Magnetic Fields Trump Oxygen in Controlling the Death of Erythro-Leukemia Cells



Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Culture Medium
2.2. Magnetic Fields
2.3. Cell Culture Measurements
3. Results
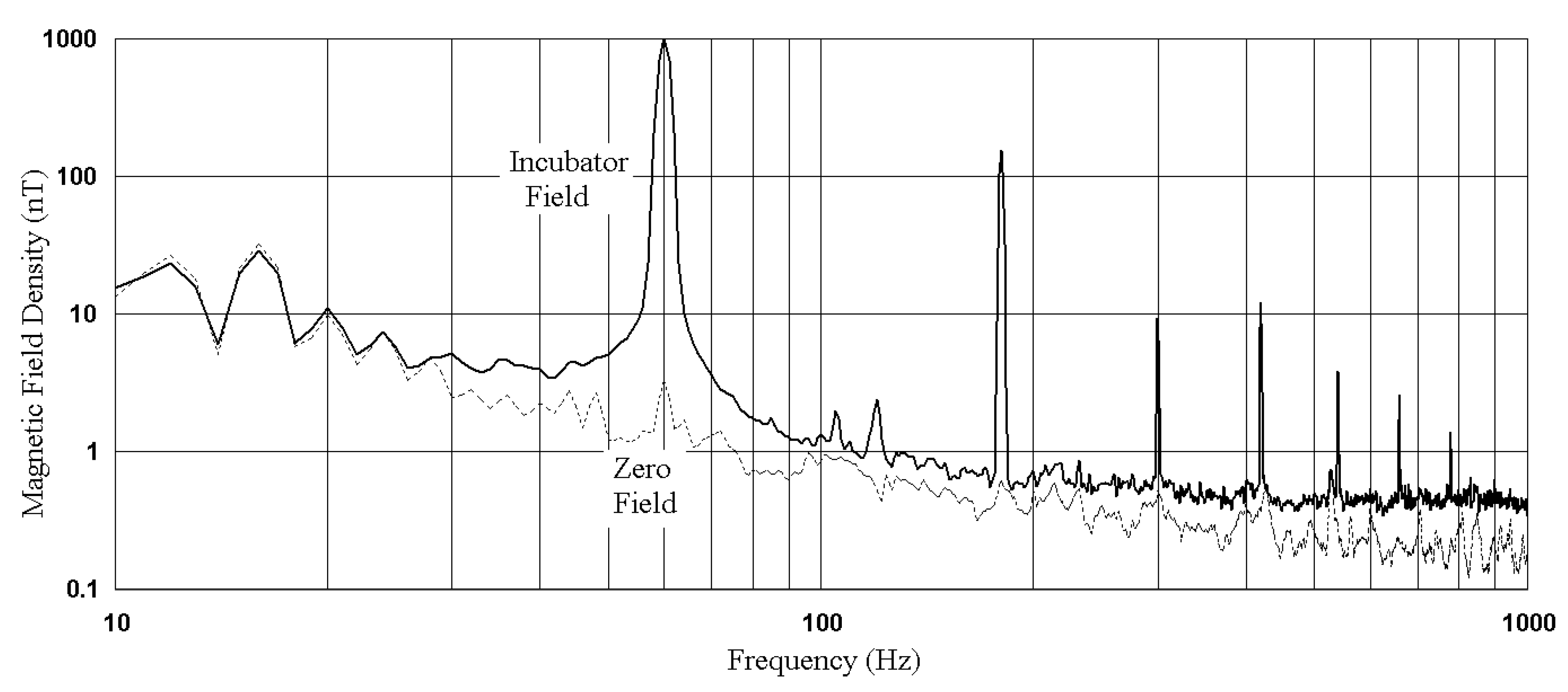
3.1. Baseline Experiments
3.2. Baseline Cell Culture Results
3.3. Transition Cell Culture Results
3.4. Necrosis
3.5. Early Apoptosis
3.6. Late Apoptosis
3.7. Late to Early Apoptosis Ratio
3.8. Necrosis vs. Apoptosis
4. Discussion
- “Zero” MF conditions (facilitated ATP synthesis) should favor a higher late/early apoptosis ratio. Table 2 shows that this is true for baseline cultures at both High (2.43 > 1.23) and even Low (1.66 > 1.51) oxygen.
