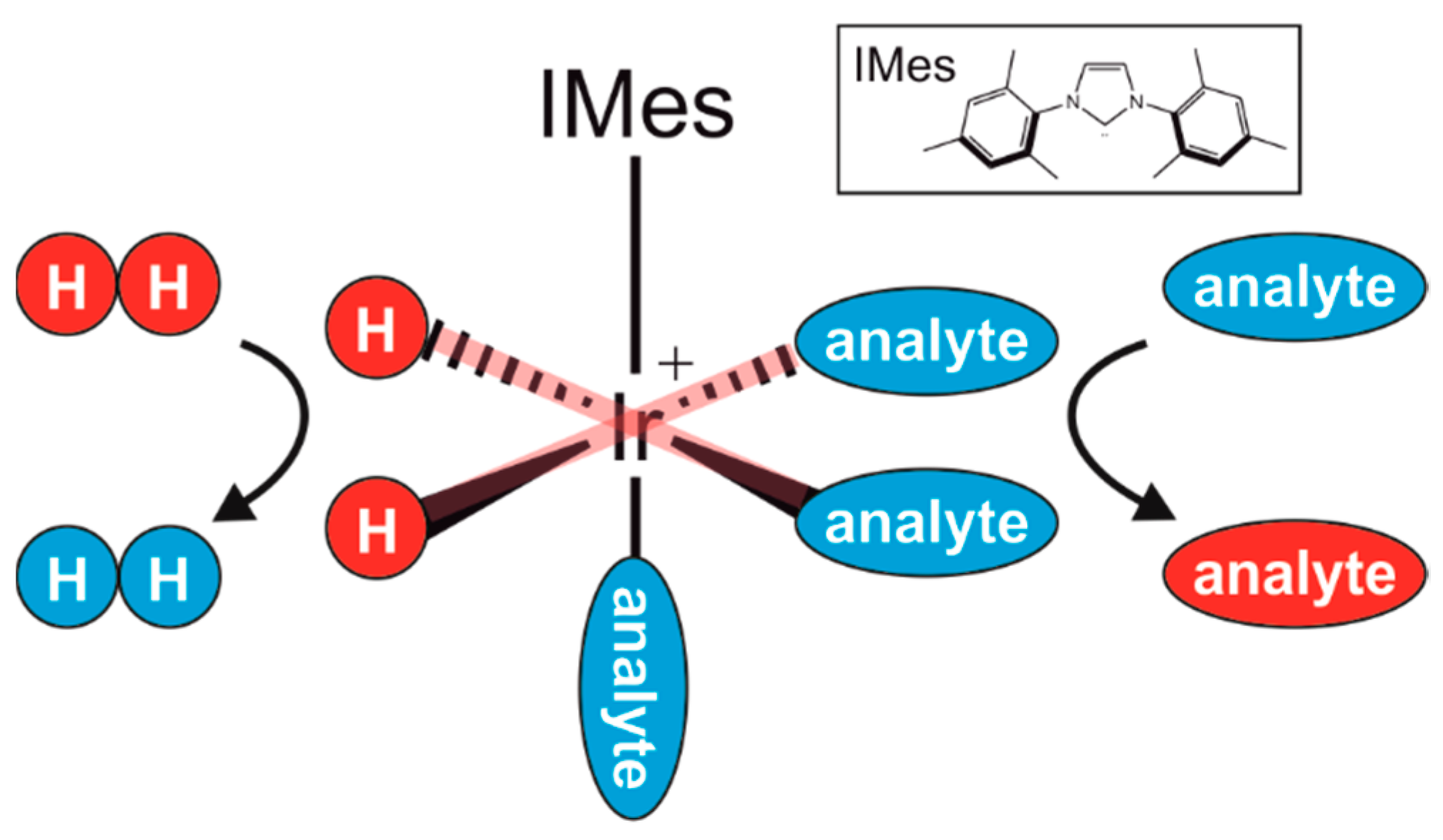
1. Introduction
2. Materials and Methods
3. Results
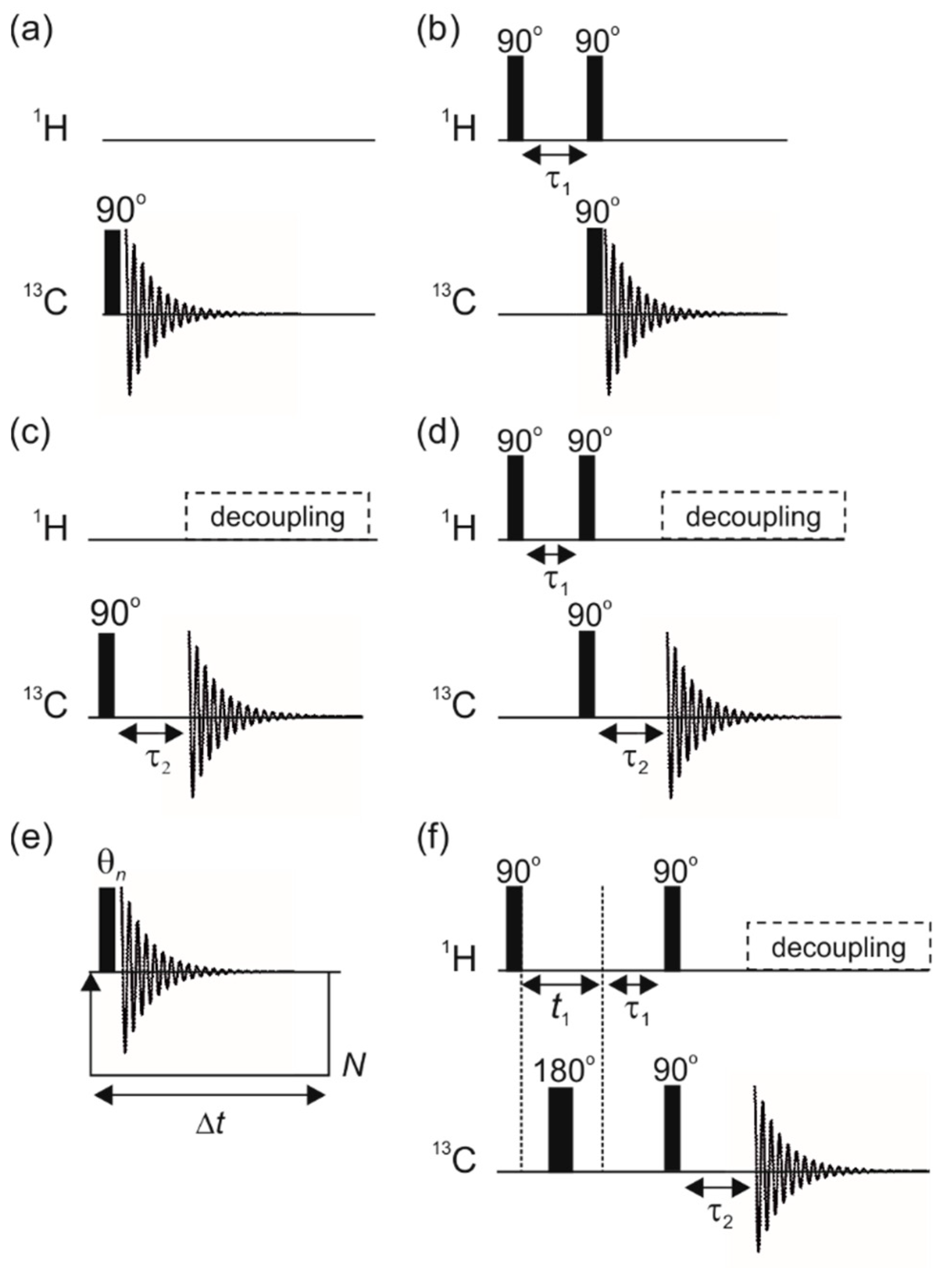
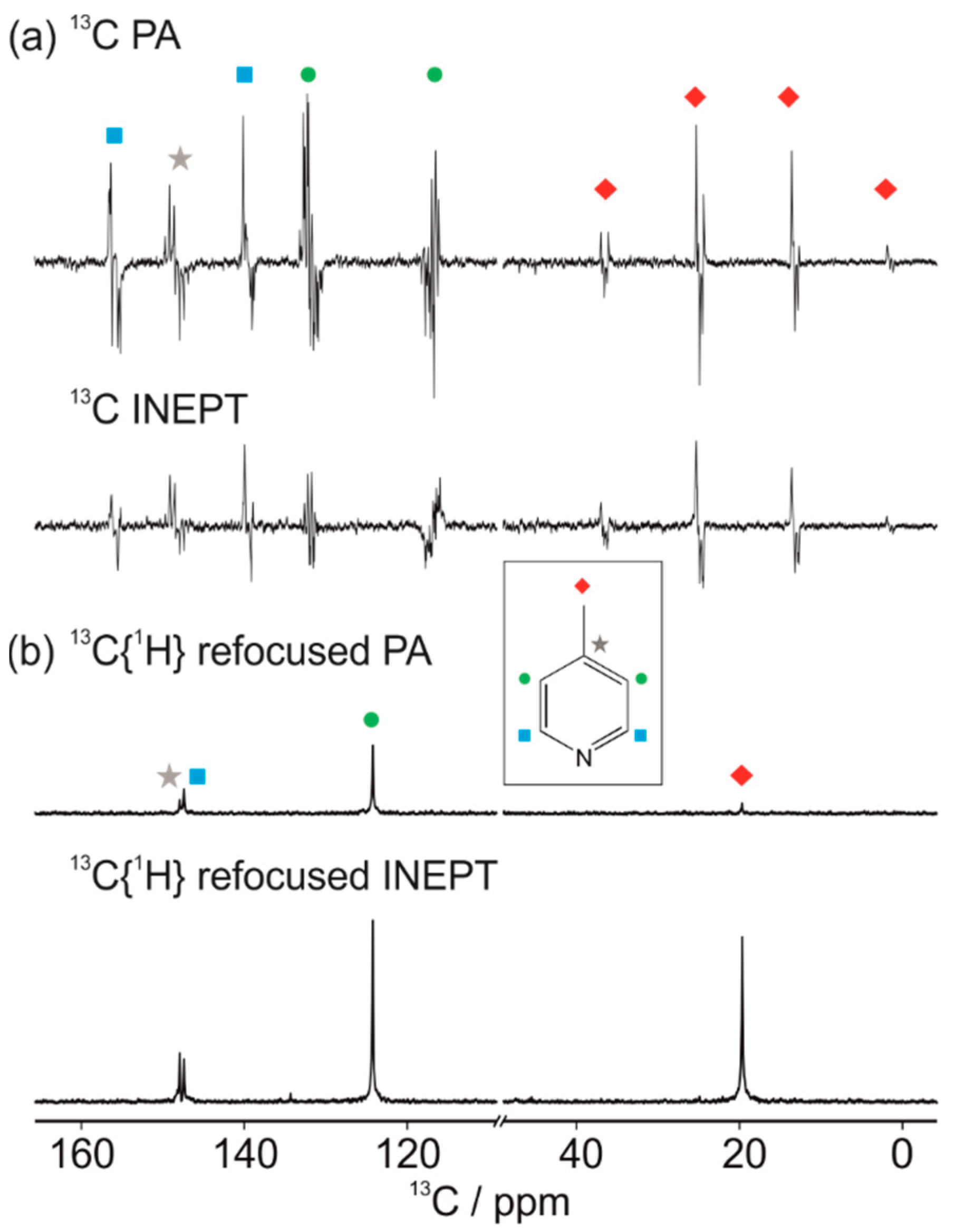
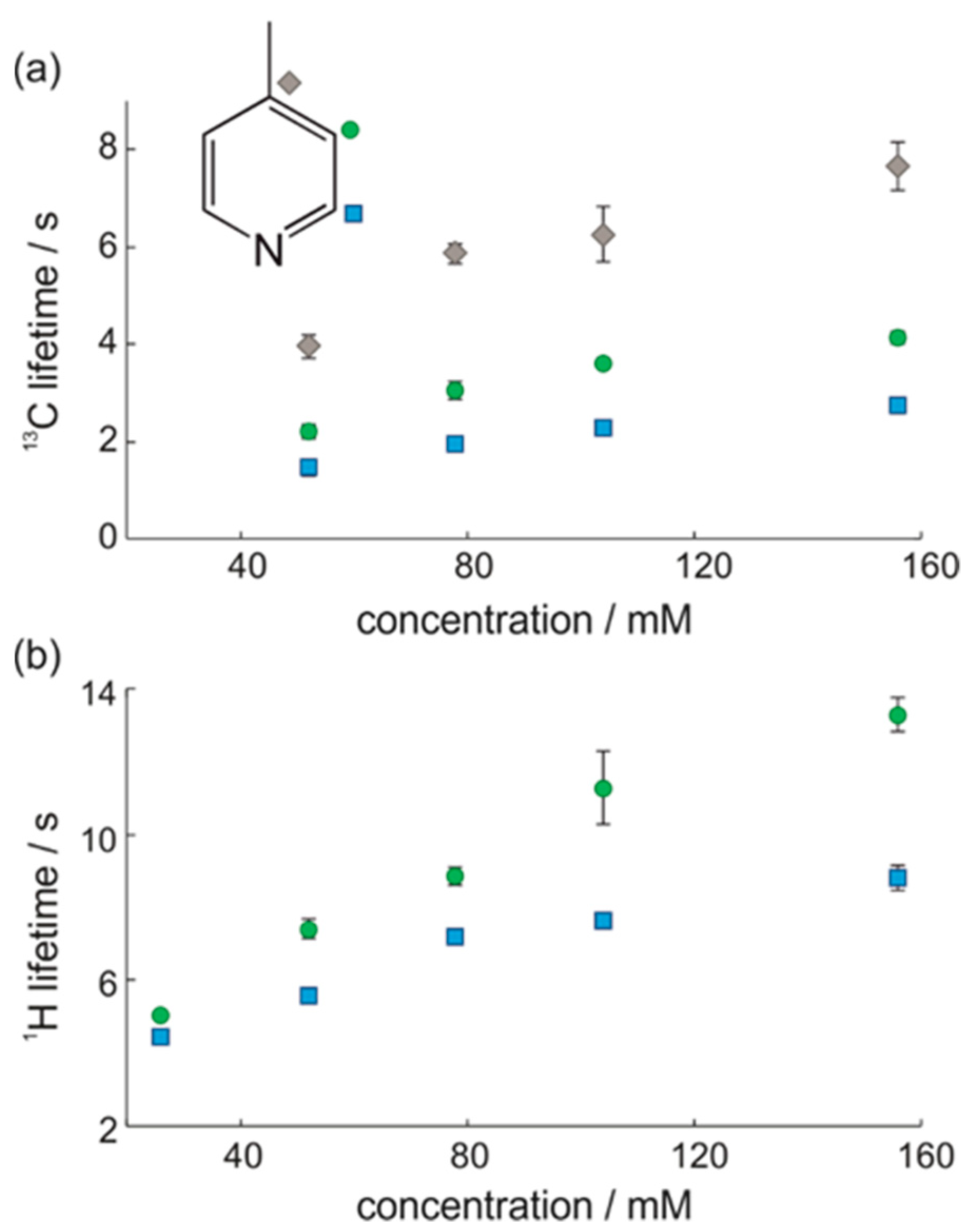
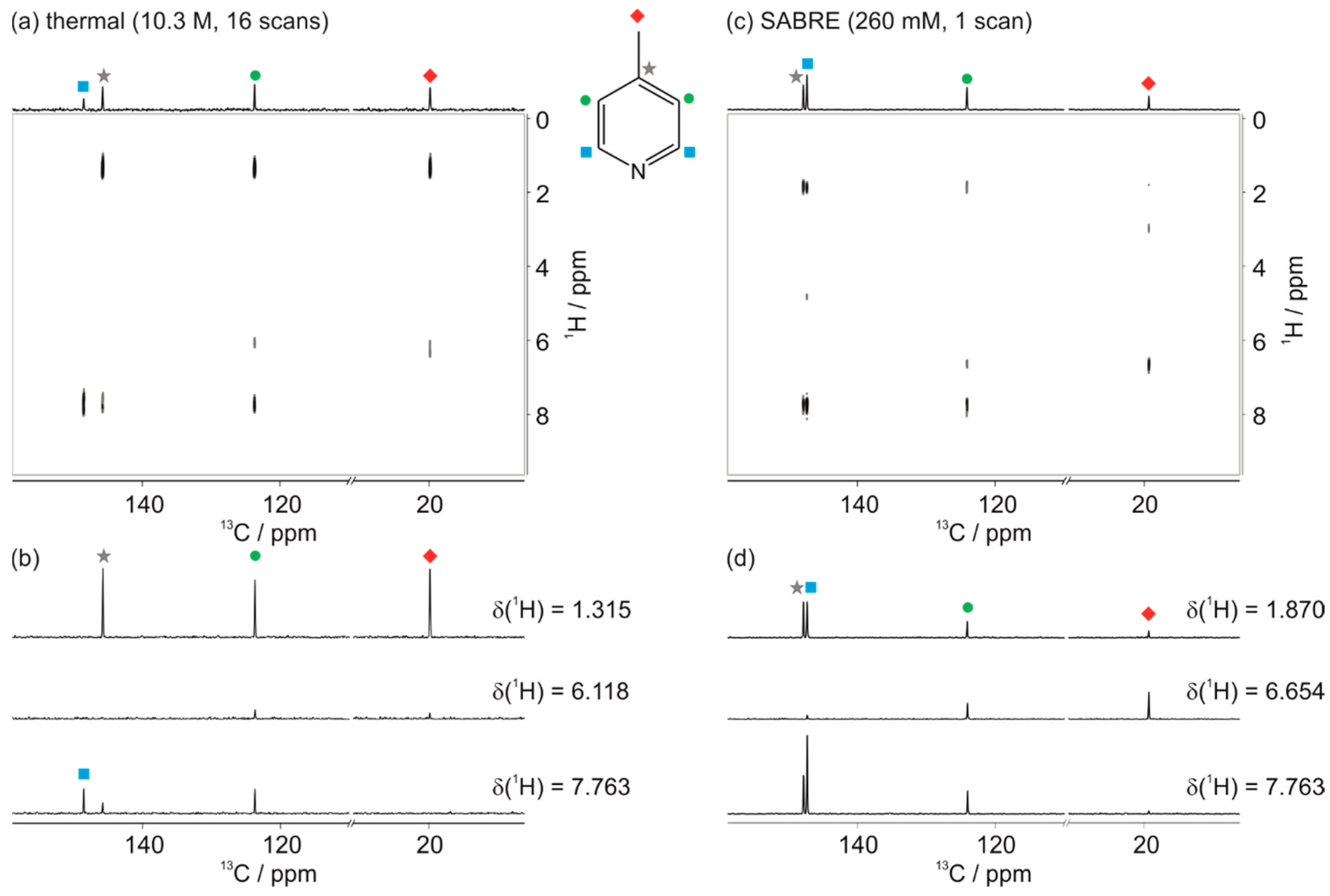
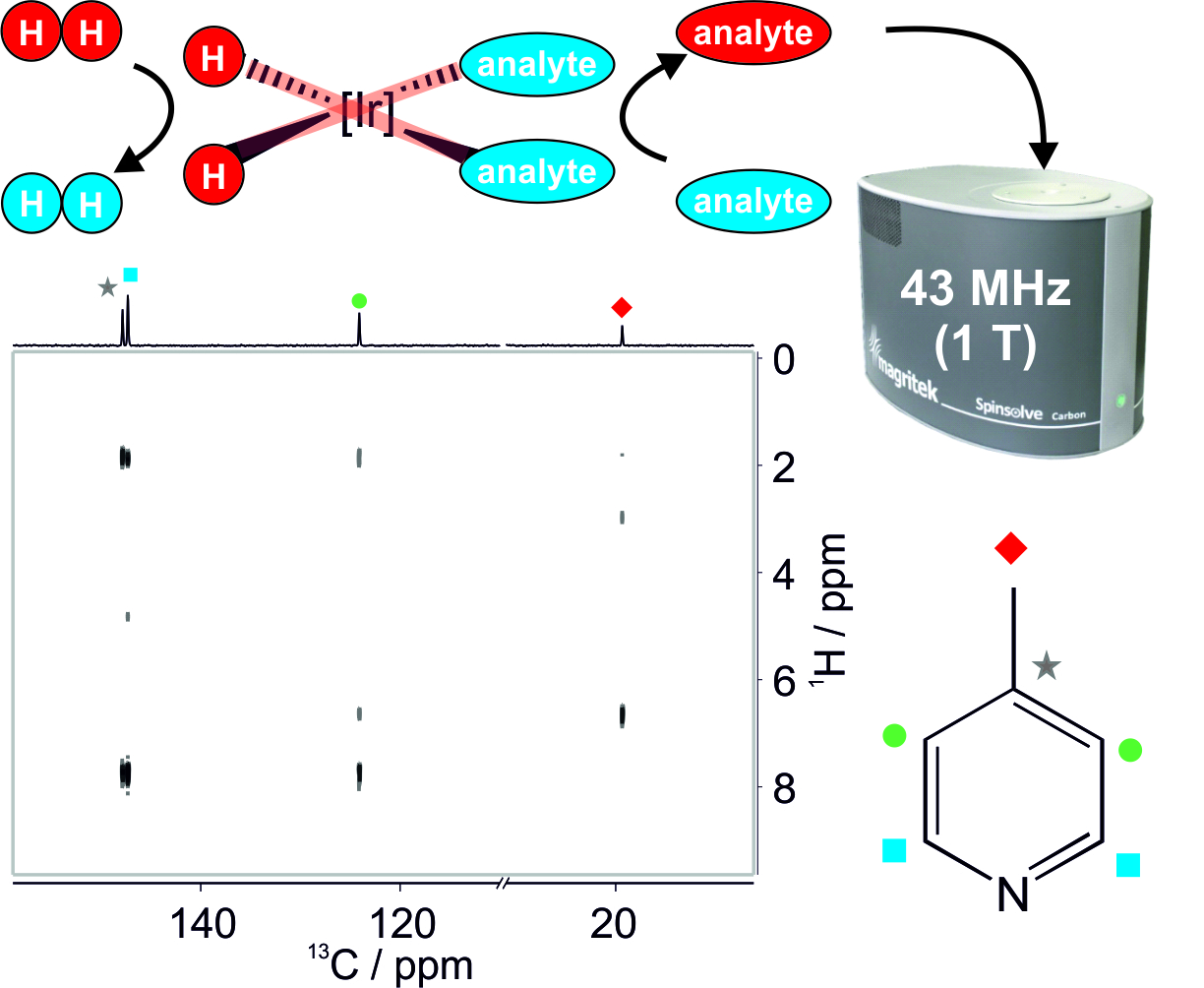
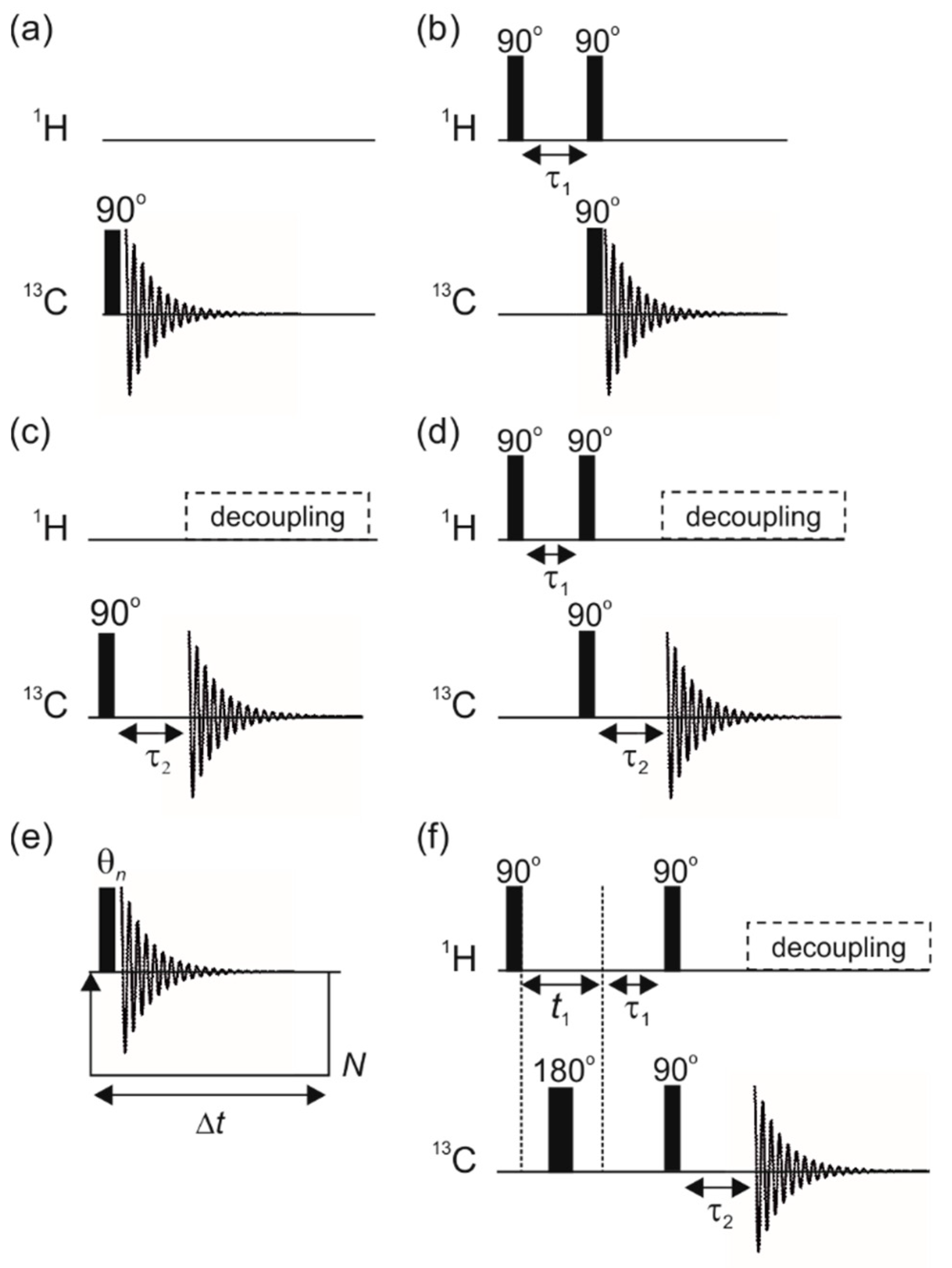
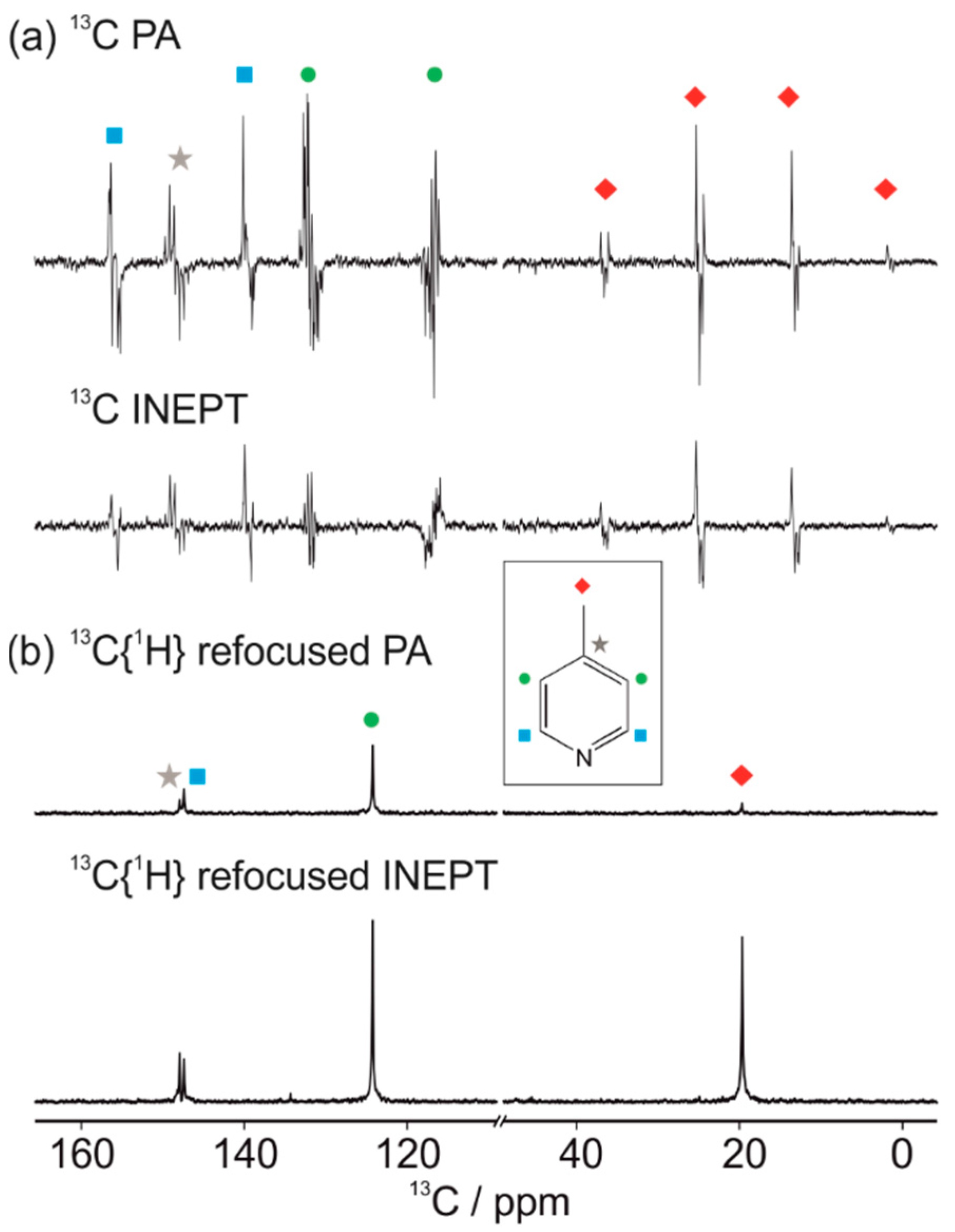
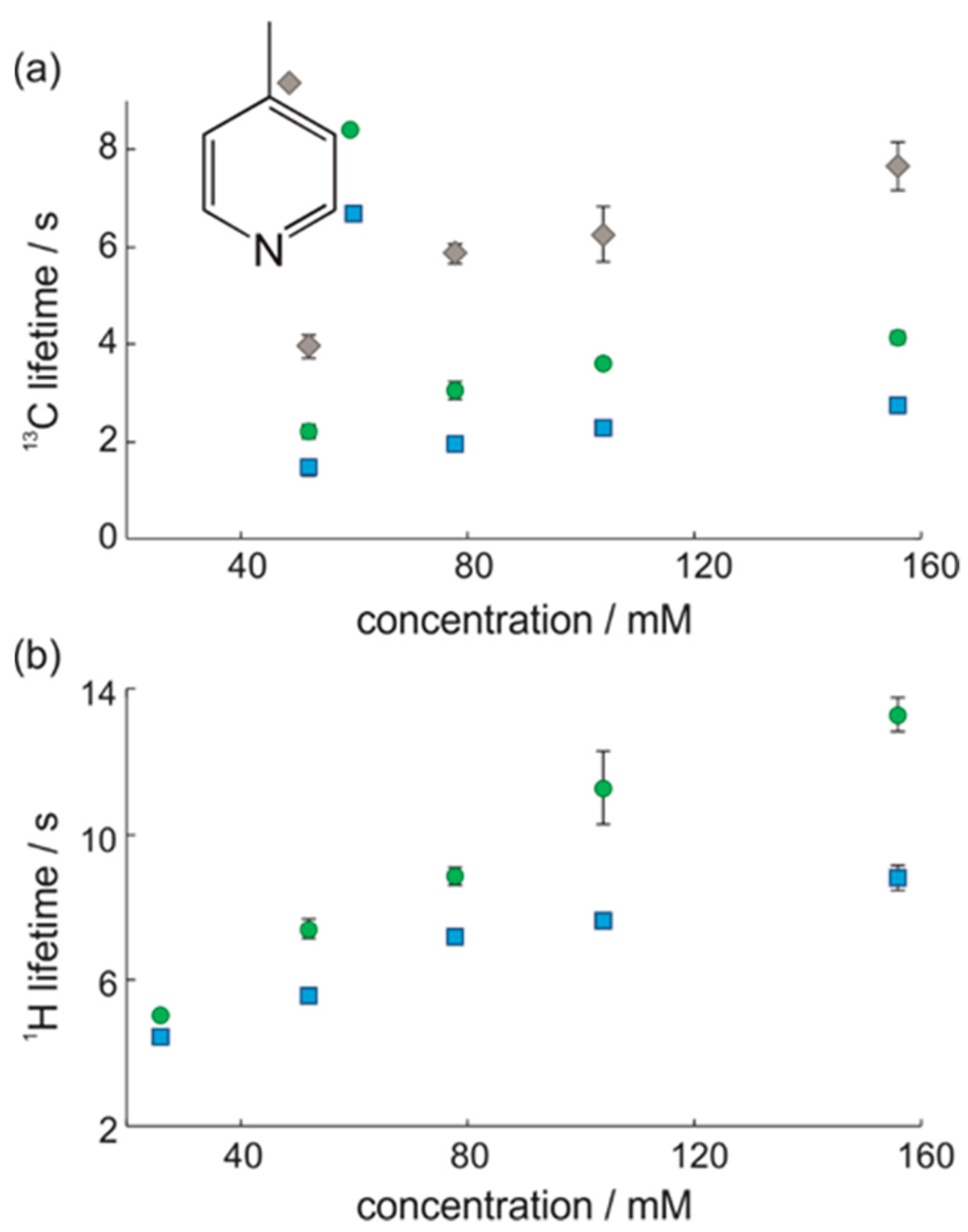
3.1. Optimal Detection of SABRE-Enhanced 13C Benchtop NMR Spectra
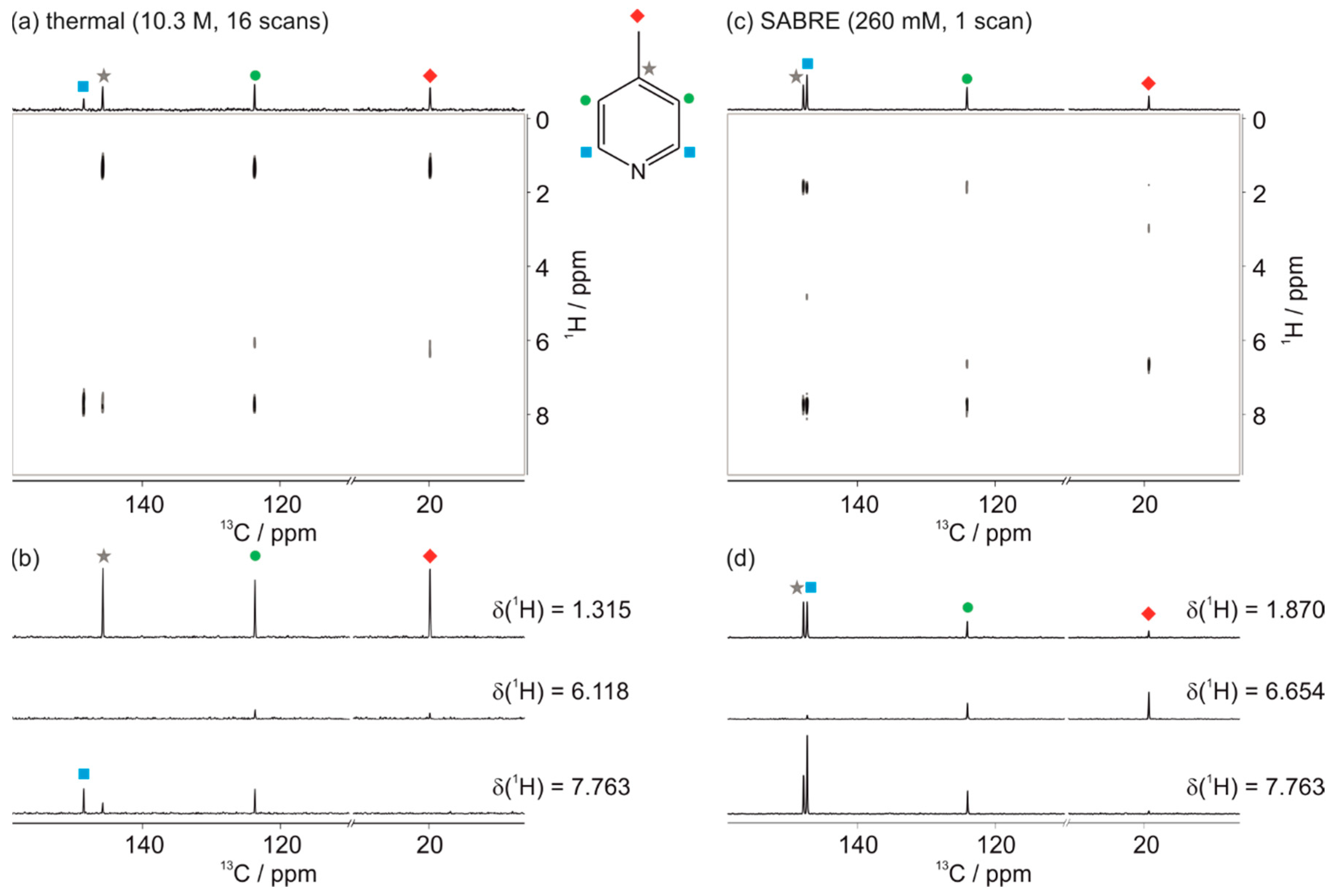
3.2. SABRE-Enhanced 2D 13C–1H Benchtop NMR Spectroscopy
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Access Statement
References
- Mitchell, J.; Gladden, L.F.; Chandrasekera, T.C.; Fordham, E.J. Low-field permanent magnets for industrial process and quality control. Prog. Nucl. Magn. Reson. Spectrosc. 2014, 76, 1–60. [Google Scholar] [CrossRef] [PubMed]
- Gutowsky, H.S.; Meyer, L.H.; McClure, R.E. Apparatus for Nuclear Magnetic Resonance. Rev. Sci. Instrum. 1953, 24, 644–652. [Google Scholar] [CrossRef]
- Leane, J.B.; Richards, R.E.; Schaefer, T.P. High-resolution nuclear resonance apparatus. J. Sci. Instrum. 1959, 36, 230–233. [Google Scholar] [CrossRef]
- Primas, H.; Günthard, H.H. Field Stabilizer for High-Resolution Nuclear Magnetic Resonance. Rev. Sci. Instrum. 1957, 28, 510–514. [Google Scholar] [CrossRef]
- Gutowsky, H.S.; Hoffman, C.J. Nuclear Magnetic Shielding in Fluorine and Hydrogen Compounds. J. Chem. Phys. 1951, 19, 1259–1267. [Google Scholar] [CrossRef]
- Gutowsky, H.S.; McCall, D.W.; Slichter, C.P. Nuclear Magnetic Resonance Multiplets in Liquids. J. Chem. Phys. 1953, 21, 279–292. [Google Scholar] [CrossRef]
- Arnold, J.T. Magnetic Resonances of Protons in Ethyl Alcohol. Phys. Rev. 1956, 102, 136–150. [Google Scholar] [CrossRef] [Green Version]
- Anderson, W.A. Nuclear Magnetic Resonance Spectra of Some Hydrocarbons. Phys. Rev. 1956, 102, 151–167. [Google Scholar] [CrossRef] [Green Version]
- Grunwald, E.; Loewenstein, A.; Meiboom, S. Rates and Mechanisms of Protolysis of Methylammonium Ion in Aqueous Solution Studied by Proton Magnetic Resonance. J. Chem. Phys. 1957, 27, 630–640. [Google Scholar] [CrossRef]
