Abstract
Phosphaturic mesenchymal tumor (PMT) is strongly related to tumor-induced osteomalacia (TIO) which brings diverse skeletal events, such as bony deformities, gait disturbance, and multiple bone fractures. Overexpressed fibroblast growth factor 23 by the tumor induces hypophosphatemia leading to the oncogenic osteomalacia, a rare paraneoplastic syndrome. PMT occurring in the oral and maxillofacial regions is extremely rare, and this case highlights the need to understand mechanisms by which local lesions can affect systemic conditions, and the importance of a combination of physical examinations, laboratory investigations, and radiologic investigations for a decisive diagnosis.
1. Introduction
PMT is rare tumor with roughly 450 cases reported, with the first case reported by Robert McCance in 1947 [1]. Most PMTs have been discovered in middle-aged adults with no gender predilection [2]. They are mostly found at appendicular sites, while only rare cases have been reported in the oral and maxillofacial region [3], including the paranasal sinuses, mandible, intracranial localization, maxilla, and oral cavity, in descending order of prevalence [4].
Clinically, patients with PMT present with systemic symptoms related to osteomalacia. TIO, also known as oncogenic osteomalacia, is a rare paraneoplastic syndrome that results in abnormal phosphate and vitamin D metabolism. It is mainly characterized by hypophosphatemia, hyperphosphaturia, reduced or inappropriately normal 1,25-dihydroxyvitamin D, elevated serum alkaline phosphatase, elevated or inappropriately normal plasma fibroblast growth factor 23 (FGF23), normal serum calcium, and normal parathyroid hormone concentrations [5]. These biochemical alterations eventually cause osteomalacia, of which clinical symptoms include diffuse body pain, muscle weakness, gait disturbance, fragility of bone, and multiple bone fractures due to defective mineralization of the osteoid [2].
Complete surgical resection with an adequately wide margin is the most appropriate treatment [6]. Postoperative normalization of laboratory values is typically observed following successful resection of the tumor. As the origin of TIO, the intraoral region is very rare. Thus, we report a case in which a tumor of oral origin affected a patient systemically.
2. Case Report
2.1. Clinical Presentation
A 50-year-old female was referred to the Department of Oral and Maxillofacial Surgery at Kyung Hee University Medical Center (Seoul, Republic of Korea) for the evaluation of gingival bulging of the left buccal and palatal areas. She had been receiving calcium supplements for 5 years for osteomalacia diagnosed at a local clinic. Clinical examination revealed a single, ovoid-shaped bulging mass on the left posterior maxilla, which expanded bilaterally on the buccal and palatal sides. Without displacement of the teeth, an expanded firm mass was found with contact overlying soft tissue (Figure 1).
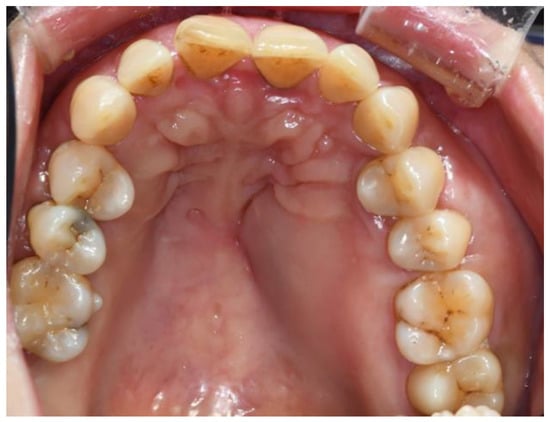
Figure 1.
Expanded solid mass with intact soft tissue Involving the left maxilla area.
Radiological examination through contrast-enhanced computed tomography showed a soft tissue mass (about 2.7 × 2.2 × 2.2 cm) in the left maxilla occupying the left maxillary sinus and left nasal middle meatus and extending to the left side of maxilla and teeth. There was an intraosseous osteolytic lesion evading the adjacent maxilla (Figure 2).
