Hypermethylation of Circulating Free DNA in Cutaneous Melanoma

 , , ,
, , ,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasma Preparation
2.2. Purification of Circulating Free DNA from Plasma
2.3. Analysis of ctDNA from Plasma
2.4. PCR Primer Design
2.5. Sample QC and Preparation
2.6. Bisulfite Conversion of DNA
2.7. PCR Amplification
2.8. Library Construction
- (1)
- No shearing of the DNA
- (2)
- No removal of large and small DNA fragments (i.e., no size selection).
2.9. Library QC
2.10. Library Pooling and Sequencing
2.11. Bioinformatics for Bisulfite Amplicon Sequencing
2.12. Statistical Analysis of Methylation Data
3. Results
3.1. Cohort and Gene Targets
3.2. Determination of the Fraction of Circulating Free DNA Methylation
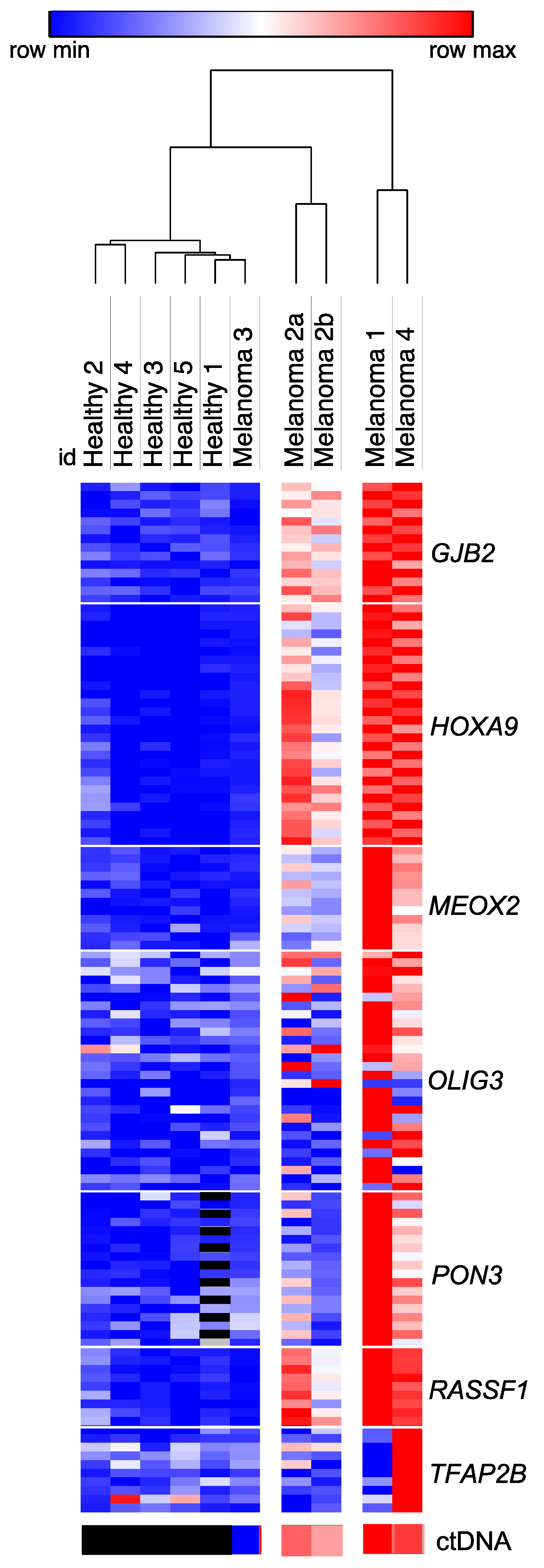
3.3. Identification of Genes Which Discriminate for Melanoma Based on Differential Methylation of Circulating Free DNA
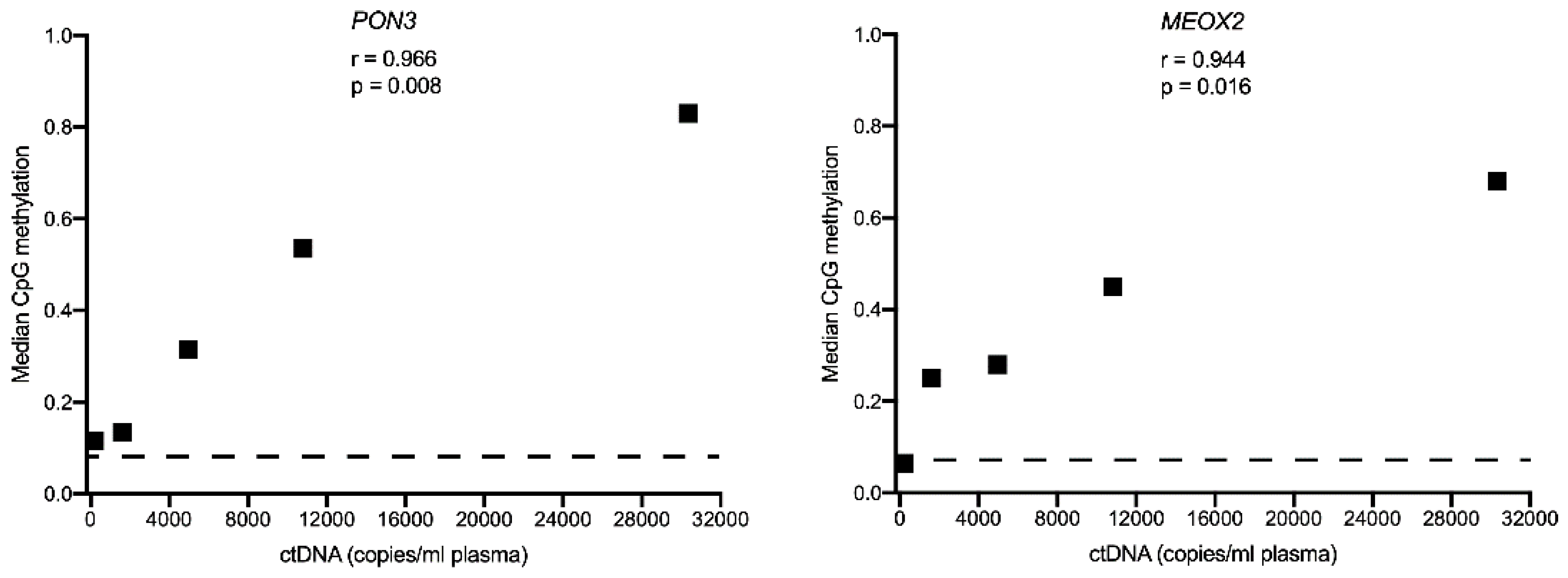
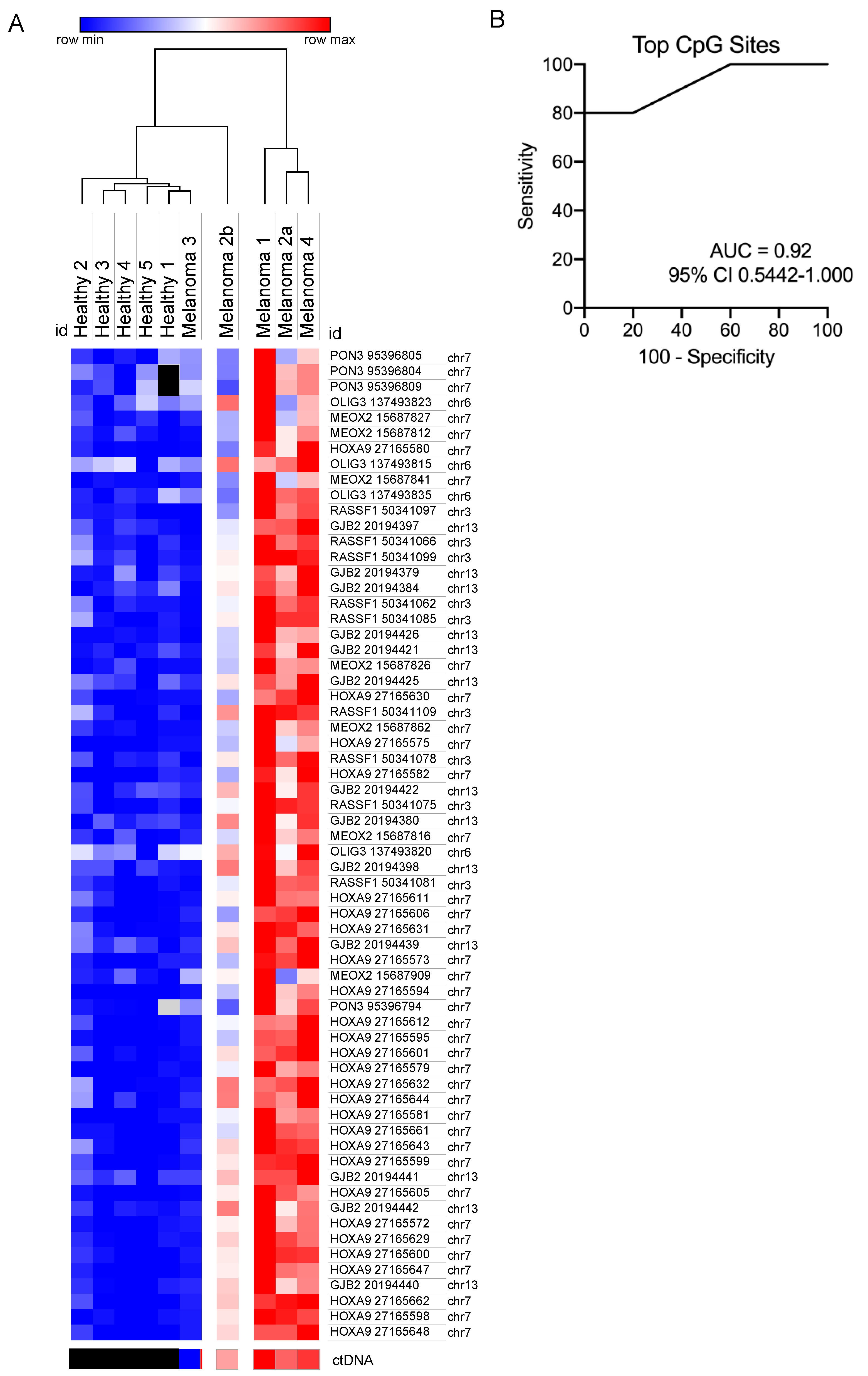
3.4. Comparison of Circulating Free DNA Methylation and Copy Number Levels in Melanoma Patients
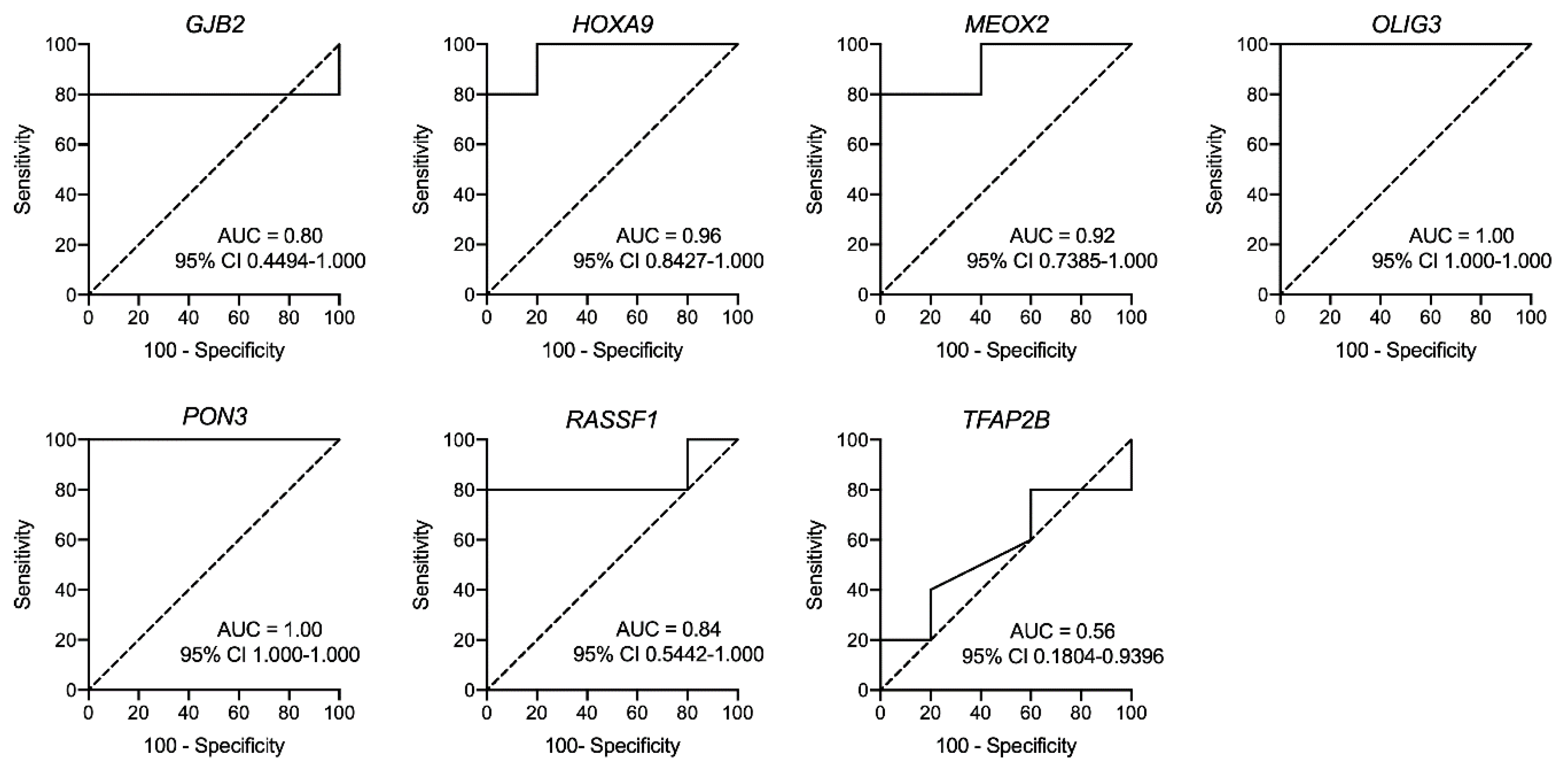
3.5. Evaluation of Methylated CpG Sites in Circulating Free DNA as a Biomarker for Detecting for Metastatic Melanoma
