2.3. Electrical Equivalent Circuit Model
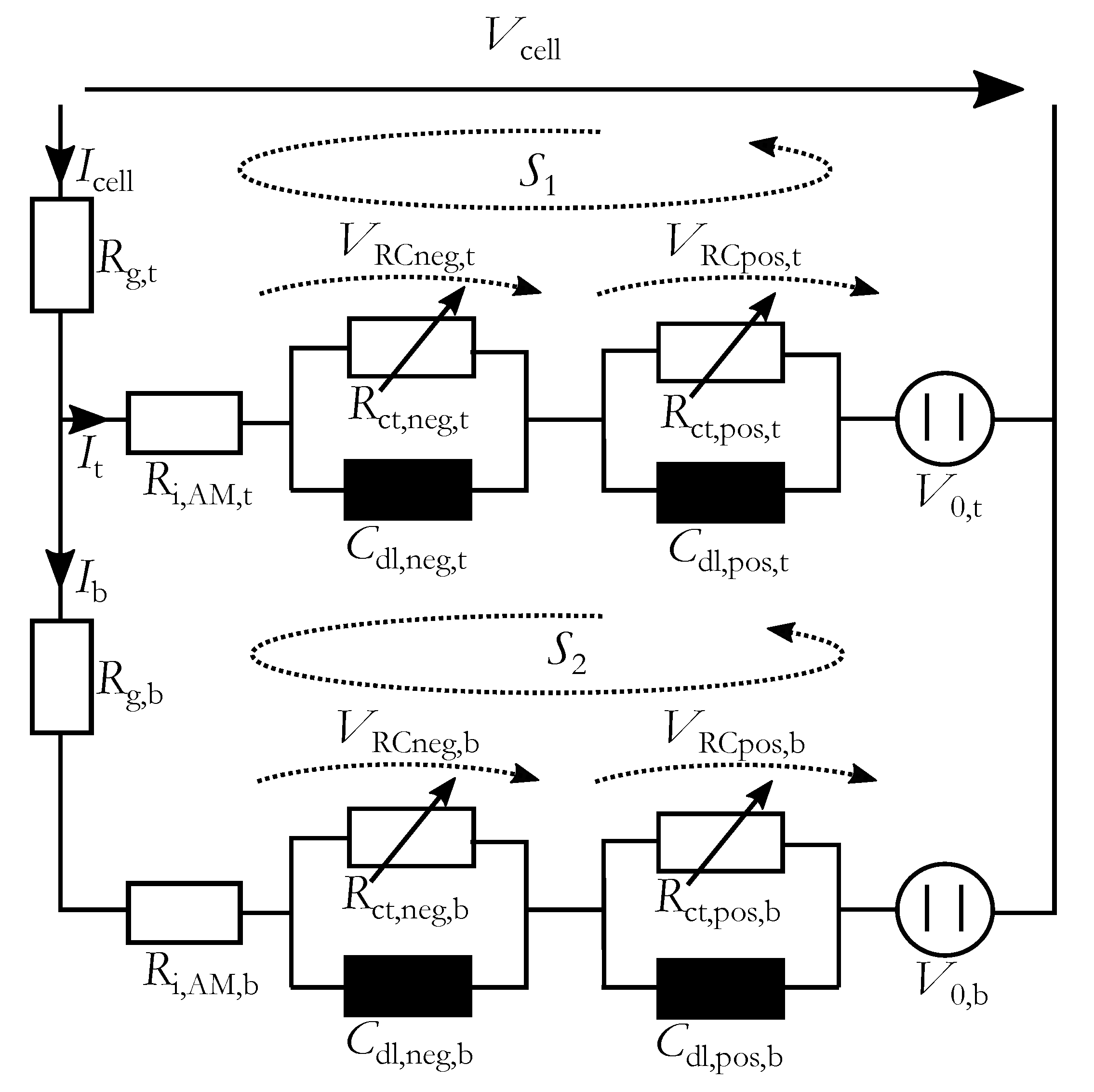
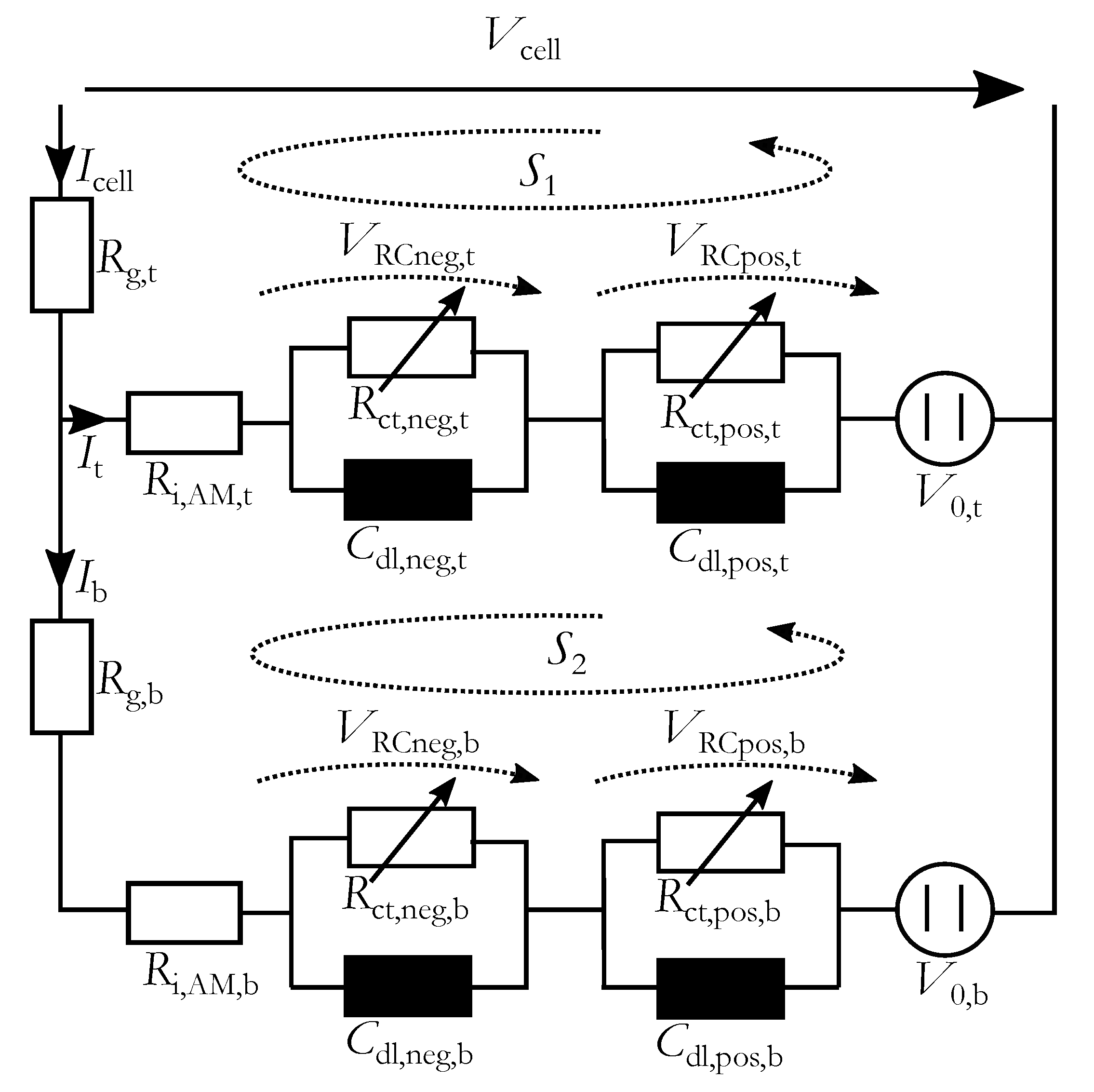
To reproduce the changes of impedance spectrum due to stratification a spatially-resolved equivalent electrical circuit (srEEC) was used. It consists of two layers to simulate inhomogeneous current distribution over the height of the electrodes, which is intensified by the stratification. Every layer contains a resistance connected in series to two RC-elements with the charge-transfer resistances and the double-layer capacitances for negative and positive electrode, respectively. The current dependency of the charge-transfer process in the lead-acid battery is significant in a wide range of charging and discharging currents and is considered here [
23]. With the corresponding double-layer capacitance the characteristic form of the impedance spectrum is generated. A voltage source indicates the open-circuit voltage (OCV), which depends on the local acid density. The resulting srEEC is shown in
Figure 3.
The relationship between OCV and the density
is assumed to be linear (see Equation (
2)), which is a good approximation for a wide range of the acid density [
8].
The ohmic resistance of a lead-acid battery comprises of the lead grid resistance, the active mass and the electrolyte resistance as well as the resistances of the intersections from grid to active mass and to the electrolyte. Here the grid resistance is considered separately and is named for the top part and for the bottom part. The other components of the ohmic resistance are summarized in the resistances and .
The charge-transfer resistances of the negative
and positive
electrodes were connected in parallel to the corresponding double-layer capacitances (
and
). On the negative electrode a second electro-chemical process is present, which can be modeled with an additional RC-element [
23]. However, this process could not be described yet [
16] and the share to the total battery impedance is small, so that this second RC-element is not considered here.
The Butler-Volmer equation describes the dependency of the charge-transfer rate
I on the potential difference to equilibrium potential of the electrode
and is presented in Equation (
3).
is the exchange current,
is the symmetry factor between charging and discharging reaction,
n is the number of electrons involved in the reaction,
F is the Faraday constant,
R is the gas constant and
T is the temperature in Kelvin [
24]. The Butler-Volmer equation provides the current rate
I through the charge-transfer resistance.