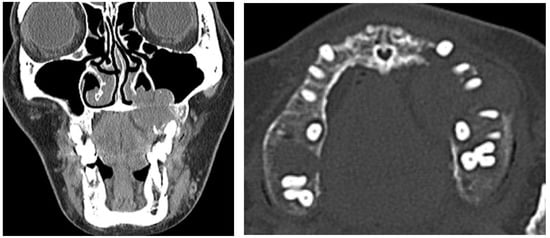
Figure 2.
Preoperative contrast-enhanced computed tomography image presenting radiolucent lesion evading adjacent bone.
Clinical and radiological examination revealed a mass suspicious for a benign tumor in the left palatal area. Routine clinical laboratory assays, including parathyroid gland function tests, were performed before the surgical treatment. Surgical excision of the tumor was performed under general anesthesia.
2.2. Differential Diagnosis
As the patient presented with a slow-growing expanded intraoral lesion with a firm mass involving the maxilla, we suspected a benign tumor in the jaw.
First, as a significant bulging mass in the palatal area was found during clinical examination, pleomorphic adenoma (PA) was suspected. In addition, a benign tumor of the jaw with radiological features which represent expanding while destroying the surrounding bone, including calcifying epithelial odontogenic tumor, calcifying odontogenic cyst, ossifying fibroma, central giant cell granuloma (CGCG) and brown tumor, could be considered. However, calcifying epithelial odontogenic tumors, calcifying odontogenic cysts, and ossifying fibromas were excluded from the provisionally diagnosed diseases because the radiopacities scattered in the radiolucent lesion were not observed in the radiological images of this patient. In addition, with the absence of a history of the rapid growth of the lesion, aggressive malignant tumors, such as osteosarcoma or chondrosarcoma, were excluded.
Finally, the differential diagnosis for benign tumors with clinical and radiographic features like the patient’s includes the following: PA, CGCG, and brown tumor.
PA, the most common benign salivary gland tumor, occurs mostly in the parotid gland, while less frequently affecting the minor salivary glands [7]. When originating from the minor salivary glands, it occurs mostly on the hard and soft palate due to the highly concentrated salivary glands [8]. Clinically, patients with PA present an asymptomatic, slow-growing, unilateral firm mass. PA may occur at any age but mainly affects middle-aged patients with a female predilection. Histological features are highly variable; classically, it is characterized by a mixture of epithelial and mesenchymal components in a variable stroma [9].
CGCG, a benign lesion of the jaw, accounts for less than 7% of all benign tumors of the jaw. Clinical features include slowly growing symptomatic swelling, and in an aggressive form it might be accompanied by pain, root resorption, displacement of teeth, and perforation of the subjacent cortical bone. CGCG usually occurs in the first three decades of life in patents with a female predilection. It occurs about three times more often in the mandible than the maxilla [10]. Radiographically, it appears with a unilocular or multilocular radiolucency with the expansion of the cortical bone plate. Histological features show numerous multinucleated giant cells embedded in a stroma of round and spindle-shaped mononuclear cells [11].
Brown tumors are uncommon focal giant cell lesions that develop in patients with hyperparathyroidism (HPT). Radiological features show a radiolucent extending mass eroding adjacent structures and loss of the lamina dura [12]. Giant cell lesions that can occur in the jaws include giant cell granuloma, cherubism, and brown tumors. Since it is difficult to distinguish brown tumors from other giant cell lesions histologically, clinical diagnosis of the brown tumor should be accompanied by the finding of HPT using laboratory tests or nuclear imaging of the parathyroid glands [13]. Treatment for hyperparathyroidism should be accompanied by normalization of parathyroid function [14].
2.3. Diagnosis and Management
Surgical excision of the tumor was performed with the patient under general anesthesia. Macroscopically, the surgically resected specimen, measuring 3.2 × 2.5 × 2.5 cm in size, shows a grayish-white solid mass (Figure 3).