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mathai, R.A.; Vidya, R.V.S.; Reddy, B.S.; Thomas, L.; Udupa, K.; Kolesar, J.; Rao, M. Potential utility of liquid biopsy as a diagnostic and prognostic tool for the assessment of solid tumors: Implications in the precision oncology. J. Clin. Med. 2019, 8, 373. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Lee, J.H.; Diefenbach, R.J.; Kefford, R.F.; Rizos, H. Liquid biomarkers in melanoma: Detection and discovery. Mol. Cancer 2018, 17, 8. [Google Scholar] [CrossRef] [PubMed]
- Barbany, G.; Arthur, C.; Lieden, A.; Nordenskjold, M.; Rosenquist, R.; Tesi, B.; Wallander, K.; Tham, E. Cell free tumour DNA testing for early detection of cancer-a potential future tool. J. Intern. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra224. [Google Scholar] [CrossRef]
- Cabel, L.; Riva, F.; Servois, V.; Livartowski, A.; Daniel, C.; Rampanou, A.; Lantz, O.; Romano, E.; Milder, M.; Buecher, B.; et al. Circulating tumor DNA changes for early monitoring of anti-PD1 immunotherapy: A proof-of-concept study. Ann. Oncol. 2017, 28, 1996–2001. [Google Scholar] [CrossRef]
- Lee, J.H.; Long, G.V.; Boyd, S.; Lo, S.; Menzies, A.M.; Tembe, V.; Guminski, A.; Jakrot, V.; Scolyer, R.A.; Mann, G.J.; et al. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann. Oncol. 2017, 28, 1130–1136. [Google Scholar] [CrossRef]
- Gray, E.S.; Rizos, H.; Reid, A.L.; Boyd, S.C.; Pereira, M.R.; Lo, J.; Tembe, V.; Freeman, J.; Lee, J.H.; Scolyer, R.A.; et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget 2015, 6, 42008–42018. [Google Scholar] [CrossRef]