The charge-transfer resistance
is the quotient of
and
I. When
I is measured or determined from a system of equations, which is the case here, Equation (
3) needs to be resolved for
. This was done numerically with the Matlab function
vpasolve.
Due to the nonlinearity of the Butler-Volmer equation, a distinction between the small-signal and the large-signal value of
was made here. The small-signal value
is measured during EIS with a small amplitude of sinusoidal current and corresponds to the derivative of the Butler-Volmer equation resolved accordingly to
(see Equation (
4)). For the calculation of the current distribution in the srEEC the large-signal value
is required (Equation (
5)), which is equal to the integration of the Butler-Volmer equation resolved accordingly to
over current
I [
16].
Equation (
3) could be parameterized for the positive electrode in charging and discharging direction, but for the negative electrode only in discharging direction. In charging direction the low concentration of Pb
-ions, caused by a low dissolution rate of PbSO
crystals, limits the charge-transfer process on the negative electrode and induced an increase of the charge-transfer resistance. The dissolution rate is affected by the surface area volume ratio of the crystals and not by any over-potentials [
16]. In discharging direction the SO
-ions are relevant for the reaction and because of their high concentration the charge-transfer process is not limited. The dissolution rate dependent concentration of Pb
-ions for charging could be modeled and integrated in the Butler-Volmer equation, as it is described by Thele and Sauer [
23,
25] However, this would make the model complex and computationally intensive, so that the measured SoC and current dependency of
in charging direction was described with a surface polynomial function (Equation (
17)).
Because of the inhomogeneous current distribution the SoC levels in the top and the bottom become differently, so that the SoC-dependency of the single elements are included in the model. The dependency on SoC is induced by the changing conductivity and density of the acid, the reduction of porosity and the augmented accumulation of lead-sulfate cystals on the surface of the electrodes towards low SoC.
The exchange current
includes the activity of the SO
-ions in the electrolyte as factor. For the activity the molar concentration of the electrolyte is used approximately [
24]. During the test procedure with stratified electrolyte the molar concentration varied between
mol L
−1 and
mol L
−1, so that due to the concentration change
increased by 47% from low to high concentration. In the parameterization measurements the acid concentration increased only by 6% from
mol L
−1 at 0% SoC to
mol L
−1 at 90% SoC. The small concentration change over SoC during parameterization is caused by the large volume of the electrolyte ( 1200 mL) in the test cell. Because of this small concentration change the dependency of
on molar concentration was separated from the measured SoC dependency. The change of the exchange current over the concentration or the SoC was assumed to be linear, as the OCV SoC relationship was modeled as linear as well (see Equation (
2)). This linear change of
was subtracted from the measured
values over SoC. The modified SoC dependency describes now the change of
caused only by transformations in the active mass and is given in
Section 2.4. The concentration dependency is modeled using Equation (
6) with the present molar concentration
c and
, the exchange current at present SoC and at
, the molar concentration at 0% SoC.
Equation (
6) is applied on
of the positive and the negative electrode in discharging direction. For
in charging direction this separation of the concentration dependency was not made, as the measured increase of the charge-transfer resistance in charging direction is significantly higher than changes generated by the acid concentration.
For the resistances
and
a separated acid concentration dependency had to be introduced as well. During parameterization measurements the conductivity of the acid in the test cell increased from low to high SoC [
8]. However, the parameterization measurements presented a decrease of the conductivity (see
Section 2.4). With 20% the decrease is lower than measured by Thele on lead-acid starter-batteries (37% for 20 °C) [
23]. So the decrease of acid concentration in the test cell extenuated the ohmic resistance increase towards low SoC and the increase is mainly generated by the accumulation of lead-sulfate crystals and the change of porosity. Therefore, a separate consideration of the concentration dependency of
was implemented. The rate of the electrolyte resistance on
could not be measured separately, but it was assumed that the electrolyte resistance provides the main part in
. Equation (
7) describes this dependency with
as the resistance corresponding to the present SoC and determined with Equation (
20),
as the relative change of specific resistance per g/cm
density change (here 3 (g cm
−3)
−1, based on data from Berndt [
8]),
as reference density (here
g cm
−3) and
as the present densities in the top and bottom of the cell.
In case of stratification this equation is used to adapt
in the top and the bottom of srEEC to the local acid densities. The influence of these separations of the acid concentration dependence from the measured SoC dependency is presented in
Figure S1 provided as supplementary material.
The SoC-dependency of the double-layer capacitances are described with simple polynomial functions (provided in
Section 2.4) and no separation of the acid concentration dependency was considered, as the capacitances does not influence the current distribution in the steady-state.
The simulation using this srEEC starts with the calculation of initial values for
and
based on initial local SoC. The capacitances are not charged at the beginning, therefore they can be assumed as short circuit and the voltages
is equal to zero. The current
is determined with Equation (
8) and
is determined with Equation (
12). The current
is the set DC-current during EIS.
In the next step the
values are determined using the Butler-Volmer equation or Equation (
17) for charging currents with
I as the currents, which flow through the resistances. At the beginning these currents are zero.
With Equation (
9) the voltage drops
over the RC-elements are calculated for every RC-element. The parameter
n is the counter for the time steps, while
specifies the values from previous time steps. For
and
the same value are used at first, which are the initial values for
and
.
With the determined
the cell voltage
is calculated with Equation (
10), which was generated with the Kirchhoff’s voltage law applied to the closed circuit
in
Figure 3.
At this point all values in the srEEC are calculated for one simulation step. At next, the local currents
and
are determined for next simulation step using the perviously calculated voltages
(Equations (
11) and (
12)).
With these new values the new local
are determined as well (Equation (
13)). Here to an half of
is used for SoC, as the local SoC corresponds to an half of the electrode, respectively.
The duration of one time step
is adapted to the dynamic change of the currents through the capacitances. Therefore these currents are determined with Equation (
14) for the present simulation step and are compared with the values from previous step.
The duration
is used to calculate
. If the changes of the currents between two steps are too big, Equation (
9) is not valid anymore and the model becomes instable. The current changes are limited to
A. If the limit was exceeded,
is divided by 10 and the values for
are calculated again for the same simulation step, until the differences are below the limit or
is equal
−5 s. A duration of
−5 s for
ensures the stability of the model with the used parameters and the adaption of
reduces the simulation duration and generated data volume.
The determined currents through the capacitances (Equation (
14)) are used in the next simulation step to determine the currents through
, which are the differences between
and
.
The model does not simulate the local acid density on the basis of diffusion, generation and consumption of sulfate ions during charging and discharging, gravity and electrolyte mixing, like in the work of Kowal et al. or Sauer [
19,
25] In the test cell a homogeneous SoC could be set before the measurements with stratification, so that the initial condition of the acid was well known for
Strat. X. Furthermore, the duration of EIS measurements, which have to be simulated, was only
h, so that diffusion processes did not change the local acid densities significantly.
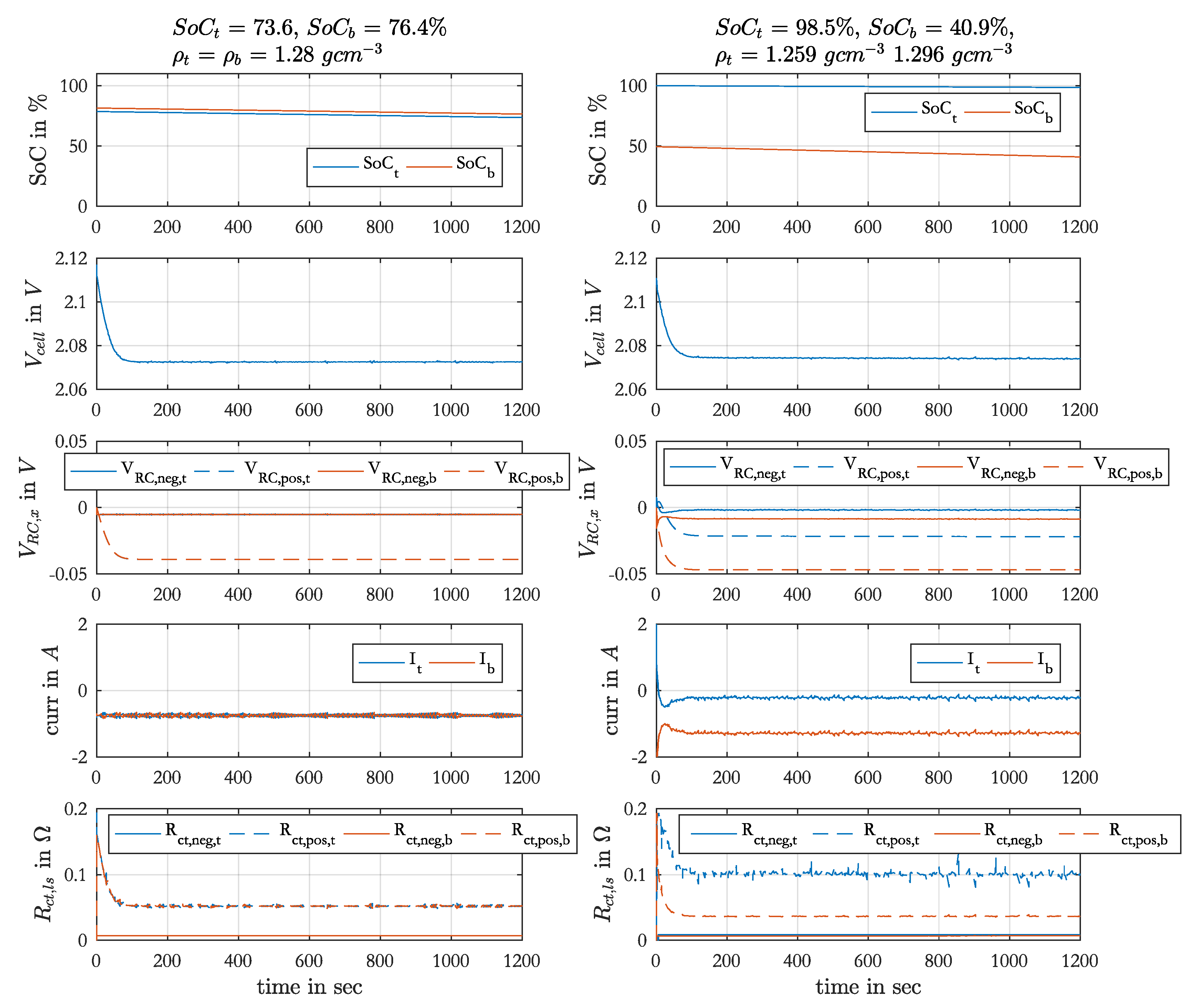
During discharging with constant current the local acid densities are reduced by the consumption of SO ions. The generated density gradient between the pores and the bulk forces diffusion, so that an equilibrium state between consumption and diffusion keeps the acid density constant, but lower than the measured value. The same holds for the charging direction. This local density change cannot be measured and is not considered here.