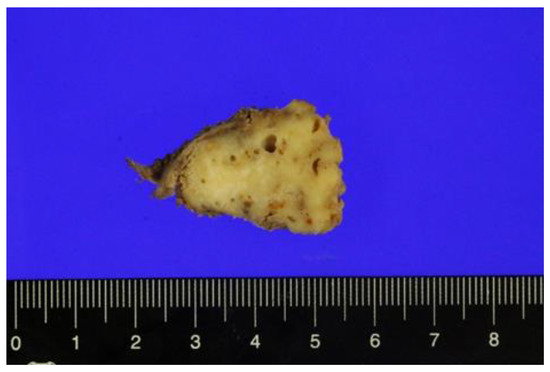
Figure 3.
Cut surface of the resected specimen.
Histologic examination of the specimen revealed a high proliferation of spindle-shaped cells in patterns of fascicles in the focal area. Also, distinct “grungy” calcifications of osteoid-like elements and osteoclast-like multinucleated giant cells were observed (Figure 4).
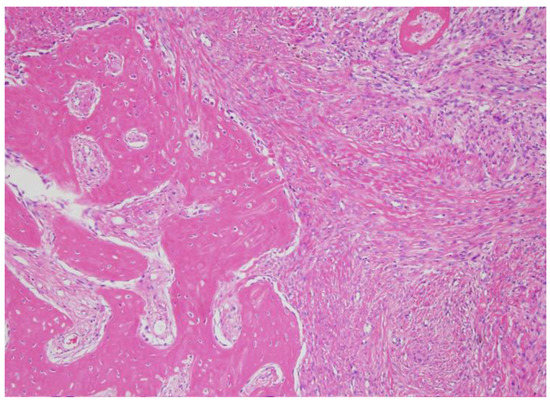
Figure 4.
Microscopic findings of phosphaturic mesenchymal tumor. Woven bone elements and an incomplete rim of membranous ossification were observed.
The histologic findings suggested a diagnosis of phosphaturic mesenchymal tumor, mixed connective tissue variant. Routine clinical laboratory assays performed before surgery revealed the following findings: hypophosphatemia (0.8 [normal, 2.5–4.5 mg/dl]), elevated alkaline phosphatase (306 [normal, 30~120U/L]). Based on the clinical and laboratory findings consistent with osteomalacia, tumor-induced osteomalacia was finally diagnosed. After complete resection of the tumor, postoperative normalization of laboratory values was confirmed. At the patient’s follow-up visit, her clinical symptoms related to osteomalacia improved, with no evidence of recurrence of tumor. Furthermore, following surgery, she had been visiting the endocrinologist regularly for postoperative follow-up.
3. Discussion
The final diagnosis in the present case was a phosphaturic mesenchymal tumor, mixed connective tissue variant (PMTMCT). Most PMTMCTs have been discovered in middle-aged adults with no gender predilection [2]. They are mostly found at appendicular sites, while only rare cases have been reported in the oral and maxillofacial region [3], including the paranasal sinuses, mandible, intracranial localization, maxilla, and oral cavity, in descending order of prevalence [4].
Among phosphaturic mesenchymal tumors (PMTs) including PMTMCT, osteoblastoma-like PMT, non-ossifying fibroma-like PMT, and ossifying fibroma-like PMT [15,16,17], over 90% of tumor-induced osteomalacia (TIO)-associated mesenchymal tumors represent a single histopathological entity, PMTMCT [3]. Histological features of PMTMCT are highly variable, representing high proliferation of spindled-shaped to stellate tumor cells with a low nuclear grade, which are embedded in myxoid to a chondromyxoid matrix that often contains “grungy” calcifications. Other features include osteoclast-like giant cells, chondroblast-like cells, mature adipocytes, microcystic changes, and fibrohistiocystic reactions, which were well observed in our case. In rare malignant PMTMCTs, sarcomatous features can be seen [3,16,18].