- Dawson, S.J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef]
- Hou, H.; Yang, X.; Zhang, J.; Zhang, Z.; Xu, X.; Zhang, X.; Zhang, C.; Liu, D.; Yan, W.; Zhou, N.; et al. Discovery of targetable genetic alterations in advanced non-small cell lung cancer using a next-generation sequencing-based circulating tumor DNA assay. Sci. Rep. 2017, 7, 14605. [Google Scholar] [CrossRef]
- Du, J.; Wu, X.; Tong, X.; Wang, X.; Wei, J.; Yang, Y.; Chang, Z.; Mao, Y.; Shao, Y.W.; Liu, B. Circulating tumor DNA profiling by next generation sequencing reveals heterogeneity of crizotinib resistance mechanisms in a gastric cancer patient with MET amplification. Oncotarget 2017, 8, 26281–26287. [Google Scholar] [CrossRef]
- Thompson, J.C.; Yee, S.S.; Troxel, A.B.; Savitch, S.L.; Fan, R.; Balli, D.; Lieberman, D.B.; Morrissette, J.D.; Evans, T.L.; Bauml, J.; et al. Detection of therapeutically targetable driver and resistance mutations in lung cancer patients by next-generation sequencing of cell-free circulating tumor DNA. Clin. Cancer Res. 2016, 22, 5772–5782. [Google Scholar] [CrossRef]
- Tan, L.; Sandhu, S.; Lee, R.J.; Li, J.; Callahan, J.; Ftouni, S.; Dhomen, N.; Middlehurst, P.; Wallace, A.; Raleigh, J.; et al. Prediction and monitoring of relapse in stage III melanoma using circulating tumor DNA. Ann. Oncol. 