- Golay, M.J.E. Field Homogenizing Coils for Nuclear Spin Resonance Instrumentation. Rev. Sci. Instrum. 1958, 29, 313–315. [Google Scholar] [CrossRef]
- Anderson, W.A. Electrical Current Shims for Correcting Magnetic Fields. Rev. Sci. Instrum. 1961, 32, 241–250. [Google Scholar] [CrossRef]
- Moser, E.; Laistler, E.; Schmitt, F.; Kontaxis, G. Ultra-High Field NMR and MRI—The Role of Magnet Technology to Increase Sensitivity and Specificity. Front. Phys. 2017, 5, 33. [Google Scholar] [CrossRef]
- Blümich, B.; Casanova, F.; Appelt, S. NMR at low magnetic fields. Chem. Phys. Lett. 2009, 477, 231–240. [Google Scholar] [CrossRef]
- Parker, A.J.; Zia, W.; Rehorn, C.W.G.; Blümich, B. Shimming Halbach magnets utilizing genetic algorithms to profit from material imperfections. J. Magn. Reson. 2016, 265, 83–89. [Google Scholar] [CrossRef]
- Halbach, K. Design of permanent multipole magnets with oriented rare earth cobalt material. Nucl. Instruments Methods 1980, 169, 1–10. [Google Scholar] [CrossRef]
- Raich, H.; Blümler, P. Design and construction of a dipolar Halbach array with a homogeneous field from identical bar magnets: NMR Mandhalas. Concepts Magn. Reson. Part B Magn. Reson. Eng. 2004, 23B, 16–25. [Google Scholar] [CrossRef]
- Danieli, E.; Mauler, J.; Perlo, J.; Blümich, B.; Casanova, F. Mobile sensor for high resolution NMR spectroscopy and imaging. J. Magn. Reson. 2009, 198, 80–87. [Google Scholar] [CrossRef]
- Danieli, E.; Perlo, J.; Blümich, B.; Casanova, F.; Danieli, E.; Perlo, J.; Blümich, B.; Casanova, F. Small Magnets for Portable NMR Spectrometers. Angew. Chem. Int. Ed. 2010, 49, 4133–4135. [Google Scholar] [CrossRef]
- Singh, K.; Blümich, B. NMR spectroscopy with compact instruments. TrAC Trends Anal. Chem. 2016, 83, 12–26. [Google Scholar] [CrossRef]
- Meyer, K.; Kern, S.; Zientek, N.; Guthausen, G.; Maiwald, M. Process control with compact NMR. TrAC Trends Anal. Chem. 2016, 83, 39–52. [Google Scholar] [CrossRef]