In patients with TIO, clinical symptoms and abnormal laboratory indices are usually related to the elevation of FGF23 levels. Overexpressed FGF23 is secreted by a benign mesenchymal tumor, such as a PMT, which is a key mechanism for abnormal phosphate metabolism. In phosphate homeostasis, FGF23 is responsible for the development of hypophosphatemia [19,20]. FGF23, known as a humoral phosphaturic factor referred to as phosphatonin, is a molecule that regulates phosphate pathway metabolism and homeostasis. FGF23 plays a critical role in regulating proximal renal tubular phosphate reabsorption, of which overexpression leads to phosphaturia and hypophosphatemia [19]. Additionally, it regulates the production of vitamin D metabolites by inhibiting hydroxylation of 25-hydroxyvitamin D to 1,25-dyhydroxyvitamin D [21]. These alterations eventually cause hypophosphatemia-related disorders, including rickets in children and osteomalacia in adults [15].
TIO should be distinguished from other forms associated with hypophosphatemia. These disorders include inherited and acquired forms. Among the inherited disorders are X-linked hypophosphatemic rickets (XLHR), autosomal dominant hypophosphatemic rickets (ADHR), and autosomal recessive hypophosphatemic rickets (ARHR) [20,22]. Although patients with these disorders display a clinical phenotype similar to that of TIO, these conditions are typically present in childhood and are associated with deformities in the lower extremities. In making a definitive diagnosis, obtaining family history is important; furthermore, genetic testing of the dentin matrix protein 1 (DMP1), phosphate-regulating gene with homologies to endopeptidases on the X chromosome (PHEX) gene can be useful [23,24]. Acquired disorders include nutritional vitamin D deficiency, nutritional phosphorus deficiency or Fanconi syndrome. In patients with these disorders, there are differences in biochemical features through the laboratory investigations from those of patients with TIO [25,26,27].
Identification and location of the associated tumors are difficult, resulting in a delayed final diagnosis. Some studies have shown that it takes an average of 5 years from symptom onset to the final diagnosis [28]. The reasons for the delay in diagnosis are as follows. First, the causative tumors are usually small, slow-growing benign tumors. Second, the location of the tumors are non-specific. As a result, a thorough examination of the head and neck would be delayed because TIO is extremely rare in the oral and maxillofacial regions. Lastly, routine laboratory examinations do not include serum phosphate, vitamin D, serum alkaline phosphatase, and plasma FGF23. As a result, important laboratory results, including the presence of hypophosphatemia, a critical clue to the diagnosis of TIO, could be neglected.
Investigations for diagnosis include physical examination, laboratory investigations, radiologic investigations, and venous sampling. Physical examination is usually not helpful for diagnosis unless profound symptoms of osteomalacia are present. If a patient presents with clinical symptoms such as bone pain, muscle weakness, gait disturbance, skeletal deformities, and multiple bone fractures, TIO should be suspected. In the present case, the patient presented with bony deformities, such as scoliosis, which helped clinicians suspect TIO. As hypophosphatemia, elevated alkaline phosphatase, and elevated FGF23 levels can be critical diagnostic clues for the diagnosis of TIO [2], laboratory investigations including these biochemical profiles should be performed in cases of suspected TIO. Measurement of FGF as a tumor marker is useful. Because of the short half-life of FGF23, between 20–50 min, it can be used for intraoperative monitoring and evaluation of postoperative outcomes [29,30]. For successful removal of tumors, it is important to accurately locate the tumors. To locate the tumors, functioning imaging techniques, including octreotide scintigraphy, anatomical imaging techniques, including computed tomography (CT), magnetic resonance imaging (MRI), and the combination of functioning and anatomical techniques, including positron emission tomography along with computed tomography (PET/CT), have been used [6,23,31,32]. Indium-111 octreotide scintigraphy (octreotide scanning) can be used as a method of detection for tumors because PMTs may express somatostatin receptors (SSRTs) [33]. Recently, venous sampling of FGF23 has been proposed as a method for definitive confirmation of the tumor secreting FGF23 [30]. However, in the absence of suspicious tumors on radiological imaging, random venous sampling cannot be successful in localizing the tumors [34].