The stratification itself generated equational local currents over the electrodes between top and bottom part, which discharged the bottom part and charged the top part, so that the density difference decreased measurably over time. This is considered in the model with a linear decrease of the density difference over time, parameterized with the measured acid densities before and after the EIS set. During Strat. 1 the acid density changed with −7 g cm−3 s−1 and during Strat. 2 with a higher rate of −7 g cm−3 s−1 because of the larger density difference.
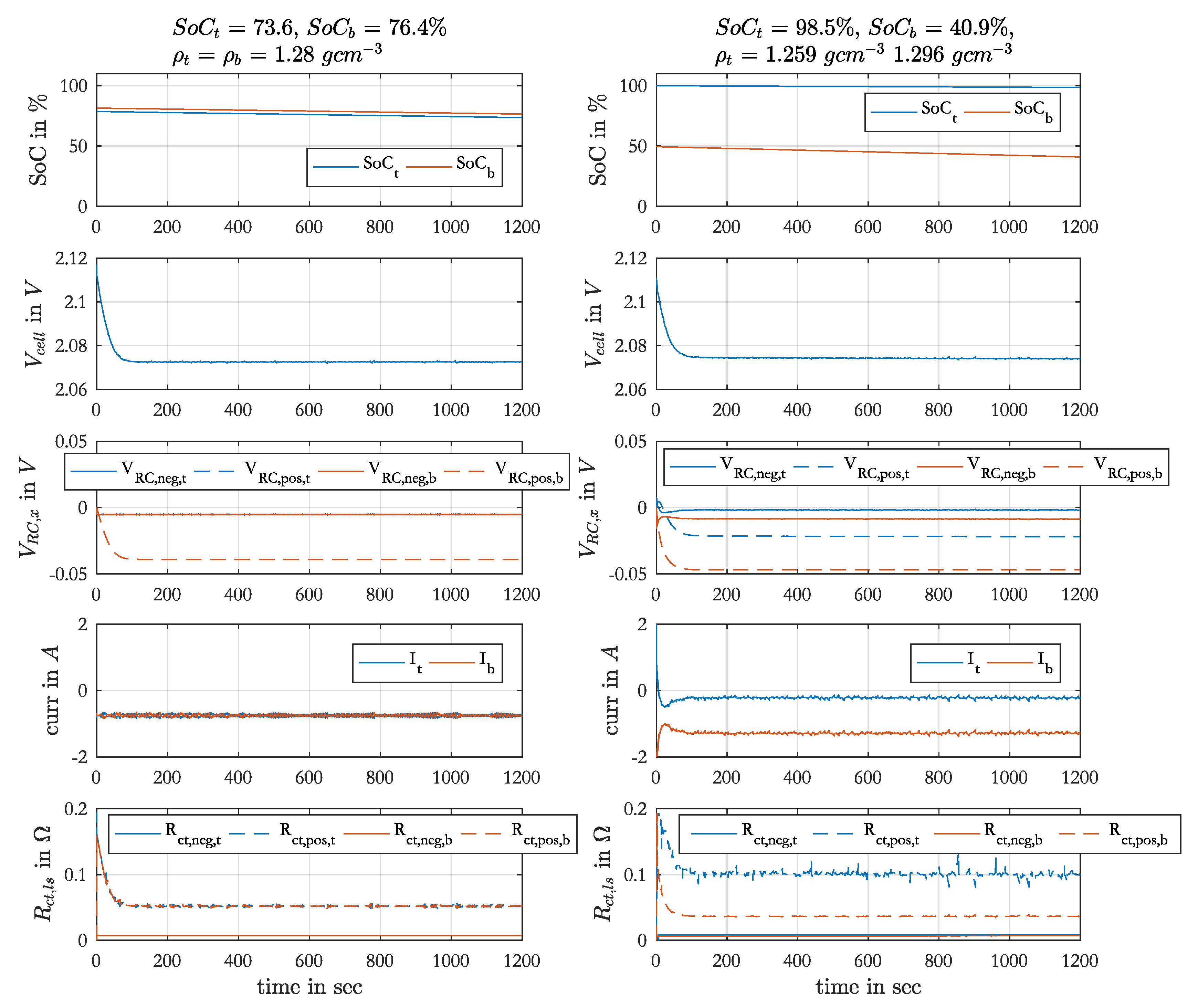
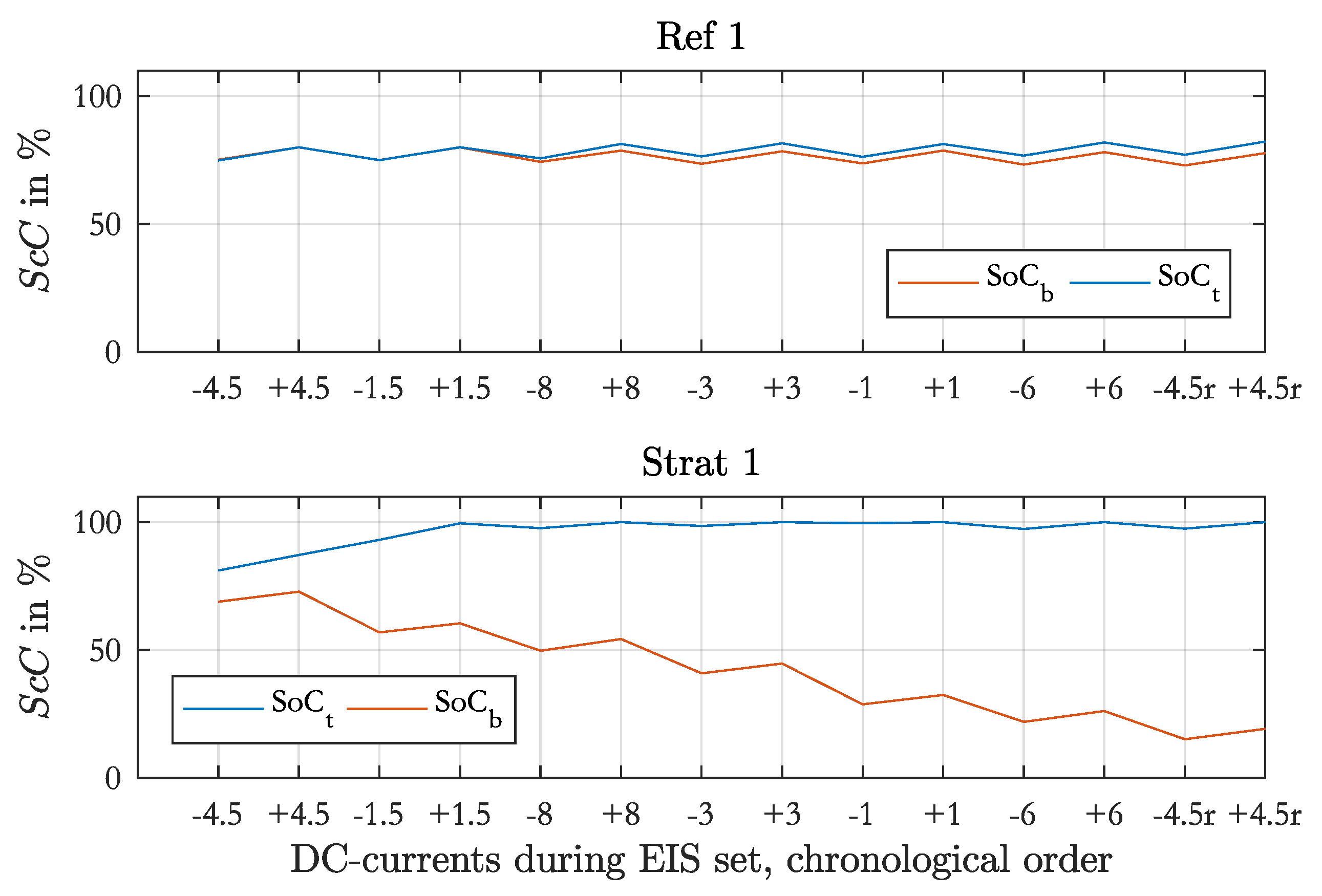
With this model the discharge-charge cycles during an EIS set were simulated. The measured acid densities before the EIS set were used as initial values. The initial local SoC were 80 % for Ref. 1 and Ref. 4. After an EIS set the local SoCs were different and for the subsequent measurements only the acid was either homogenized and stratified and not the local SoC. In these cases the calculated local SoC from the end of the simulated previous measurement were used as initial values for the next simulation.