- The hypoxic Zero MF Transition of Table 4 had the highest late/early apoptosis ratio (10) of any Transition. When Incubator fields were applied to the same hypoxic Transition, the late/early apoptosis ratio fell by 23%.
- Although hypoxia was effective at inducing apoptosis (5.2, 5.3, 3.3, 2.71 in Table 4), to induce it quickly, as shown by a high late/early ratio, the MF must be unchanging (10, 7.74). This rapid apoptosis was almost completely inhibited (1.26, 1.04 in Table 4) if a MF change was superposed, constraining ATP resources.
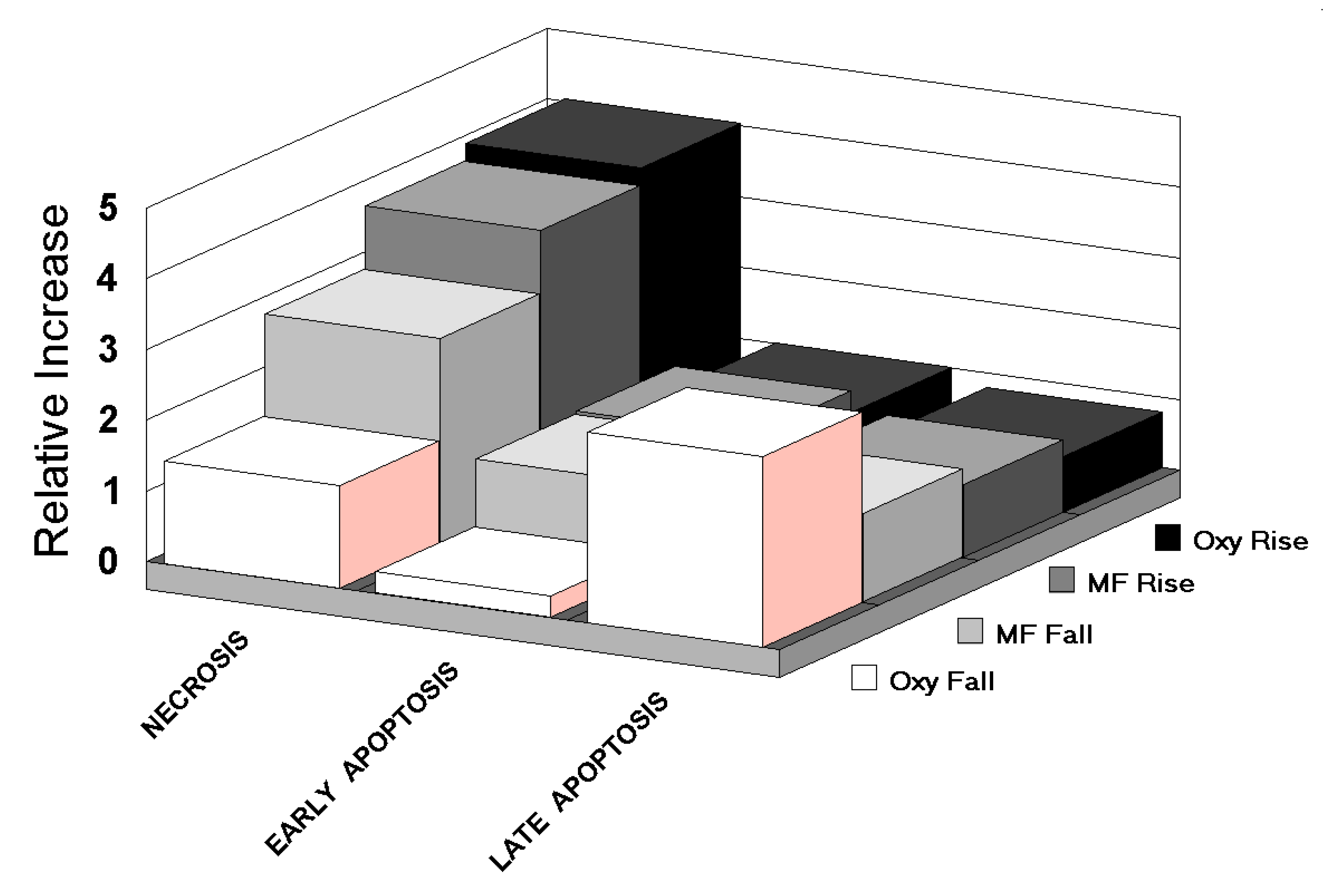
- Transitions columns in Table 4 are ordered from left to right by decreasing necrosis rating. In the left half of the Table (high necrosis), all MFs are changing, except in two cases, which correspond to hyperoxic rises. Larger shifts in cellular environments, as simultaneous oxygen and MF changes, placed a heavier adaptive load on cells, requiring 5′-adenosine monophosphate-activated protein kinase alpha (AMPK to manage more adjustments [33]. These requirements slowed apoptosis, and increased necrosis (left side of Table 4).