Complete surgical resection with an adequately wide margin is the most appropriate treatment. There is a risk of recurrence of these tumors if they do not have a wide margin [35]. Postoperative normalization of laboratory values is typically observed following successful resection of the tumor. However, considering anatomical and functional factors, it could be difficult to perform complete surgical resection in all cases due to concerns about surgical morbidity with complete resection. If resection is not complete, the patients with a persistent syndrome can be partially managed with oral phosphate supplements and 1,25-dihydroxyvitamin D (calcitriol) administration for symptomatic improvement [36]. Furthermore, periodic postoperative assessment of laboratory examinations has been recommended as a means to monitor for recurrences of tumors.
4. Conclusions
A definitive diagnosis of TIO can be challenging for clinicians. Successful diagnosis and treatment are dependent on a combination of clinical examination, laboratory investigations, and radiologic investigations. Additionally, a periodic assessment of laboratory values is necessary.
Author Contributions
Conceptualization, J.-Y.O.; methodology, M.K.; investigation, M.K., J.J. and S.-W.K.; data curation, M.K.; writing and original draft preparation, M.K.; writing, review, and editing, J.-Y.O. and J.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Written informed consent has been obtained from the patient to publish this paper.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Folpe, A.L. Phosphaturic mesenchymal tumors: A review and update. Semin. Diagn. Pathol. 2019, 36, 260–268. [Google Scholar] [CrossRef]
- Chong, W.H.; Molinolo, A.A.; Chen, C.C.; Collins, M.T. Tumor-induced osteomalacia. Endocr. Relat. Cancer 2011, 18, R53–R77. [Google Scholar] [CrossRef]
- Folpe, A.L.; Fanburg-Smith, J.C.; Billings, S.D.; Bisceglia, M.; Bertoni, F.; Cho, J.Y.; Econs, M.J.; Inwards, C.Y.; de Beur, S.M.J.; Mentzel, T.; et al. Most osteomalacia-associated mesenchymal tumors are a single histopathologic entity—An analysis of 32 cases and a comprehensive review of the literature. Am. J. Surg. Pathol. 2004, 28, 1–30. [Google Scholar] [CrossRef]
- Shah, R.; Lila, A.R.; Jadhav, R.S.; Patil, V.; Mahajan, A.; Sonawane, S.; Thadani, P.; Dcruz, A.; Pai, P.; Bal, M.; et al. Tumor induced osteomalacia in head and neck region: Single center experience and systematic review. Endocr. Connect. 2019, 8, 1330–1353. [Google Scholar] [CrossRef]
- Kumar, R.; Folpe, A.L.; Mullan, B.P. Tumor-induced osteomalacia. Transl. Endocrinol. Metab. 2015, 7, R53–R77. [Google Scholar]
- Clunie, G.P.; Fox, P.E.; Stamp, T.C. Four cases of acquired hypophosphataemic (‘oncogenic’) osteomalacia. Problems of diagnosis, treatment and long-term management. Rheumatology 2000, 39, 1415–1421. [Google Scholar] [CrossRef]
- Mendenhall, W.M.; Mendenhall, C.M.; Werning, J.W.; Malyapa, R.S.; Mendenhall, N.P. Salivary gland pleomorphic adenoma. Am. J. Clin. Oncol. 2008, 31, 95–99. [Google Scholar] [CrossRef]
- Ledesma-Montes, C.; Garces-Ortiz, M. Salivary gland tumours in a Mexican sample. A retrospective study. Med. Oral Organo Of. Soc. Esp. Med. Oral Acad. Iberoam. Patol. Med. Bucal 2002, 7, 324–330. [Google Scholar]
- Stennert, E.; Guntinas-Lichius, O.; Klussmann, J.P.; Arnold, G. Histopathology of pleomorphic adenoma in the parotid gland: A prospective unselected series of 100 cases. Laryngoscope 2001, 111, 2195–2200. [Google Scholar] [CrossRef]