The output of the simulation is the cell voltage, the local currents, the local SoCs and the values of the srEEC elements over time. Furthermore, one spectrum per discharge or charge phase with constant current is calculated. The impedance of the srEEC is determined at defined times of the simulation with the current srEEC element values using the Kirchhoff’s voltage and current laws. The used equipment for EIS measured the impedances on the test cell at defined frequencies, which can be recalculated to a time, at which the single impedances were determined during EIS measurements with consideration of the three performed sinusoidal waves per frequency. In the simulation the number of frequencies was limited by the maximum SoC change of 4% during EIS as it was the case in the measurements. To generate a shape of the spectra, which is comparable with the measured spectra, the inductance
L of the cell (
H) was included and the double-layer capacitances are expected as Constant-Phase-Elements (CPE) in the impedance calculation. The impedance spectrum of a CPE in parallel to a resistance has the shape of a depressed semi-cycle. The depression is defined by the parameter
from Equation (
23). With Equation (
15) the impedance of the double-layer capacitances were calculated in the simulation, using
from Equation (
22) or the fixed value for positive electrode (see
Section 2.4).
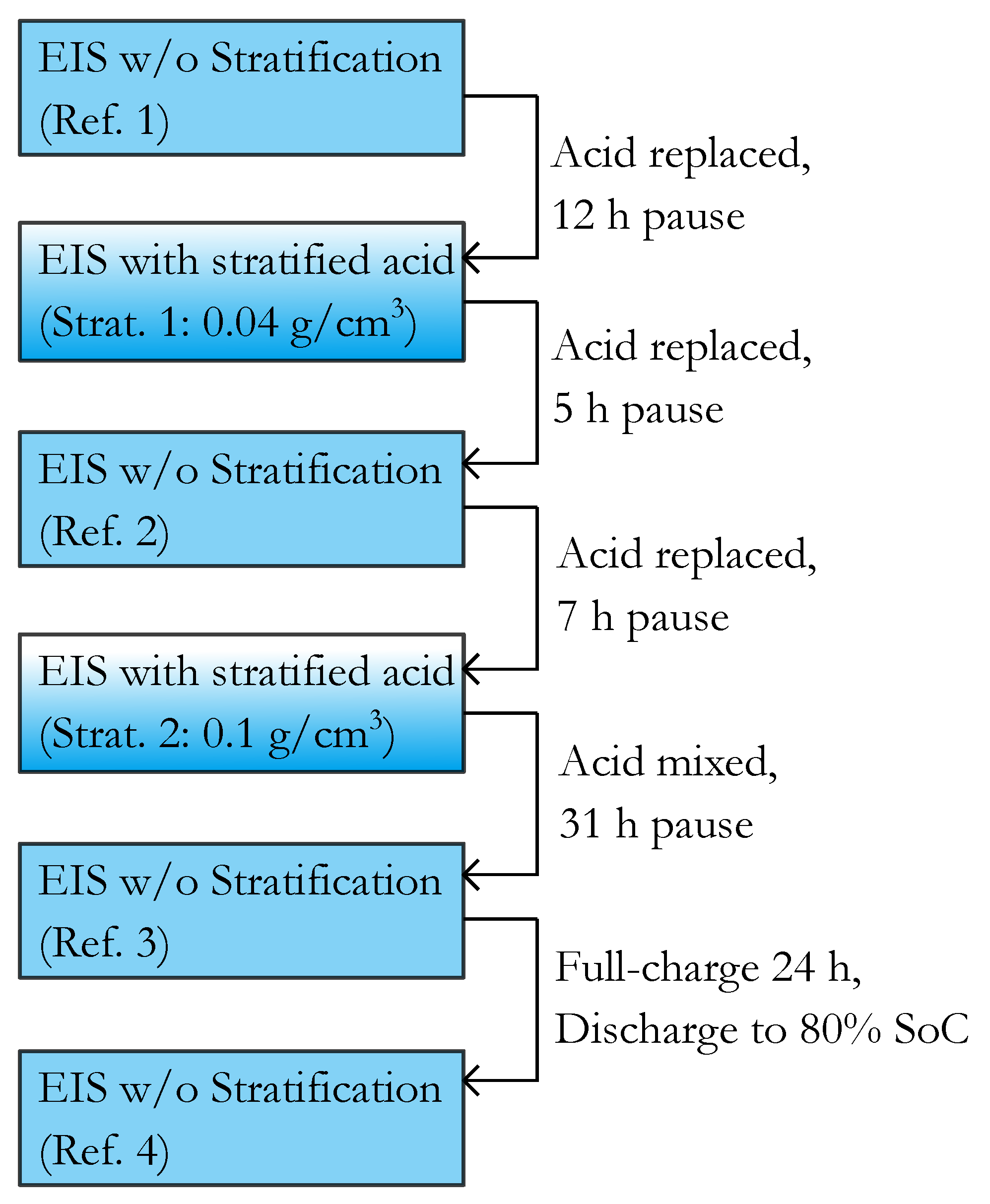
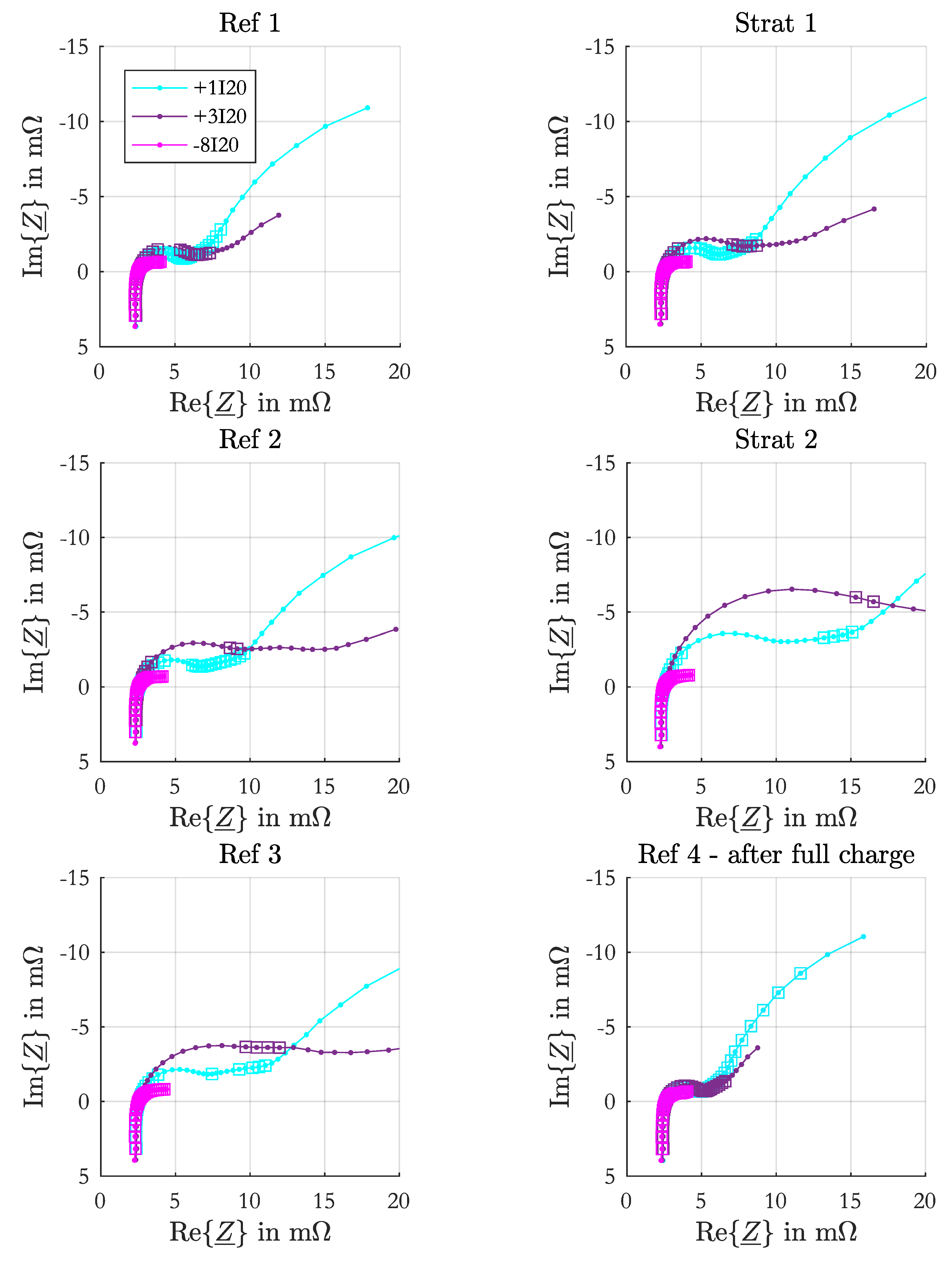
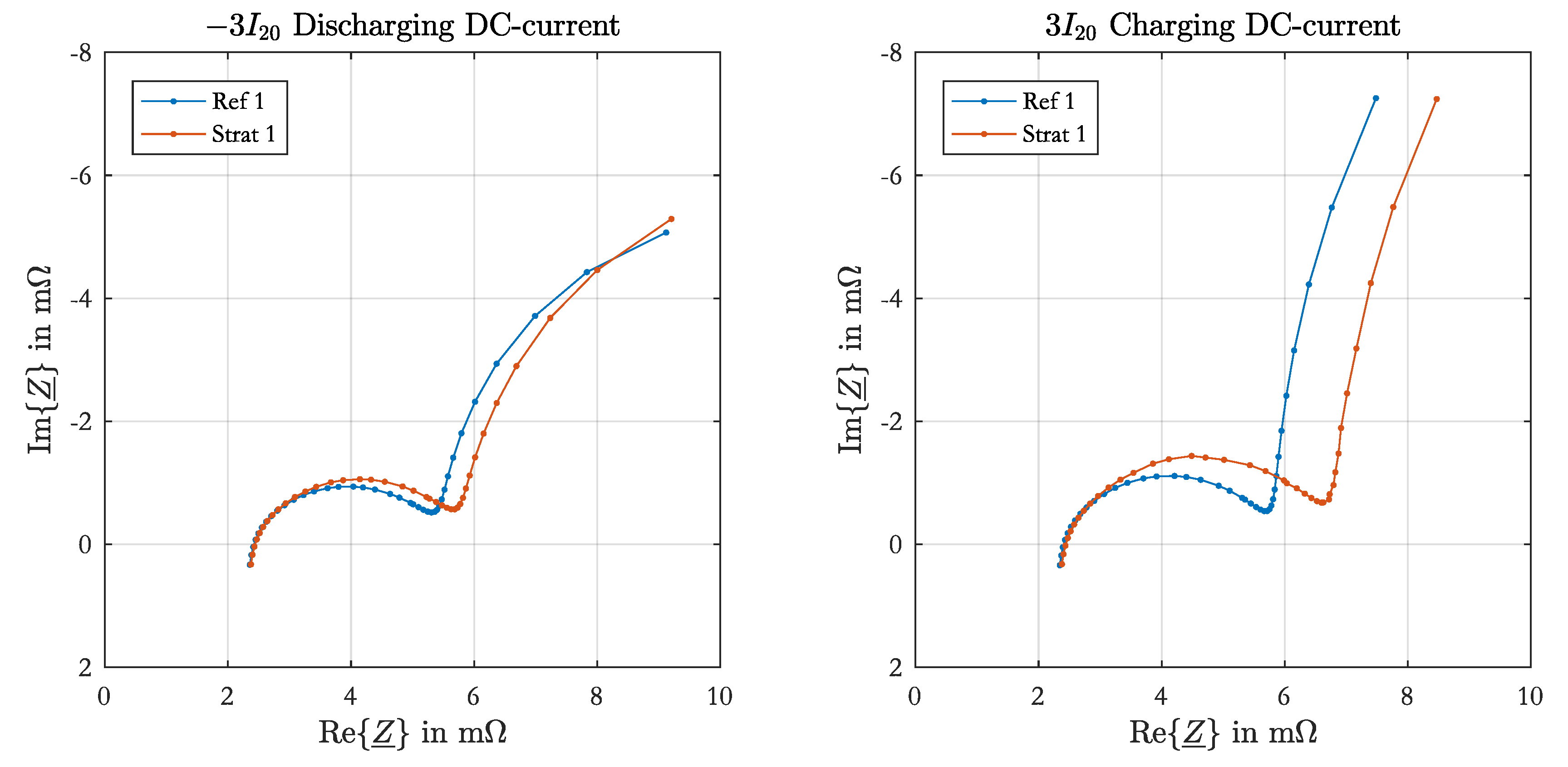
2.4. Parameterization
For the parameterization of the srEEC the same EIS set as denoted in
Section 2.1 was performed at 90%, 80%, 60%, 40%, 20%, 10% and 0% SoC. At the beginning the test cell was charged for 24 h with constant-current-constant-voltage charging ( 5 I
20,
V) followed by a pause of 24 h. The single SoC levels were set by discharging with
I
20 and a subsequent pause of 5 h. During the measurement the acid was mixed to avoid stratification. These parameterization measurements were performed in parallel to the stratification measurements on another test cell with the same configuration.
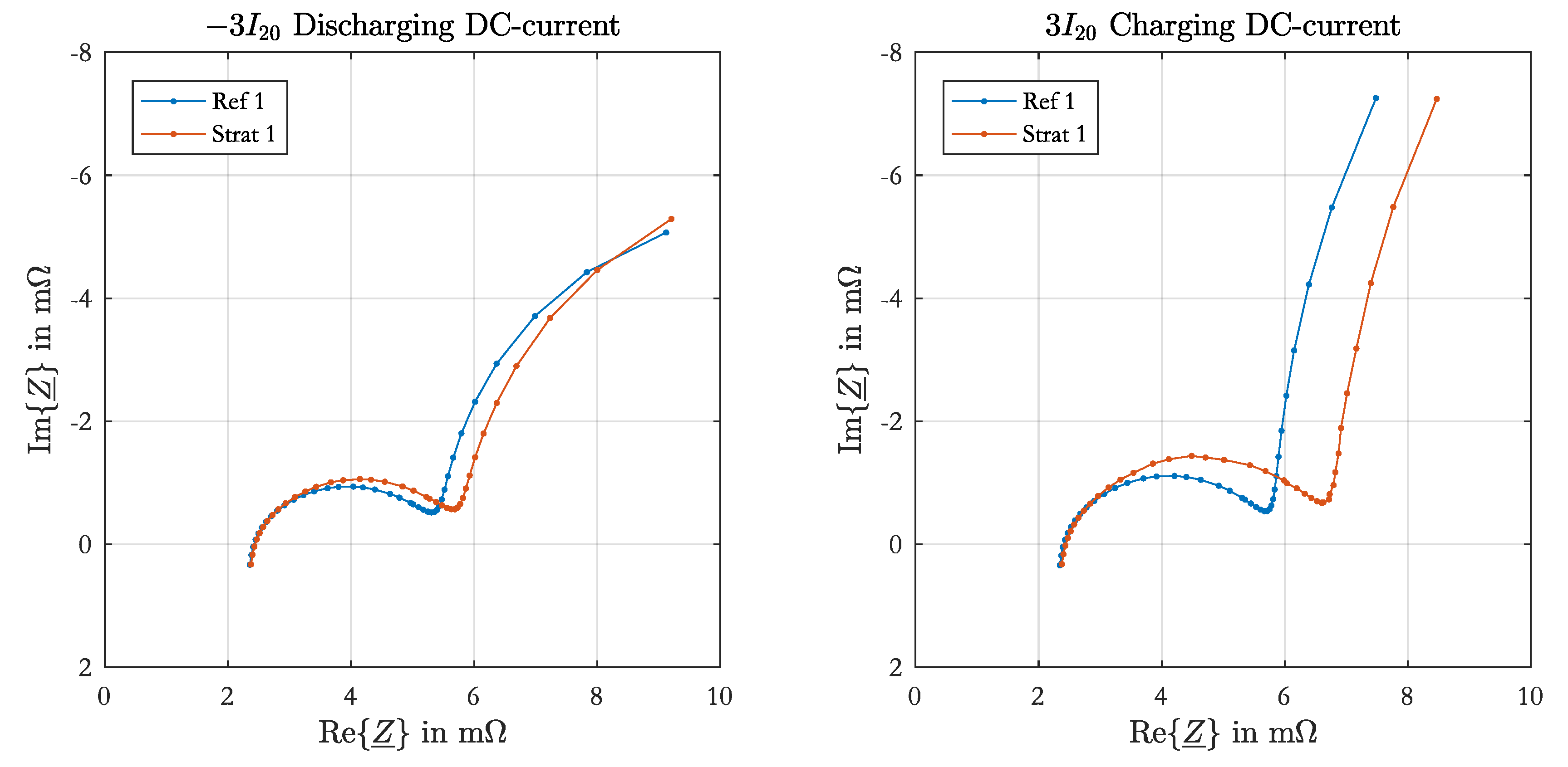
All impedance spectra of the parameterization measurement were fitted to an equivalent electrical circuit with one resistance connected in series to one RC-element containing the charge-transfer resistance and the double-layer capacitance of the negative electrode. The fitting process is equally performed as described in [
17]. The positive electrode parameters were not determined from the spectra, as in most impedance spectra the half-circle of the positive electrode was not adequately visible or affected by time-variant processes.