- Dalitz, F.; Cudaj, M.; Maiwald, M.; Guthausen, G. Process and reaction monitoring by low-field NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 2012, 60, 52–70. [Google Scholar] [CrossRef]
- Parker, T.; Limer, E.; Watson, A.D.; Defernez, M.; Williamson, D.; Kemsley, E.K. 60 MHz 1H NMR spectroscopy for the analysis of edible oils. TrAC Trends Anal. Chem. 2014, 57, 147–158. [Google Scholar] [CrossRef]
- Jakes, W.; Gerdova, A.; Defernez, M.; Watson, A.D.; McCallum, C.; Limer, E.; Colquhoun, I.J.; Williamson, D.C.; Kemsley, E.K. Authentication of beef versus horse meat using 60 MHz 1H NMR spectroscopy. Food Chem. 2015, 175, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Defernez, M.; Wren, E.; Watson, A.D.; Gunning, Y.; Colquhoun, I.J.; Le Gall, G.; Williamson, D.; Kemsley, E.K. Low-field 1H NMR spectroscopy for distinguishing between arabica and robusta ground roast coffees. Food Chem. 2017, 216, 106–113. [Google Scholar] [CrossRef] [Green Version]
- Isaac-Lam, M.F. Determination of Alcohol Content in Alcoholic Beverages Using 45 MHz Benchtop NMR Spectrometer. Int. J. Spectrosc. 2016, 2016, 1–8. [Google Scholar] [CrossRef]
- Pagès, G.; Gerdova, A.; Williamson, D.; Gilard, V.; Martino, R.; Malet-Martino, M. Evaluation of a Benchtop Cryogen-Free Low-Field 1 H NMR Spectrometer for the Analysis of Sexual Enhancement and Weight Loss Dietary Supplements Adulterated with Pharmaceutical Substances. Anal. Chem. 2014, 86, 11897–11904. [Google Scholar] [CrossRef]
- Riegel, S.D.; Leskowitz, G.M. Benchtop NMR spectrometers in academic teaching. TrAC Trends Anal. Chem. 2016, 83, 27–38. [Google Scholar] [CrossRef]
- Iler, H.D.; Justice, D.; Brauer, S.; Landis, A. Discovering 13C NMR, 1H NMR, and IR Spectroscopy in the General Chemistry Laboratory through a Sequence of Guided-Inquiry Exercises. J. Chem. Educ. 2012, 89, 1178–1182. [Google Scholar] [CrossRef]
- Van Draanen, N.A.; Page, R. Structure Determination of Benzene-Containing C9H12 Isomers Using Symmetry, Peak Heights, and Chemical Shifts in 13C NMR. J. Chem. Educ. 2009, 86, 849. [Google Scholar] [CrossRef]