2019, 30, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Saw, R.P.; Thompson, J.F.; Lo, S.; Spillane, A.J.; Shannon, K.F.; Stretch, J.R.; Howle, J.; Menzies, A.M.; Carlino, M.S.; et al. Pre-operative ctDNA predicts survival in high-risk stage III cutaneous melanoma patients. Ann. Oncol. 2019, 30, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, R.J.; Lee, J.H.; Rizos, H. Monitoring melanoma using circulating free DNA. Am. J. Clin. Dermatol. 2019, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bronkhorst, A.J.; Ungerer, V.; Holdenrieder, S. The emerging role of cell-free DNA as a molecular marker for cancer management. Biomol. Detect. Quantif. 2019, 17, 100087. [Google Scholar] [CrossRef] [PubMed]
- Chin, R.I.; Chen, K.; Usmani, A.; Chua, C.; Harris, P.K.; Binkley, M.S.; Azad, T.D.; Dudley, J.C.; Chaudhuri, A.A. Detection of solid tumor molecular residual disease (MRD) using circulating tumor DNA (ctDNA). Mol. Diagn. Ther. 2019, 23, 311–331. [Google Scholar] [CrossRef]
- Andersen, R.F. Tumor-specific methylations in circulating cell-free DNA as clinically applicable markers with potential to substitute mutational analyses. Expert Rev. Mol. Diagn. 2018, 18, 1–9. [Google Scholar] [CrossRef]
- Gai, W.; Sun, K. Epigenetic Biomarkers in Cell-Free DNA and Applications in Liquid Biopsy. Genes 2019, 10, 32. [Google Scholar] [CrossRef]
- Zeng, H.; He, B.; Yi, C.; Peng, J. Liquid biopsies: DNA methylation analyses in circulating cell-free DNA. J. Genet. Genom. 2018, 45, 185–192. [Google Scholar] [CrossRef]