- Table 3 shows that that the rush to late apoptosis of the hypoxic Transitions with no MF change (2.4, 3 in Table 3) attenuated necrosis (0.48, 0.49 in Table 4) by simple apoptosis to necrosis competition. This indicates that MF variations switch cell death from apoptosis to (inflammatory) necrosis. But under hyperoxic “Zero” MF, the hyperoxia actually attenuated apoptosis (necrosis/apoptosis = 2.6), which means that hyperoxia, through ROS, was quickly toxic to the apoptotic pathway [34], shifting cells towards necrosis.
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Héroux, P. A Dosimeter for Assessment of Exposures to ELF Fields. Bioelectromagnetics 1991, 12, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Van der Vorst, A.; Rosen, A.; Kotsuka, Y. RF/Microwave Interaction with Biological Tissues; Wiley-Interscience: Hoboken, NJ, USA, 2006; p. 119. [Google Scholar]
- Perentos, N.; Iskra, S.; McKenzie, R.J.; Cosic, I. Simulation of pulsed ELF magnetic fields generated by GSM mobile phone handsets for human electromagnetic bioeffects research. Australas. Phys. Eng. Sci. 2008, 31, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Blackman, C. Cell phone radiation: Evidence from ELF and RF studies supporting more inclusive risk identification and assessment. Pathophysiology 2009, 16, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Höckel, M.; Vaupel, P. Tumor hypoxia: Definitions and current clinical, biologic, and molecular aspects. J. Nat. Cancer Inst. 2001, 93, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Héroux, P.; Kyrychenko, I. Metabolic restriction of cancer cells in vitro causes karyotype contraction—An indicator of cancer promotion? Tumor Biol. 2012, 33, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Héroux, P. Extra-Low-Frequency Magnetic Fields alter Cancer Cells through Metabolic Restriction. Electromagn. Biol. Med. 2014, 33, 264–275. [Google Scholar] [CrossRef]
- Wright, W.E.; Shay, J.W. Inexpensive low-oxygen incubators. Nat. Protoc. 2006, 1, 2088–2090. [Google Scholar] [CrossRef]
- Su, D.; Héroux, P. Survey of Extra-Low Frequency and Very-Low Frequency Magnetic Fields in Cell Culture Incubators. arXiv 2012. Available online: http://arxiv.org/abs/1211.2458 (accessed on 18 April 2019).