- Dávila, R.M.; Widener, R.K. Structure and Nuclear Magnetic Resonance. An Experiment for the General Chemistry Laboratory. J. Chem. Educ. 2002, 79, 997. [Google Scholar] [CrossRef]
- Isaac-Lam, M.F. Analysis of Bromination of Ethylbenzene Using a 45 MHz NMR Spectrometer: An Undergraduate Organic Chemistry Laboratory Experiment. J. Chem. Educ. 2014, 91, 1264–1266. [Google Scholar] [CrossRef]
- Pelter, M.W.; Walker, N.M. A Discovery-Based Hydrochlorination of Carvone Utilizing a Guided-Inquiry Approach to Determine the Product Structure from 13C NMR Spectra. J. Chem. Educ. 2012, 89, 1183–1185. [Google Scholar] [CrossRef]
- Küster, S.K.; Danieli, E.; Blümich, B.; Casanova, F. High-resolution NMR spectroscopy under the fume hood. Phys. Chem. Chem. Phys. 2011, 13, 13172. [Google Scholar] [CrossRef] [PubMed]
- Silva Elipe, M.V.; Milburn, R.R. Monitoring chemical reactions by low-field benchtop NMR at 45 MHz: Pros and cons. Magn. Reson. Chem. 2016, 54, 437–443. [Google Scholar] [CrossRef]
- Danieli, E.; Perlo, J.; Duchateau, A.L.L.; Verzijl, G.K.M.; Litvinov, V.M.; Blümich, B.; Casanova, F. On-Line Monitoring of Chemical Reactions by using Bench-Top Nuclear Magnetic Resonance Spectroscopy. ChemPhysChem 2014, 15, 3060–3066. [Google Scholar] [CrossRef]
- Kern, S.; Meyer, K.; Guhl, S.; Gräßer, P.; Paul, A.; King, R.; Maiwald, M. Online low-field NMR spectroscopy for process control of an industrial lithiation reaction—Automated data analysis. Anal. Bioanal. Chem. 2018, 410, 3349–3360. [Google Scholar] [CrossRef] [PubMed]
- Picard, B.; Gouilleux, B.; Lebleu, T.; Maddaluno, J.; Chataigner, I.; Penhoat, M.; Felpin, F.-X.; Giraudeau, P.; Legros, J. Oxidative Neutralization of Mustard-Gas Simulants in an On-Board Flow Device with In-Line NMR Monitoring. Angew. Chemie 2017, 129, 7676–7680. [Google Scholar] [CrossRef]
- Archambault, C.M.; Leadbeater, N.E. A benchtop NMR spectrometer as a tool for monitoring mesoscale continuous-flow organic synthesis: Equipment interface and assessment in four organic transformations. RSC Adv. 2016, 6, 101171–101177. [Google Scholar] [CrossRef]