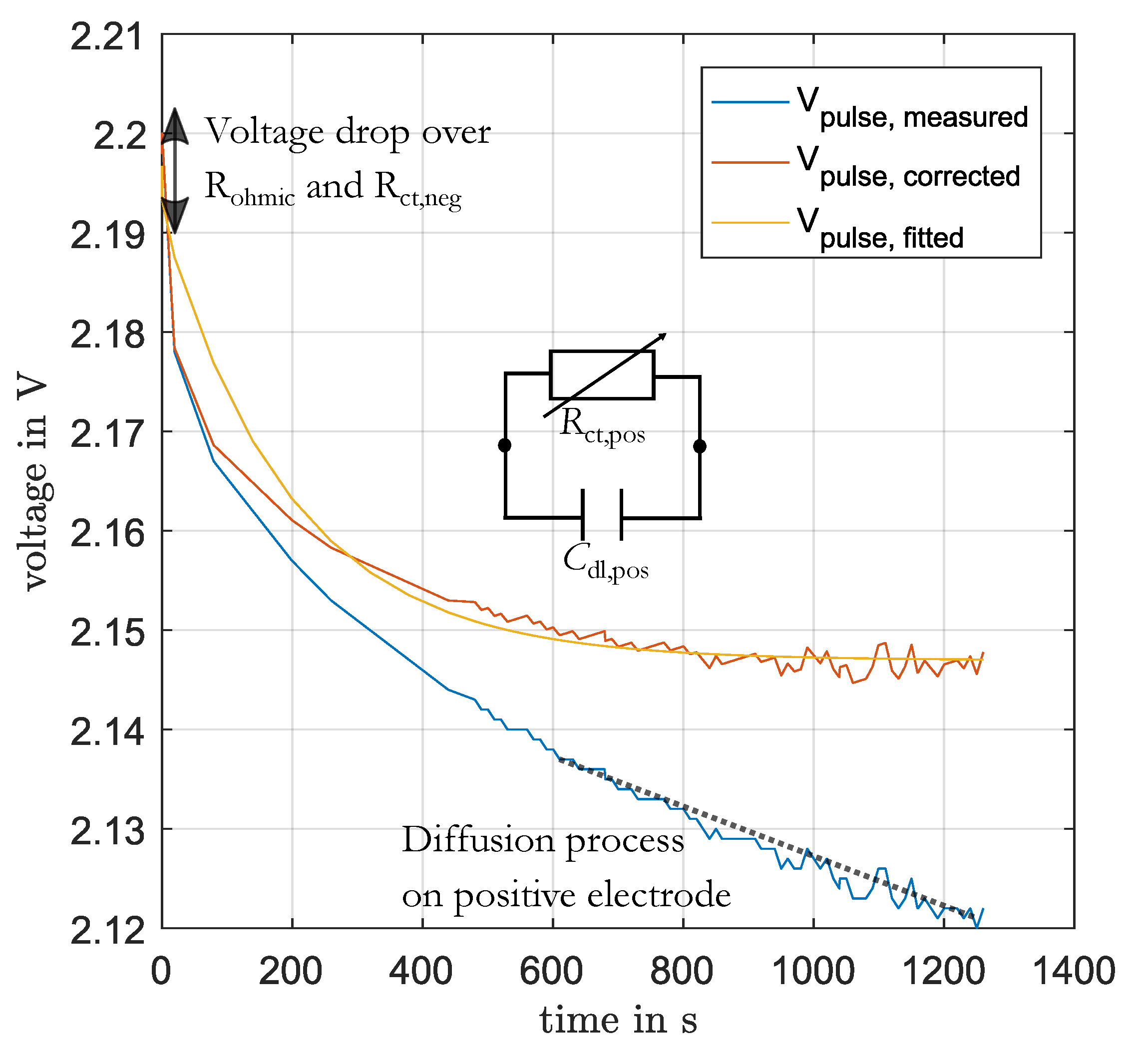
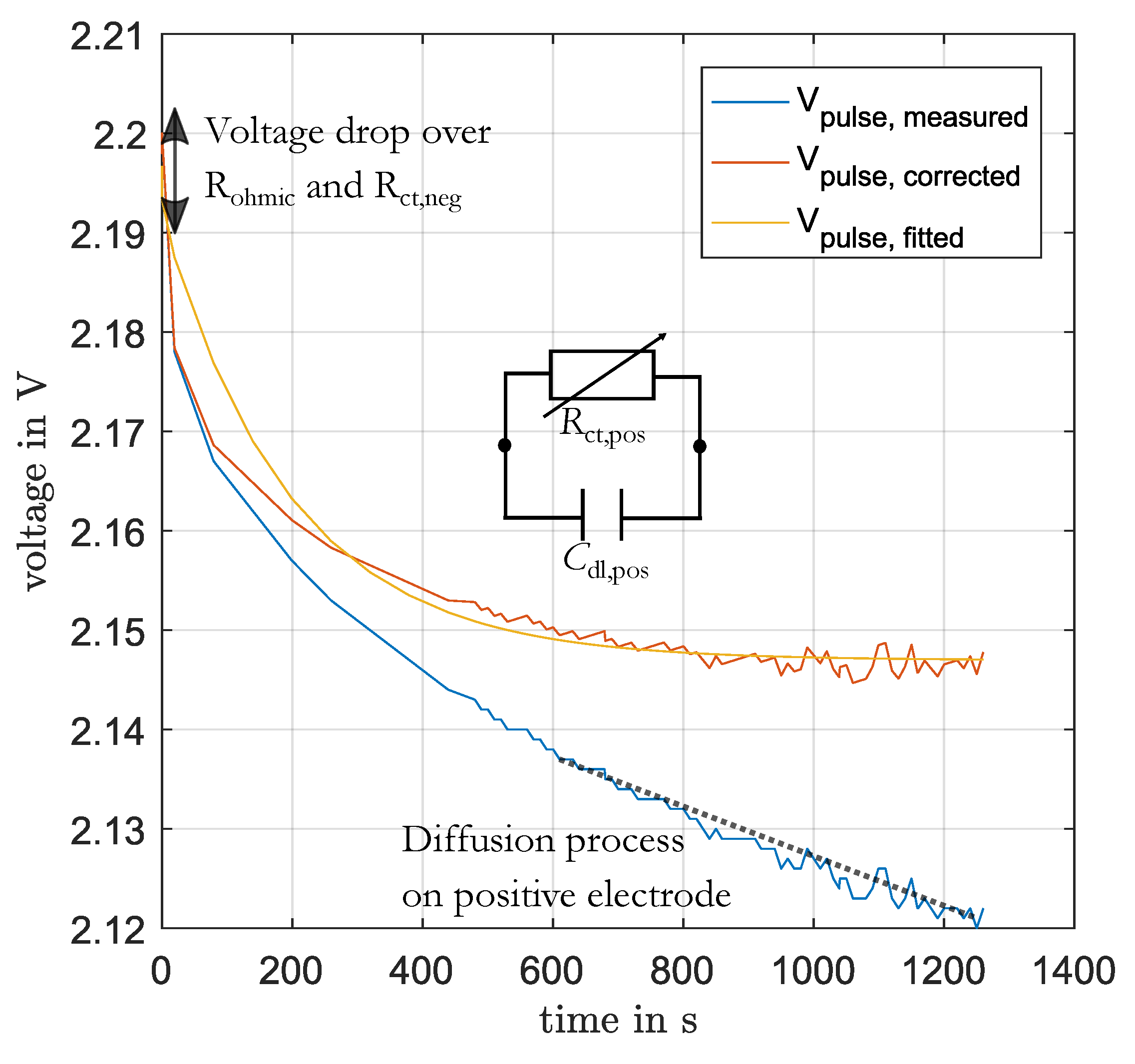
The charge-transfer process and double-layer capacitance of the positive electrode were parameterized from the voltage response during the charging and discharging of 1% SoC before EIS. This voltage response was fitted to an RC-element in time-domain using the function
fit in Matlab with default settings. Before, the linear change of voltage (with slope
m), generated by diffusion to the end of the pulse, was subtracted as well as the voltage drop over the ohmic resistance
and the charge-transfer resistance of the negative electrode
(see Equation (
16)). In
Figure 4 this process is illustrated. The values for
and
were determined for the present DC-current and SoC from the previous parameterization of these elements. The value for the slope
m was determined by the fitting of a linear function to the last part of the pulse.
Because of the current dependency of the charge-transfer resistance this approach is not able to determine the double-layer capacitance correctly [
3]. The voltage curve is nonlinear and cannot be described by a linear model. However, for the simulation of the current distribution the resistances are relevant and not the capacitances. This also means that the simulated impedance spectra will differ from the measured spectra in the part of positive electrode.
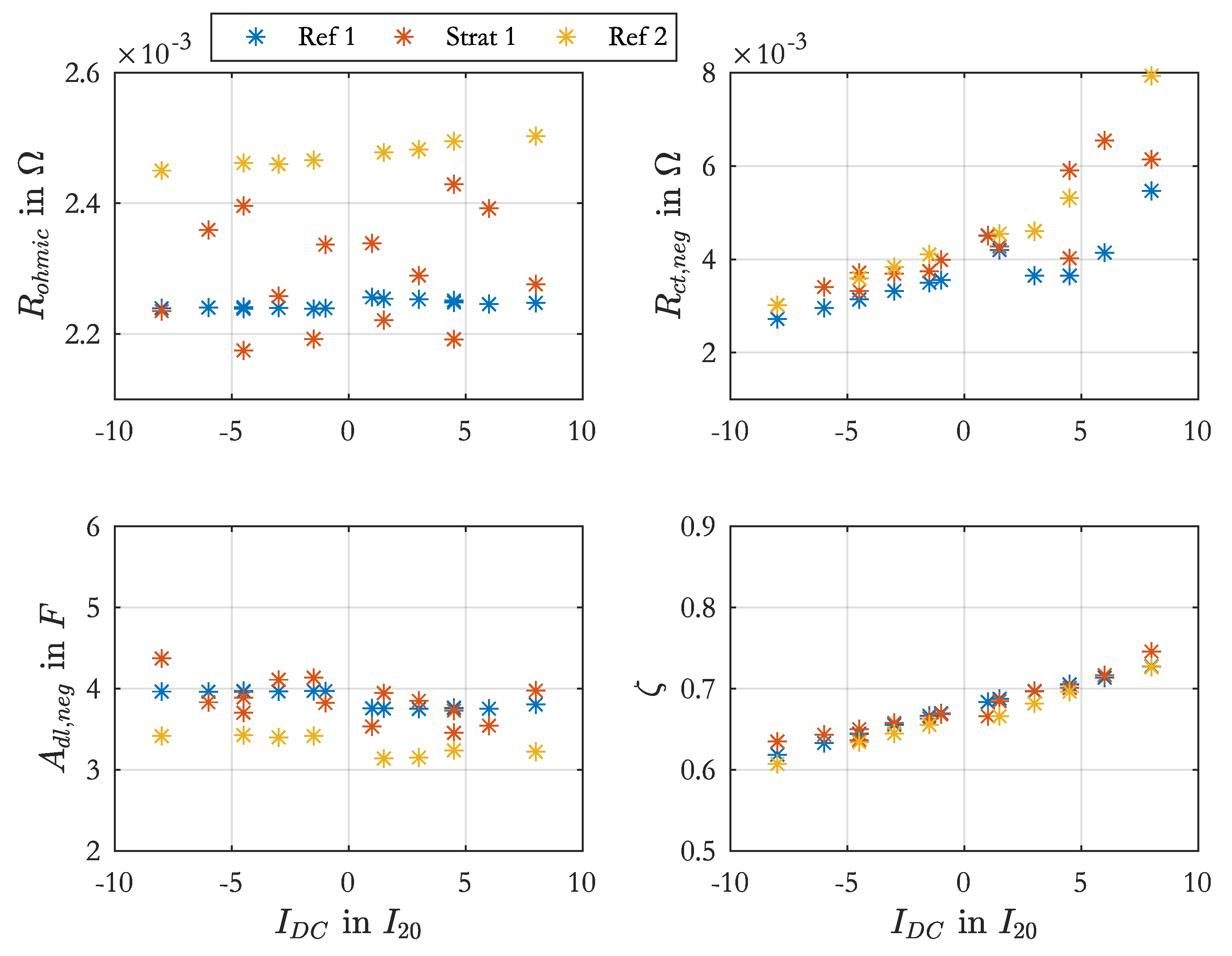
The
values at different DC-currents were used to determine
of Equation (
3) for the positive and negative electrodes, while
was set fix to
for the positive electrode and
for the negative electrode. The values for
were determined from previous parameterization process of Butler-Volmer equation with variable
and
. However, the resulting
values were not constant over the DC-currents, which should be the case. Therefore the average of the single
values were used as fixed value.