- Xu, R.H.; Wei, W.; Krawczyk, M.; Wang, W.; Luo, H.; Flagg, K.; Yi, S.; Shi, W.; Quan, Q.; Li, K.; et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat. Mater. 2017, 16, 1155–1161. [Google Scholar] [CrossRef]
- Wen, L.; Li, J.; Guo, H.; Liu, X.; Zheng, S.; Zhang, D.; Zhu, W.; Qu, J.; Guo, L.; Du, D.; et al. Genome-scale detection of hypermethylated CpG islands in circulating cell-free DNA of hepatocellular carcinoma patients. Cell Res. 2015, 25, 1250–1264. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Paredes, M.; Esteller, M. Cancer epigenetics reaches mainstream oncology. Nat. Med. 2011, 17, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Worm Orntoft, M.B.; Jensen, S.O.; Hansen, T.B.; Bramsen, J.B.; Andersen, C.L. Comparative analysis of 12 different kits for bisulfite conversion of circulating cell-free DNA. Epigenetics 2017, 12, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Wouters, J.; Vizoso, M.; Martinez-Cardus, A.; Carmona, F.J.; Govaere, O.; Laguna, T.; Joseph, J.; Dynoodt, P.; Aura, C.; Foth, M.; et al. Comprehensive DNA methylation study identifies novel progression-related and prognostic markers for cutaneous melanoma. BMC Med. 2017, 15, 101. [Google Scholar] [CrossRef] [PubMed]