- Singh, K.; Danieli, E.; Blümich, B. Desktop NMR spectroscopy for real-time monitoring of an acetalization reaction in comparison with gas chromatography and NMR at 9.4 T. Anal. Bioanal. Chem. 2017, 409, 7223–7234. [Google Scholar] [CrossRef]
- Levitt, M.H. Spin Dynamics, 2nd ed.; John Wiley & Sons Ltd.: Chichester, UK, 2008; pp. 23–36. [Google Scholar]
- Ardenkjaer-Larsen, J.H.; Fridlund, B.; Gram, A.; Hansson, G.; Hansson, L.; Lerche, M.H.; Servin, R.; Thaning, M.; Golman, K. Increase in signal-to-noise ratio of > 10,000 times in liquid-state NMR. Proc. Natl. Acad. Sci. USA 2003, 100, 10158–10163. [Google Scholar] [CrossRef] [Green Version]
- Bowen, S.; Hilty, C. Rapid sample injection for hyperpolarized NMR spectroscopy. Phys. Chem. Chem. Phys. 2010, 12, 5766. [Google Scholar] [CrossRef]
- Merritt, M.E.; Harrison, C.; Storey, C.; Jeffrey, F.M.; Sherry, A.D.; Malloy, C.R. Hyperpolarized 13C allows a direct measure of flux through a single enzyme-catalyzed step by NMR. Proc. Natl. Acad. Sci. USA 2007, 104, 19773–19777. [Google Scholar] [CrossRef] [Green Version]
- Bowers, C.R.; Weitekamp, D.P. Transformation of symmetrization order to nuclear-spin magnetization by chemical reaction and nuclear magnetic resonance. Phys. Rev. Lett. 1986, 57, 2645–2648. [Google Scholar] [CrossRef]
- Natterer, J.; Bargon, J. Parahydrogen induced polarization. Prog. Nucl. Magn. Reson. Spectrosc. 1997, 31, 293–315. [Google Scholar] [CrossRef]
- Duckett, S.B.; Mewis, R.E. Application of Para hydrogen Induced Polarization Techniques in NMR Spectroscopy and Imaging. Acc. Chem. Res. 2012, 45, 1247–1257. [Google Scholar] [CrossRef]
- Adams, R.W.; Aguilar, J.A.; Atkinson, K.D.; Cowley, M.J.; Elliott, P.I.P.; Duckett, S.B.; Green, G.G.R.; Khazal, I.G.; López-Serrano, J.; Williamson, D.C. Reversible interactions with para-hydrogen enhance NMR sensitivity by polarization transfer. Science 2009, 323, 1708–1711. [Google Scholar] [CrossRef]