- Portelli, L.A.; Schomay, T.E.; Barnes, F.S. Inhomogeneous Background Magnetic Field in Biological Incubators is a Potential Confounder for Experimental Variability and Reproducibility. Bioelectromagnetics 2013, 34, 337–348. [Google Scholar] [CrossRef]
- Lantow, M.; Viergutz, T.; Weiss, D.G.; Simkó, M. Comparative Study of Cell Cycle Kinetics and Induction of Apoptosis or Necrosis after Exposure of Human Mono Mac 6 Cells to Radiofrequency Radiation. Radiat. Res. 2006, 166, 539–543. [Google Scholar] [CrossRef]
- Kaszuba-Zwoinska, J.; Wojcik, K.; Bereta, M.; Ziomber, A.; Pierzchalski, P.; Rokita, E.; Marcinkiewicz, J.; Zaraska, W.; Thor, P. Pulsating electromagnetic field stimulation prevents cell death of puromycin treated u937 cell line. J. Physiol. Pharmacol. 2010, 61, 201–205. [Google Scholar] [PubMed]
- Emre, M.; Cetiner, S.; Zencir, S.; Unlukurt, I.; Kahraman, I.; Topcu, Z. Oxidative Stress and Apoptosis in Relation to Exposure to Magnetic Field. Cell Biochem. Biophys. 2011, 59, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Juszczak, K.; Kaszuba-Zwoinska, J.; Thor, P.J. Pulsating electromagnetic field stimulation of urothelial cells induces apoptosis and diminishes necrosis: New insight to magnetic therapy in urology. J. Physiol. Pharmacol. 2012, 63, 397–401. [Google Scholar] [PubMed]
- Filipovic, N.; Djukic, T.; Radovic, M.; Cvetkovic, D.; Curcic, M.; Markovic, S.; Peulic, A.; Jeremic, B. Electromagnetic field investigation on different cancer cell lines. Cancer Cell Int. 2014, 14, 84. Available online: http://www.cancerci.com/content/14/1/84 (accessed on 18 April 2019). [CrossRef]
- Wang, H.; Zhang, X. Magnetic fields and reactive oxygen species. Int. J. Mol. Sci. 2017, 18, 2175. [Google Scholar] [CrossRef]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF Transcription Factors, Inflammation, and Immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef]
- Sepehrimanesh, M.; Kazemipour, N.; Saeb, M.; Nazifi, S.; Davis, D.L. Proteomic analysis of continuous 900-MHz radiofrequency electromagnetic field exposure in testicular tissue: A rat model of human cell phone exposure. Environ. Sci. Pollut. Res. 2017, 24, 13666–13673. [Google Scholar] [CrossRef]
- Bagkos, G.; Koufopoulos, K.; Piperi, C. A new model for mitochondrial membrane potential production and storage. Med. Hypotheses 2014, 83, 175–181. [Google Scholar] [CrossRef]
- Bagkos, G.; Koufopoulos, K.; Piperi, C. Mitochondrial emitted electromagnetic signals mediate retrograde signaling. Med. Hypotheses 2015, 85, 810–818. [Google Scholar] [CrossRef]
- Walter, P.B.; Knutson, M.D.; Paler-Martinez, A.; Lee, S.; Xu, Y.; Viteri, F.E.; Ames, B.N. Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats. Proc. Natl. Acad. Sci. USA 2002, 99, 2264–2269. [Google Scholar] [CrossRef] [PubMed]
- Yakymenko, I.; Tsybulin, O.; Sidorik, E.; Henshel, D.; Kyrylenko, O.; Kyrylenko, S. Oxidative mechanisms of biological activity of low-intensity radiofrequency radiation. Electromagn. Biol. Med. 2016, 35, 186–202. [Google Scholar] [CrossRef] [PubMed]
- Kivrac, E.F.; Yurt, K.K.; Kaplan, A.A.; Alkan, I.; Gamze, A. Effects of electromagnetic fields exposure on the antioxidant defense system. J. Microsc. Ultrastruct. 2017, 5, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, Y.; Shimizu, S.; Tsujimoto, Y. Intracellular ATP Levels Determine Cell Death Fate by Apoptosis or Necrosis. Cancer Res. 1997, 57, 1835–1840. [Google Scholar] [PubMed]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Sanders, A.P.; Joines, W.T.; Allis, J.W. Effects of Continuous-Wave, Pulsed, and Sinusoidal-Amplitude-Modulated Microwaves on Brain Energy Metabolism. Bioelectromagnetics 1985, 6, 89–97. [Google Scholar] [CrossRef]
- Sanders, A.P.; Schaefer, D.J.; Joines, W.T. Microwave Effects on Energy Metabolism of Rat Brain. Bioelectromagnetics 1980, 1, 171–181. [Google Scholar] [CrossRef]
- BioInitiative Working Group. A Rationale for Biologically-based Exposure Standards for Low-Intensity Electromagnetic Radiation 2012. Available online: Bioinitiative.org (accessed on 18 April 2019).