- Tanemura, A.; Terando, A.M.; Sim, M.S.; van Hoesel, A.Q.; de Maat, M.F.; Morton, D.L.; Hoon, D.S. CpG island methylator phenotype predicts progression of malignant melanoma. Clin. Cancer Res. 2009, 15, 1801–1807. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Toung, J.M.; Jassowicz, A.F.; Vijayaraghavan, R.; Kang, H.; Zhang, R.; Kruglyak, K.M.; Huang, H.J.; Hinoue, T.; Shen, H.; et al. Targeted methylation sequencing of plasma cell-free DNA for cancer detection and classification. Ann. Oncol. 2018, 29, 1445–1453. [Google Scholar] [CrossRef]
- Widschwendter, M.; Evans, I.; Jones, A.; Ghazali, S.; Reisel, D.; Ryan, A.; Gentry-Maharaj, A.; Zikan, M.; Cibula, D.; Eichner, J.; et al. Methylation patterns in serum DNA for early identification of disseminated breast cancer. Genome Med. 2017, 9, 115. [Google Scholar] [CrossRef]
- Widschwendter, M.; Zikan, M.; Wahl, B.; Lempiainen, H.; Paprotka, T.; Evans, I.; Jones, A.; Ghazali, S.; Reisel, D.; Eichner, J.; et al. The potential of circulating tumor DNA methylation analysis for the early detection and management of ovarian cancer. Genome Med. 2017, 9, 116. [Google Scholar] [CrossRef]
- Calapre, L.; Warburton, L.; Millward, M.; Ziman, M.; Gray, E.S. Circulating tumour DNA (ctDNA) as a liquid biopsy for melanoma. Cancer Lett. 2017, 404, 62–69. [Google Scholar] [CrossRef]
- Moran, B.; Silva, R.; Perry, A.S.; Gallagher, W.M. Epigenetics of malignant melanoma. Semin. Cancer Biol. 2018, 51, 80–88. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 28 May 2018).