for negative electrode is only valid for discharging, because the measured
in charging direction could not be described by the Butler-Volmer equation, as already described. The measured SoC and current dependency of
in charging direction is described with the surface polynomial function in Equation (
17) with the local currents
and local SoC values
. The
values together with the fitting curves are presented in
Figure A2.
Equation (
17) describes the polynomial function for the small-signal value of
. This value was used to calculate the impedance spectrum during simulation. For the calculation of the voltages and current distribution in the srEEC, the large-signal values are required, as already described in
Section 2.3. The large-signal value was determined from the integration of Equation (
17) from 0 A to the present local currents
and
.
The measured SoC-dependency of the ohmic resistance is described by a polynomial function given in Equation (
18).
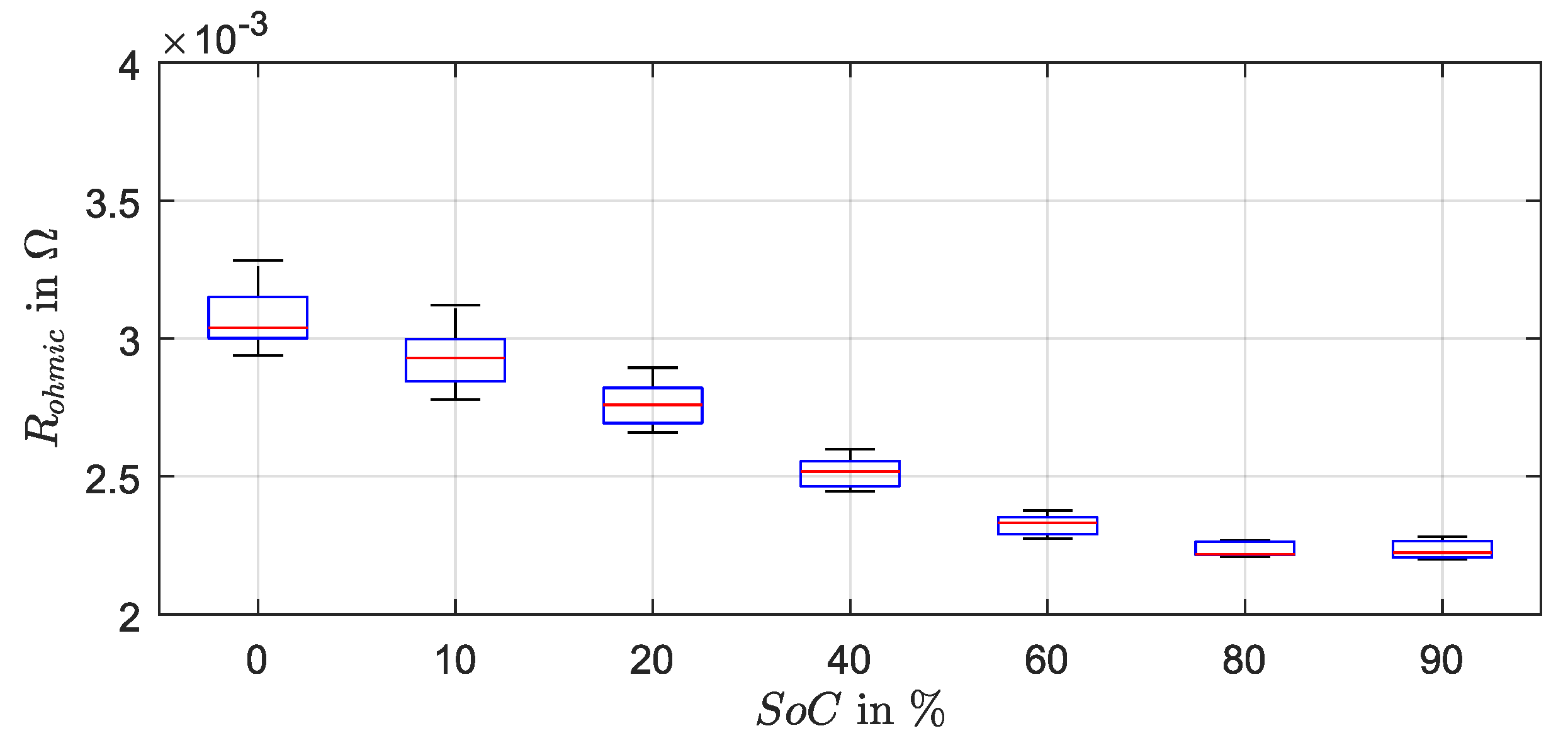
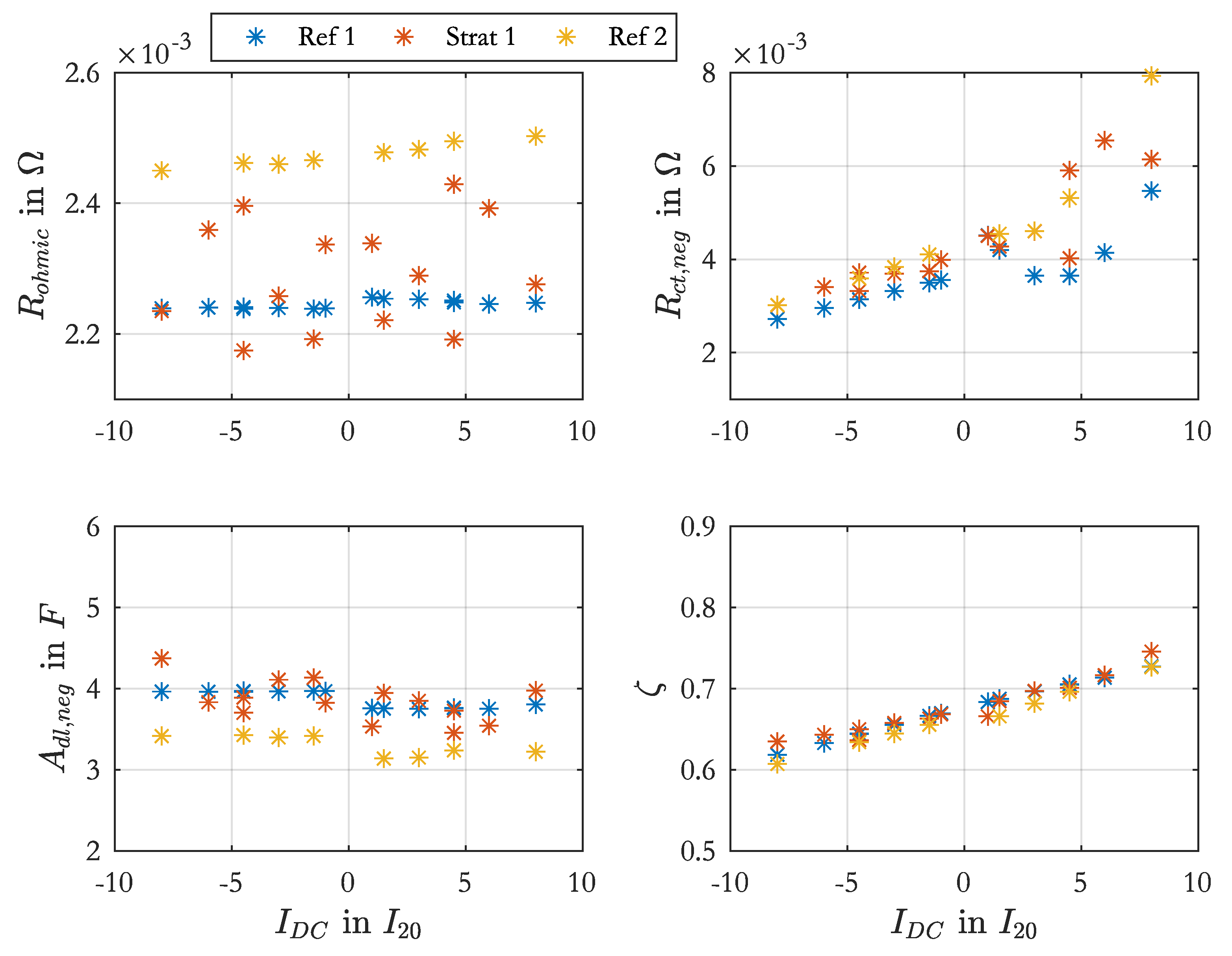
Figure A1 shows the determined resistance values over SoC as boxplot.