- Halse, M.E. Perspectives for hyperpolarisation in compact NMR. Trends Anal. Chem. 2016, 83, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Richardson, P.M.; Parrott, A.J.; Semenova, O.; Nordon, A.; Duckett, S.B.; Halse, M.E. SABRE hyperpolarization enables high-sensitivity 1H and 13C benchtop NMR spectroscopy. Analyst 2018, 143, 3442–3450. [Google Scholar] [CrossRef]
- Adams, R.W.; Duckett, S.B.; Green, R.A.; Williamson, D.C.; Green, G.G.R. A theoretical basis for spontaneous polarization transfer in non-hydrogenative parahydrogen-induced polarization. J. Chem. Phys. 2009, 131, 194505. [Google Scholar] [CrossRef]
- Rayner, P.J.; Duckett, S.B. Signal Amplification by Reversible Exchange (SABRE): From Discovery to Diagnosis. Angew. Chemie Int. Ed. 2018, 57, 6742–6753. [Google Scholar] [CrossRef]
- Kiryutin, A.S.; Yurkovskaya, A.V.; Zimmermann, H.; Vieth, H.-M.; Ivanov, K.L. Complete magnetic field dependence of SABRE-derived polarization. Magn. Reson. Chem. 2018, 56, 651–662. [Google Scholar] [CrossRef]
- Pravdivtsev, A.N.; Yurkovskaya, A.V.; Zimmermann, H.; Vieth, H.-M.; Ivanov, K.L. Transfer of SABRE-derived hyperpolarization to spin-1/2 heteronuclei. RSC Adv. 2015, 5, 63615–63623. [Google Scholar] [CrossRef]
- Theis, T.; Truong, M.L.; Coffey, A.M.; Shchepin, R.V.; Waddell, K.W.; Shi, F.; Goodson, B.M.; Warren, W.S.; Chekmenev, E.Y. Microtesla SABRE Enables 10% Nitrogen-15 Nuclear Spin Polarization. J. Am. Chem. Soc. 2015, 137, 1404–1407. [Google Scholar] [CrossRef] [PubMed]
- Barskiy, D.A.; Shchepin, R.V.; Tanner, C.P.N.; Colell, J.F.P.; Goodson, B.M.; Theis, T.; Warren, W.S.; Chekmenev, E.Y. The Absence of Quadrupolar Nuclei Facilitates Efficient 13C Hyperpolarization via Reversible Exchange with Parahydrogen. ChemPhysChem 2017, 18, 1493–1498. [Google Scholar] [CrossRef]
- Shchepin, R.V.; Goodson, B.M.; Theis, T.; Warren, W.S.; Chekmenev, E.Y. Toward Hyperpolarized 19F Molecular Imaging via Reversible Exchange with Parahydrogen. ChemPhysChem 2017, 18, 1961–1965. [Google Scholar] [CrossRef]