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Karolchik, D.; Hinrichs, A.S.; Kent, W.J. The UCSC genome browser. Curr. Protoc. Hum. Genet. 2011. [Google Scholar] [CrossRef]
- Shao, C.; Dai, W.; Li, H.; Tang, W.; Jia, S.; Wu, X.; Luo, Y. The relationship between RASSF1A gene promoter methylation and the susceptibility and prognosis of melanoma: A meta-analysis and bioinformatics. PLoS ONE 2017, 12, e0171676. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Long, J.; Lei, S. Promoter methylation as biomarkers for diagnosis of melanoma: A systematic review and meta-analysis. J. Cell. Physiol. 2019, 234, 7356–7367. [Google Scholar] [CrossRef]
- de Unamuno Bustos, B.; Murria Estal, R.; Perez Simo, G.; Simarro Farinos, J.; Pujol Marco, C.; Navarro Mira, M.; Alegre de Miquel, V.; Ballester Sanchez, R.; Sabater Marco, V.; Llavador Ros, M.; et al. Aberrant DNA methylation is associated with aggressive clinicopathological features and poor survival in cutaneous melanoma. Br. J. Dermatol. 2018, 179, 394–404. [Google Scholar] [CrossRef]
- Gao, L.; Smit, M.A.; van den Oord, J.J.; Goeman, J.J.; Verdegaal, E.M.; van der Burg, S.H.; Stas, M.; Beck, S.; Gruis, N.A.; Tensen, C.P.; et al. Genome-wide promoter methylation analysis identifies epigenetic silencing of MAPK13 in primary cutaneous melanoma. Pigment. Cell Melanoma Res. 2013, 26, 542–554. [Google Scholar] [CrossRef]
- Salvianti, F.; Orlando, C.; Massi, D.; De Giorgi, V.; Grazzini, M.; Pazzagli, M.; Pinzani, P. Tumor-related methylated cell-free DNA and circulating tumor cells in melanoma. Front. Mol. Biosci. 2015, 2, 76. [Google Scholar] [CrossRef]
- Tellez, C.S.; Shen, L.; Estecio, M.R.; Jelinek, J.; Gershenwald, J.E.; Issa, J.P. CpG island methylation profiling in human melanoma cell lines. Melanoma Res. 2009, 19, 146–155. [Google Scholar] [CrossRef]
- Micevic, G.; Theodosakis, N.; Bosenberg, M. Aberrant DNA methylation in melanoma: Biomarker and therapeutic opportunities. Clin. Epigenet. 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Olova, N.; Krueger, F.; Andrews, S.; Oxley, D.; Berrens, R.V.; Branco, M.R.; Reik, W. Comparison of whole-genome bisulfite sequencing library preparation strategies identifies sources of biases affecting DNA methylation data. Genome Biol. 2018, 19, 33. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Okamoto, A. Degradation of DNA by bisulfite treatment. Bioorg. Med. Chem. Lett. 2007, 17, 1912–1915. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Chung, W.Y.; Qian, M.; Pellegrini, M.; Zhang, M.Q. Characterizing the strand-specific distribution of non-CpG methylation in human pluripotent cells. Nucleic Acids Res. 2014, 42, 3009–3016. [Google Scholar] [CrossRef]
- Laird, C.D.; Pleasant, N.D.; Clark, A.D.; Sneeden, J.L.; Hassan, K.M.; Manley, N.C.; Vary, J.C., Jr.; Morgan, T.; Hansen, R.S.; Stoger, R. Hairpin-bisulfite PCR: Assessing epigenetic methylation patterns on complementary strands of individual DNA molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 204–209. [Google Scholar] [CrossRef]