- Houston, M.A.; Augenlicht, L.H.; Heerdt, B.G. Intrinsic Mitochondrial Membrane Potential and Associated Tumor Phenotype are Independent of MUC1 Over-Expression. PLoS ONE 2011, 6, e25207. [Google Scholar] [CrossRef]
- Forrest, M.D. Why cancer cells have a more hyperpolarised mitochondrial membrane potential and emergent prospects for therapy. bioRxiv 2015, 025197. [Google Scholar] [CrossRef]
- Solaini, G.; Sgarbi, G.; Baracca, A. Oxidative phosphorylation in cancer cells. Biochim. Biophys. Acta 2011, 1807, 534–542. [Google Scholar] [CrossRef]
- Cai, Y.; Martens, G.A.; Hinke, S.A.; Heimberg, H.; Pipeleers, D.; Van de Casteele, M. Increased oxygen radical formation and mitochondrial dysfunction mediate beta cell apoptosis under conditions of AMP-activated protein kinase stimulation. Free Radic. Biol. Med. 2007, 42, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Baigi, M.G.; Brault, L.; Néguesque, A.; Beley, M.; El Hilali, R.; Gaüzère, F.; Bagrel, D. Apoptosis/necrosis switch in two different cancer cell lines: Influence of benzoquinone- and hydrogen peroxide-induced oxidative stress intensity, and glutathione. Toxicol. In Vitro 2008, 22, 1547–1554. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Araki, E. Impact of Mitochondrial ROS Production in the Pathogenesis of Diabetes Mellitus and its Complications. Antioxid. Redox Signal. 2007, 9, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Fernyhough, P.; Roy Chowdhury, S.K.; Schmidt, R.E. Mitochondrial stress and the pathogenesis of diabetic neuropathy. Expert Rev. Endocrinol. Metab. 2010, 5, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Pulakat, L.; Whaley-Connell, A.; Sowers, J.R. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J. Mol. Med. 2010, 88, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.; Apostolova, N.; Hernandez-Mijares, A.; Herance, R.; Victor, V.M. Oxidative stress and endothelial dysfunction in cardiovascular disease: Mitochondria-targeted therapeutics. Curr. Med. Chem. 2010, 17, 3827–3841. [Google Scholar] [CrossRef] [PubMed]
- Camara, A.K.S.; Bienengraeber, M.; Stowe, D.F. Mitochondrial approaches to protect against cardiac ischemia and reperfusion injury. Front. Physiol. 2011, 2, 13. [Google Scholar] [CrossRef]
- Kones, R. Parkinson’s disease: Mitochondrial molecular pathology, inflammation, statins, and therapeutic neuroprotective nutrition. Nutr. Clin. Pract. 2010, 25, 371–389. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Chesselet, M.-F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef]
- Ferreira, I.L.; Resende, R.; Ferreiro, E.; Rego, A.C.; Pereira, C.F. Multiple defects in energy metabolism in Alzheimer’s disease. Curr. Drug Targets 2010, 11, 1193–1206. [Google Scholar] [CrossRef]