- Zhou, Z.; Yu, J.; Colell, J.F.P.; Laasner, R.; Logan, A.; Barskiy, D.A.; Shchepin, R.V.; Chekmenev, E.Y.; Blum, V.; Warren, W.S.; et al. Long-Lived 13C2 Nuclear Spin States Hyperpolarized by Parahydrogen in Reversible Exchange at Microtesla Fields. J. Phys. Chem. Lett. 2017, 8, 3008–3014. [Google Scholar] [CrossRef]
- Shchepin, R.V.; Jaigirdar, L.; Theis, T.; Warren, W.S.; Goodson, B.M.; Chekmenev, E.Y. Spin Relays Enable Efficient Long-Range Heteronuclear Signal Amplification by Reversible Exchange. J. Phys. Chem. C 2017, 121, 28425–28434. [Google Scholar] [CrossRef]
- Mewis, R.E.; Atkinson, K.D.; Cowley, M.J.; Duckett, S.B.; Green, G.G.R.; Green, R.A.; Highton, L.A.R.; Kilgour, D.; Lloyd, L.S.; Lohman, J.A.B.; et al. Probing signal amplification by reversible exchange using an NMR flow system. Magn. Reson. Chem. 2014, 52, 358–369. [Google Scholar] [CrossRef] [Green Version]
- Dücker, E.B.; Kuhn, L.T.; Münnemann, K.; Griesinger, C. Similarity of SABRE field dependence in chemically different substrates. J. Magn. Reson. 2012, 214, 159–165. [Google Scholar] [CrossRef]
- Cowley, M.J.; Adams, R.W.; Atkinson, K.D.; Cockett, M.C.R.; Duckett, S.B.; Green, G.G.R.; Lohman, J.A.B.; Kerssebaum, R.; Kilgour, D.; Mewis, R.E. Iridium N-Heterocyclic Carbene Complexes as Efficient Catalysts for Magnetization Transfer from para-Hydrogen. J. Am. Chem. Soc. 2011, 133, 6134–6137. [Google Scholar] [CrossRef]
- Lloyd, L.S.; Adams, R.W.; Bernstein, M.; Coombes, S.; Duckett, S.B.; Green, G.G.R.; Lewis, R.J.; Mewis, R.E.; Sleigh, C.J. Utilization of SABRE-Derived Hyperpolarization to Detect Low-Concentration Analytes via 1D and 2D NMR Methods. J. Am. Chem. Soc. 2012, 134, 12904–12907. [Google Scholar] [CrossRef]
- Shaver, R.; Van Wallendael, S.; Rillema, D.P. A rapid method for degassing samples. J. Chem. Educ. 1991, 68, 604. [Google Scholar] [CrossRef]
- Richardson, P.M.; John, R.O.; Parrott, A.J.; Rayner, P.J.; Iali, W.; Nordon, A.; Halse, M.E.; Duckett, S.B. Quantification of hyperpolarisation efficiency in SABRE and SABRE-Relay enhanced NMR spectroscopy. Phys. Chem. Chem. Phys. 2018, 20, 26362–26371. [Google Scholar] [CrossRef]