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Kin Sung, K.W.; Rigoutsos, I.; Loring, J.; et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010, 20, 320–331. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Minor, D.; Ribas, A.; Lebbe, C.; O’Hagan, A.; Arya, N.; Guckert, M.; Schadendorf, D.; Kefford, R.F.; Grob, J.J.; et al. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J. Clin. Oncol. 2013, 31, 3205–3211. [Google Scholar] [CrossRef]
- Huang, D.; Wang, Y.; He, Y.; Wang, G.; Wang, W.; Han, X.; Sun, Y.; Lin, L.; Shan, B.; Shen, G.; et al. Paraoxonase 3 is involved in the multi-drug resistance of esophageal cancer. Cancer Cell Int. 2018, 18, 168. [Google Scholar] [CrossRef]
- Shui, I.M.; Wong, C.J.; Zhao, S.; Kolb, S.; Ebot, E.M.; Geybels, M.S.; Rubicz, R.; Wright, J.L.; Lin, D.W.; Klotzle, B.; et al. Prostate tumor DNA methylation is associated with cigarette smoking and adverse prostate cancer outcomes. Cancer 2016, 122, 2168–2177. [Google Scholar] [CrossRef]
- Baharudin, R.; Ab Mutalib, N.S.; Othman, S.N.; Sagap, I.; Rose, I.M.; Mohd Mokhtar, N.; Jamal, R. Identification of predictive DNA methylation biomarkers for chemotherapy response in colorectal cancer. Front. Pharmacol. 2017, 8, 47. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject | Age (years) | Current Treatment at Sampling | Tumor Mutation | ctDNA (copies/mL plasma) # | wtDNA (copies/mL plasma)# | ctDNA Fraction (MAF%) | Input Circulating Free DNA for Bisulfite Conversion (ng) |
|---|---|---|---|---|---|---|---|
| Healthy 1 | 32 | No treatment | ND | ND | 3238 | ND | 23.56 |
| Healthy 2 | 53 | No treatment | ND | ND | 2188 | ND | 18.22 |
| Healthy 3 | 37 | No treatment | ND | ND | 2045 | ND | 17.33 |
| Healthy 4 | 30 | No treatment | ND | ND | 1477 | ND | 14.67 |
| Healthy 5 | 54 | No treatment | ND | ND | 1420 | ND | 15.11 |
| Melanoma 1 | 76 | Pembro | BRAF V600E | 30,350 | 56,250 | 35 | 21.78 |
| Melanoma 2a (PRE *) | 61 | Combi-DT | BRAF V600K | 4960 | 5800 | 46.1 | 15.00 |
| Melanoma 2b (EDT **) | 61 | Ipi + nivo | BRAF V600K | 1615 | 5800 | 21.8 | 20.80 |
| Melanoma 3 | 75 | No treatment. Pre-ipi | NRAS Q61R | 200 | 252,200 | 0.08 | 20.00 |
| Melanoma 4 | 38 | Ipi + pembro | NRAS Q61K | 10,800 | 10,250 | 51.3 | 24.89 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diefenbach, R.J.; Lee, J.H.; Chandler, D.; Wang, Y.; Pflueger, C.; Long, G.V.; Scolyer, R.A.; Carlino, M.S.; Menzies, A.M.; Kefford, R.F.; et al. Hypermethylation of Circulating Free DNA in Cutaneous Melanoma. Appl. Sci. 2019, 9, 5074. https://doi.org/10.3390/app9235074
Diefenbach RJ, Lee JH, Chandler D, Wang Y, Pflueger C, Long GV, Scolyer RA, Carlino MS, Menzies AM, Kefford RF, et al. Hypermethylation of Circulating Free DNA in Cutaneous Melanoma. Applied Sciences. 2019; 9(23):5074. https://doi.org/10.3390/app9235074
Chicago/Turabian StyleDiefenbach, Russell J., Jenny H. Lee, David Chandler, Yinan Wang, Christian Pflueger, Georgina V. Long, Richard A. Scolyer, Matteo S. Carlino, Alexander M. Menzies, Richard F. Kefford, and et al. 2019. "Hypermethylation of Circulating Free DNA in Cutaneous Melanoma" Applied Sciences 9, no. 23: 5074. https://doi.org/10.3390/app9235074
APA StyleDiefenbach, R. J., Lee, J. H., Chandler, D., Wang, Y., Pflueger, C., Long, G. V., Scolyer, R. A., Carlino, M. S., Menzies, A. M., Kefford, R. F., & Rizos, H. (2019). Hypermethylation of Circulating Free DNA in Cutaneous Melanoma. Applied Sciences, 9(23), 5074. https://doi.org/10.3390/app9235074