- Sas, K.; Pardutz, A.; Toldi, J.; Vecsei, L. Dementia, stroke and migraine–some common pathological mechanisms. J. Neurol. Sci. 2010, 299, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.L. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta 2010, 1802, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, H.; Manfredi, G. Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech. Ageing Dev. 2010, 131, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Haas, R.H. Autism and mitochondrial disease. Dev. Disabil. Res. Rev. 2010, 16, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Scaglia, F. The role of mitochondrial dysfunction in psychiatric disease. Dev. Disabil. Res. Rev. 2010, 16, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Exner, N.; Lutz, A.K.; Haass, C.; Winklhofe, K.F. Mitochondrial dysfunction in Parkinson’s disease: Molecular mechanisms and pathophysiological consequences. EMBO J. 2012, 31, 3038–3062. [Google Scholar] [CrossRef] [PubMed]
- Ravera, S.; Panfoli, I. Role of myelin sheath energy metabolism in neurodegenerative diseases. Neural Regen. Res. 2015, 10, 1570–1571. [Google Scholar] [CrossRef]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef]
- Besse-Patin, A.; Estall, J.L. An Intimate Relationship between ROS and Insulin Signalling: Implications for Antioxidant Treatment of Fatty Liver Disease. Int. J. Cell Biol. 2014, 2014, 519153. [Google Scholar] [CrossRef]
- Wang, X.; Roper, M.G. Measurement of DCF fluorescence as a measure of reactive oxygen species in murine islets of Langerhans. Anal. Methods 2014, 6, 3019–3024. [Google Scholar] [CrossRef]
- Meinhardt, A.; Wilhelm, B.; Seitz, J. Expression of mitochondrial marker proteins during spermatogenesis. Hum. Reprod. Update 1999, 5, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [PubMed]
- Lane, N. The Vital Question. Energy, Evolution, and the Origins of Complex Life; WW Norton & Co: New York, NY, USA; London, UK, 2015. [Google Scholar]
- Wolff, J.N.; Ladoukakis, E.D.; Enríquez, J.A.; Dowling, D.K. Mitonuclear interactions: Evolutionary consequences over multiple biological scales. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130443. [Google Scholar] [CrossRef] [PubMed]
- Lionaki, E.; Gkikas, I.; Tavernarakis, N. Differential Protein Distribution between the Nucleus and Mitochondria: Implications in Aging. Front. Genet. 2016, 7, 162. [Google Scholar] [CrossRef]
- Winkler, J.R.; Di Bilio, A.J.; Farrow, N.A.; Richards, J.H.; Gray, H.B. Electron tunneling in biological molecules. Pure Appl. Chem. 1999, 71, 1753–1764. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cell Categories | Annexin V Result | 7-AAD Result |
|---|---|---|
| Died via necrosis, not by apoptosis | − | + |
| In early stages of apoptosis | + | − |
| In late stages, or dead by apoptosis | + | + |
| Early | Late | Total | ||
|---|---|---|---|---|
| Magnetic Field | Necrosis (%) | Apoptosis (%) | Apoptosis (%) | Death (%) |
| Oxygen (n) | SD (%) | SD (%) | SD (%) | SD (%) |
| Incubator Field | 0.45 | 6.35 | 7.83 | 14.63 |
| High O2 (6) | 38 | 50 | 9.7 | 19 |
| Incubator Field | 0.67 | 5.77 | 8.7 | 15.13 |
| Low O2 (6) | 32 | 45 | 14 | 24 |
| Zero Field | 0.5 | 3.83 | 9.3 | 13.63 |
| High O2 (4) | 35 | 32 | 28 | 20 |
| Zero Field | 0.47 | 3.77 | 6.27 | 10.5 |
| Low O2 (4) | 32 | 52 | 63 | 55 |
| Average (20) | 0.52 | 4.93 | 8.03 | 13.47 |