The resistance of the lead grid is not SoC-dependent and was separated from the measured ohmic resistance, like presented in the srEEC with and . From the design of the lead grid the grid resistance on the top and the bottom was estimated. The grid was cm wide, 11 cm high and mm thick. The grid bars were 2 mm wide in the middle part and 1 mm in the border area. With 11 bars of 2 mm and 8 bars of 1 mm and a specific resistance of lead of mm2/m a resistance value of m was calculated for the top part (). For the bottom part with 2 bars of 2 mm and 13 bars of 1 mm the grid resistance amounted to m.
The SoC-dependency of the measured ohmic resistance was attributed to
. To parameterize this dependency of
Equation (
19) was solved with the Matlab function
vpasolve. The equation describes the total impedance of the EEC in
Figure 3 for high frequencies, when the RC-elements can be assumed as short-circuits.
After Equation (
19) was solved for various values of SoC, the determined values for
over SoC were used to parameterize the polynomial function of
given in Equation (
20).
The SoC dependency of
in charging direction is covered by Equation (
17). For discharging the determined
of
over SoC can be described with a polynomial function (Equation (
21)).
According to the separation of acid concentration dependency of
, described in
Section 2.3, the parameterized factor of the second term in Equation (
21) is adapted.
is the molar concentration at 100% SoC (
mol/L) and
equals to the molar concentration at 0% SoC (
mol/L).
of the positive electrode did not change significantly over SoC (see
Figure A2), so that an average value of
A was used over the whole SoC range.
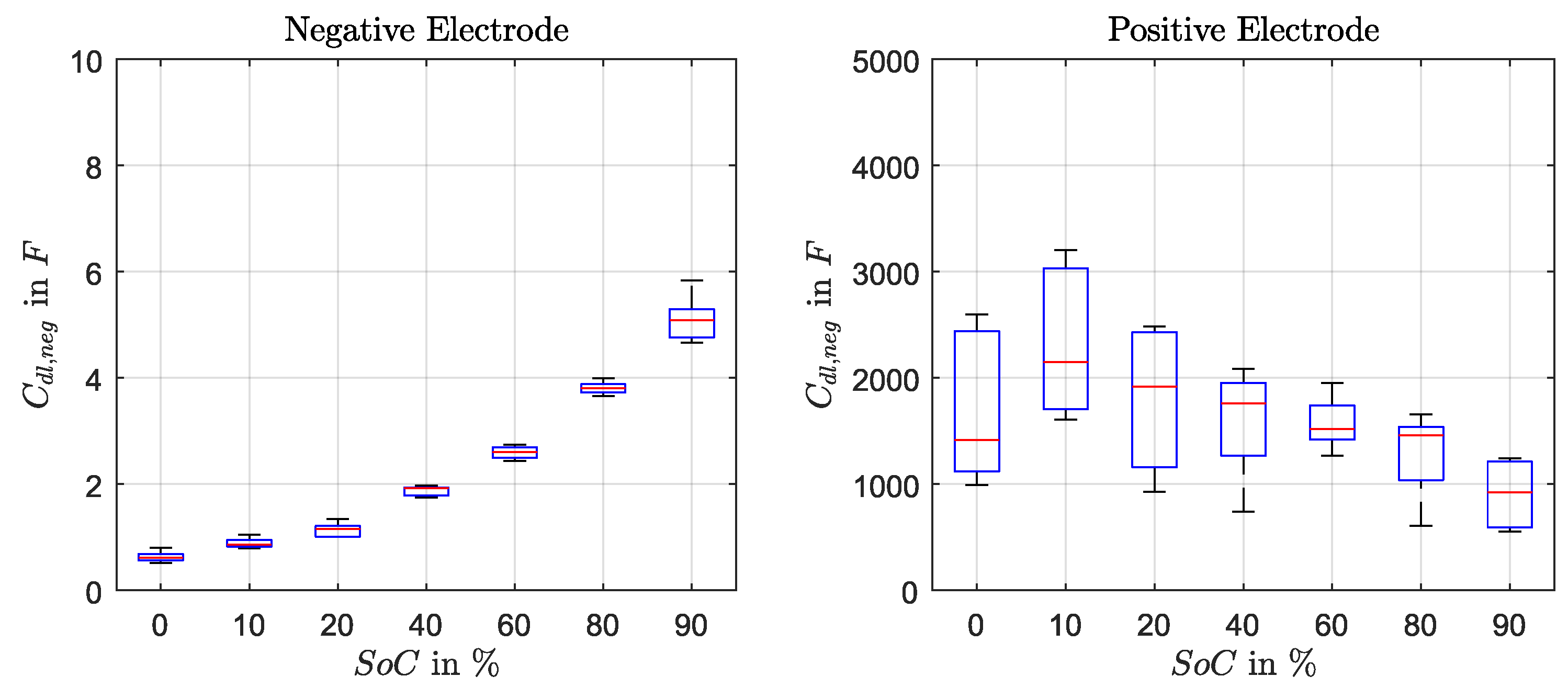
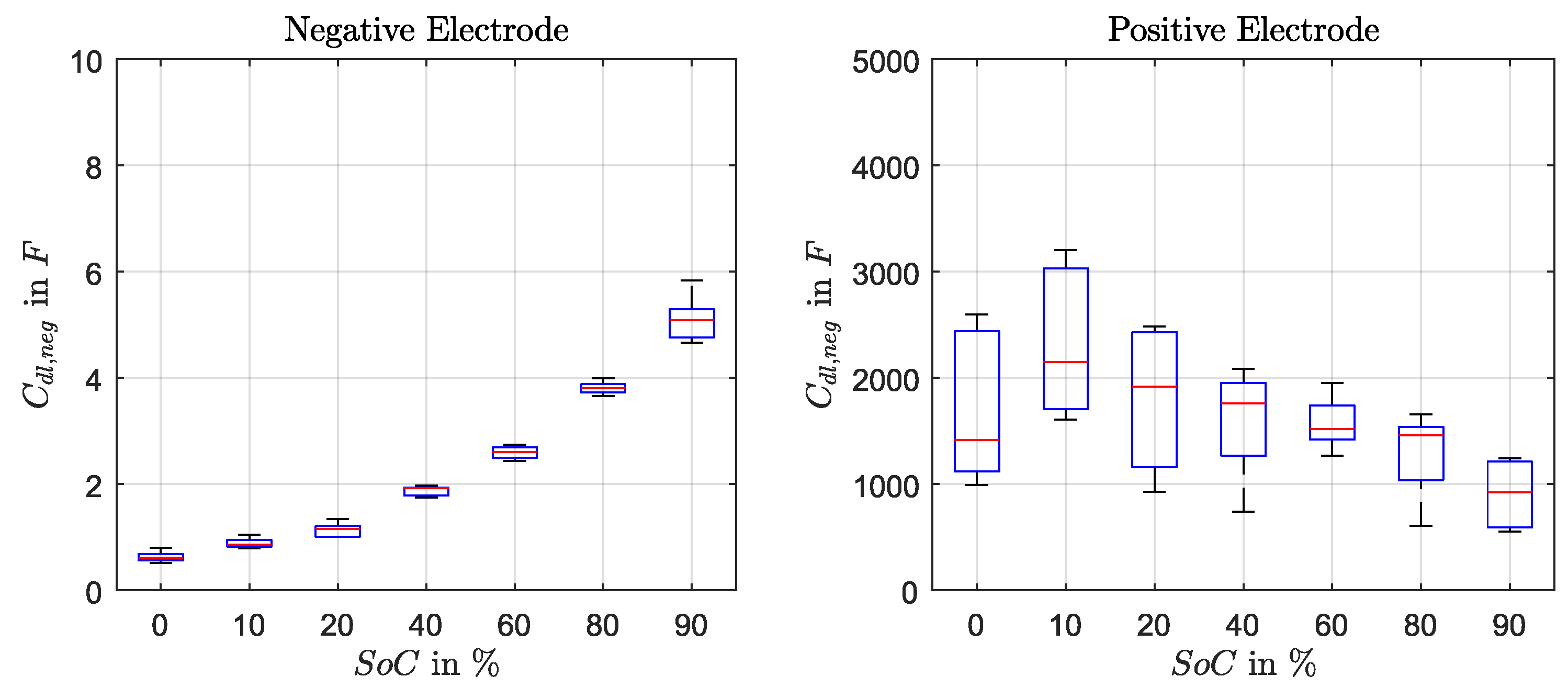
The double-layer capacitance of negative electrode
decreased with the SoC and is described as polynomial function (Equation (
22)). For
of the positive electrode a fix value of 1500 F was used, although an increase of the capacity towards low SoC was observed. However, due to the fact that the determination method using discharge pulses in time-domain is not accurate enough this SoC-dependency was not implemented.
Figure A3 presents the capacitance values as box plots.
To generate a comparable shape of the simulated impedance spectra, the double-layer capacitances were used as CPE with the parameter
(Equation (
15)). The SoC-dependency of
was also determined from the parameterization measurements are is given in Equation (
23).
The parameters of
are already adapted to the srEEC. The determined capacitance value using Equation (
22) and the fix value of
are valid for a one-layer-EEC. In the srEEC, however, one capacitance corresponds to the half of an electrode. Therefore the determined values have to be divided by two.
The exchange currents are surface area dependent, so that the values are divided by two as well for the srEEC. for charging direction is multiplied with 2 to be adapted for the srEEC, based on the general dependency of ohmic resistance on the surface area, which is an approximation for the charge-transfer resistance.
Impedance spectra were generated with an 1D-model and the srEEC using the determined parameters with the described adaption for the srEEC. These spectra, presented in the
supplementary material, show no differences.

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}