- Richardson, P.M.; Jackson, S.; Parrott, A.J.; Nordon, A.; Duckett, S.B.; Halse, M.E. A simple hand-held magnet array for efficient and reproducible SABRE hyperpolarisation using manual sample shaking. Magn. Reson. Chem. 2018, 56, 641–650. [Google Scholar] [CrossRef]
- Mewis, R.E.; Green, R.A.; Cockett, M.C.R.; Cowley, M.J.; Duckett, S.B.; Green, G.G.R.; John, R.O.; Rayner, P.J.; Williamson, D.C. Strategies for the Hyperpolarization of Acetonitrile and Related Ligands by SABRE. J. Phys. Chem. B 2014, 119, 1416–1424. [Google Scholar] [CrossRef]
- Giraudeau, P.; Frydman, L. Ultrafast 2D NMR: An emerging tool in analytical spectroscopy. Annu. Rev. Anal. Chem. (Palo Alto. Calif.) 2014, 7, 129–161. [Google Scholar] [CrossRef]
- Daniele, V.; Legrand, F.-X.; Berthault, P.; Dumez, J.-N.; Huber, G. Single-Scan Multidimensional NMR Analysis of Mixtures at Sub-Millimolar Concentrations by using SABRE Hyperpolarization. ChemPhysChem 2015, 16, 3413–3417. [Google Scholar] [CrossRef]
- Iali, W.; Rayner, P.J.; Alshehri, A.; Holmes, A.J.; Ruddlesden, A.J.; Duckett, S.B. Direct and indirect hyperpolarisation of amines using para hydrogen. Chem. Sci. 2018, 9, 3677–3684. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 13C Resonance | Refocused PA | Refocused INEPT |
|---|---|---|
| para | 9.9 ± 0.5 | 31 ± 2 |
| ortho | 18.9 ± 0.9 | 23 ± 2 |
| meta | 43 ± 2 | 91 ± 7 |
| methyl | 6.4 ± 0.6 | 73 ± 7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robinson, A.D.; Richardson, P.M.; Halse, M.E. Hyperpolarised 1H–13C Benchtop NMR Spectroscopy. Appl. Sci. 2019, 9, 1173. https://doi.org/10.3390/app9061173
Robinson AD, Richardson PM, Halse ME. Hyperpolarised 1H–13C Benchtop NMR Spectroscopy. Applied Sciences. 2019; 9(6):1173. https://doi.org/10.3390/app9061173
Chicago/Turabian StyleRobinson, Alastair D., Peter M. Richardson, and Meghan E. Halse. 2019. "Hyperpolarised 1H–13C Benchtop NMR Spectroscopy" Applied Sciences 9, no. 6: 1173. https://doi.org/10.3390/app9061173
APA StyleRobinson, A. D., Richardson, P. M., & Halse, M. E. (2019). Hyperpolarised 1H–13C Benchtop NMR Spectroscopy. Applied Sciences, 9(6), 1173. https://doi.org/10.3390/app9061173