| 17 | 23 | 14 | 13 |
| Transferred to | Incubator Field High O2 | Incubator Field Low O2 | Zero Field High O2 | Zero Field Low O2 | NECROSIS | ||
| Starting Culture | |||||||
| Incubator Field/High O2 | 1 | 1.3 ± 0.5 | 2.7 ± 1.3 | 3.5 ± 1.9 | |||
| Incubator Field/Low O2 | 4.2 ± 2.8 | 1 | 4.46 ± 2.2 | 3.1 ± 1.5 | |||
| Zero Field/High O2 | 3.9 ± 2.6 | 4.21 ± 3.1 | 1 | 1.62 ± 0.7 | |||
| Zero Field/Low O2 | 4.1 ± 2.37 | 3.7 ± 2.1 | 3.9 ± 1.8 | 1 | |||
| Transferred to | Incubator Field High O2 | Incubator Field Low O2 | Zero Field High O2 | Zero Field Low O2 | EARLY APOPTOSIS | ||
| Starting culture | |||||||
| Incubator Field/High O2 | 1 | 0.31 ± 0.12 | 1.05 ± 0.2 | 2.6 ± 1.1 | |||
| Incubator Field/Low O2 | 1.2 ± 0.33 | 1 | 2.5 ± 0.9 | 1.5 ± 1.1 | |||
| Zero Field/High O2 | 1.33 ± 0.5 | 2.3 ± 1.2 | 1 | 0.3 ± 0.19 | |||
| Zero Field/Low O2 | 1.65 ± 0.82 | 1.28 ± 0.45 | 0.8 ± 0.45 | 1 | |||
| Transferred to | Incubator Field High O2 | Incubator Field Low O2 | Zero Field High O2 | Zero Field Low O2 | LATE APOPTOSIS | ||
| Starting culture | |||||||
| Incubator Field/High O2 | 1 | 2.4 ± 1.12 | 1.1 ± 0.65 | 2.7 ± 0.89 | |||
| Incubator Field/Low O2 | 0.9 ± 0.46 | 1 | 1.85 ± 0.81 | 1.4 ± 0.62 | |||
| Zero Field/High O2 | 1.06 ± 0.83 | 2.9 ± 1.21 | 1 | 3 ± 1.51 | |||
| Zero Field/Low O2 | 2.2 ± 0.72 | 1.02 ± 0.58 | 0.7 ± 0.53 | 1 | |||
| Oxygen | Rise | Fall | Rise | Rise | H% | Rise | L% | Fall | L% | H% | Fall | Fall | Δ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MF | Fall | Rise | Incu | Rise | Rise | Zero | Rise | Fall | Fall | Fall | Zero | Incu | |
| Necrosis | 4.46 | 4.21 | 4.2 | 4.1 | 3.9 | 3.9 | 3.7 | 3.5 | 3.1 | 2.7 | 1.62 | 1.3 | 3.4 |
| Late Apoptosis | 1.85 | 2.9 | 0.9 | 2.2 | 1.06 | 0.7 | 1.02 | 2.7 | 1.4 | 1.85 | 3 | 2.4 | 4.3 |
| Early Apoptosis | 2.5 | 2.3 | 1.2 | 1.65 | 1.33 | 0.8 | 1.28 | 2.6 | 1.5 | 0.3 | 0.3 | 0.31 | 8.7 |
| All Apoptosis | 4.35 | 5.2 | 2.1 | 3.85 | 2.39 | 1.5 | 2.3 | 5.3 | 2.9 | 2.15 | 3.3 | 2.71 | 3.5 |
| Late/Early Apoptosis | 0.74 | 1.26 | 0.75 | 1.33 | 0.80 | 0.88 | 0.80 | 1.04 | 0.93 | 6.17 | 10.00 | 7.74 | 13.5 |
| Total Damage | 8.81 | 9.41 | 6.3 | 7.95 | 6.29 | 5.4 | 6 | 8.8 | 6 | 4.85 | 4.92 | 4.01 | 2.4 |
| Necrosis/Apoptosis | 1.03 | 0.81 | 2 | 1.06 | 1.63 | 2.6 | 1.61 | 0.66 | 1.07 | 1.26 | 0.49 | 0.48 | 5.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Héroux, P. Magnetic Fields Trump Oxygen in Controlling the Death of Erythro-Leukemia Cells. Appl. Sci. 2019, 9, 5318. https://doi.org/10.3390/app9245318
Li Y, Héroux P. Magnetic Fields Trump Oxygen in Controlling the Death of Erythro-Leukemia Cells. Applied Sciences. 2019; 9(24):5318. https://doi.org/10.3390/app9245318
Chicago/Turabian StyleLi, Ying, and Paul Héroux. 2019. "Magnetic Fields Trump Oxygen in Controlling the Death of Erythro-Leukemia Cells" Applied Sciences 9, no. 24: 5318. https://doi.org/10.3390/app9245318
APA StyleLi, Y., & Héroux, P. (2019). Magnetic Fields Trump Oxygen in Controlling the Death of Erythro-Leukemia Cells. Applied Sciences, 9(24), 5318. https://doi.org/10.3390/app9245318