- Kruse-Lösler, B.; Diallo, R.; Gaertner, C.; Mischke, K.-L.; Joos, U.; Kleinheinz, J. Central giant cell granuloma of the jaws: A clinical, radiologic, and histopathologic study of 26 cases. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontology 2006, 101, 346–354. [Google Scholar] [CrossRef]
- de Lange, J.; van den Akker, H.P.; van den Berg, H. Central giant cell granuloma of the jaw: A review of the literature with emphasis on therapy options. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontology 2007, 104, 603–615. [Google Scholar] [CrossRef]
- Silverman Jr, S.; Ware, W.H.; Gillooly Jr, C. Dental aspects of hyperparathyroidism. Oral Surg. Oral Med. Oral Pathol. 1968, 26, 184–189. [Google Scholar] [CrossRef]
- Düsünsel, R.; Güney, E.; de Gündüz, Z.; Poyrazoglu, M.; Yigitbasi, O.; Kontas, O. Maxillary brown tumor caused by secondary hyperparathyroidism in a boy. Pediatr. Nephrol. 2000, 14, 529–530. [Google Scholar]
- Keyser, J.S.; Postma, G.N. Brown tumor of the mandible. Am. J. Otolaryngol. 1996, 17, 407–410. [Google Scholar] [CrossRef]
- Weidner, N.; Cruz, D.S. Phosphaturic mesenchymal tumors. A polymorphous group causing osteomalacia or rickets. Cancer 1987, 59, 1442–1454. [Google Scholar] [CrossRef]
- Drezner, M.K. Tumor-induced osteomalacia. Rev. Endocr. Metab. Disord. 2001, 2, 175–186. [Google Scholar] [CrossRef]
- Shane, E.; Parisien, M.; Henderson, J.E.; Dempster, D.W.; Feldman, F.; Hardy, M.A.; Tohme, J.F.; Karaplis, A.C.; Clemens, T.L. Tumor-induced osteomalacia: Clinical and basic studies. J. Bone Miner. Res. 1997, 12, 1502–1511. [Google Scholar] [CrossRef]
- Reyes-Múgica, M.; Arnsmeier, S.L.; Backeljauw, P.F.; Persing, J.; Ellis, B.; Carpenter, T.O. Phosphaturic mesenchymal tumor-induced rickets. Pediatr. Dev. Pathol. 2000, 3, 61–69. [Google Scholar] [CrossRef]
- Shimada, T.; Mizutani, S.; Muto, T.; Yoneya, T.; Hino, R.; Takeda, S.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Yamashita, T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc. Natl. Acad. Sci. USA 2001, 98, 6500–6505. [Google Scholar] [CrossRef]
- Jonsson, K.B.; Zahradnik, R.; Larsson, T.; White, K.E.; Sugimoto, T.; Imanishi, Y.; Yamamoto, T.; Hampson, G.; Koshiyama, H.; Ljunggren, Ö. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N.Engl. J. Med. 2003, 348, 1656–1663. [Google Scholar] [CrossRef]
- Shimada, T.; Hasegawa, H.; Yamazaki, Y.; Muto, T.; Hino, R.; Takeuchi, Y.; Fujita, T.; Nakahara, K.; Fukumoto, S.; Yamashita, T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J. Bone Miner. Res. 2004, 19, 429–435. [Google Scholar] [CrossRef] [PubMed]
- White, K.E.; Evans, W.E.; O’Riordan, J.L.; Speer, M.C.; Econs, M.J.; Lorenz-Depiereux, B.; Grabowski, M.; Meitinger, T.; Strom, T.M. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat. Genet. 2000, 26, 345–348. [Google Scholar] [CrossRef] [PubMed]
- De Beur, S.M.J. Tumor-induced osteomalacia. JAMA 2005, 294, 1260–1267. [Google Scholar] [CrossRef]
- Feng, J.Q.; Ward, L.M.; Liu, S.; Lu, Y.; Xie, Y.; Yuan, B.; Yu, X.; Rauch, F.; Davis, S.I.; Zhang, S. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat. Genet. 2006, 38, 1310–1315. [Google Scholar] [CrossRef]
- Audran, M.; Kumar, R. The physiology and pathophysiology of vitamin D. In Proceedings of the Mayo Clinic Proceedings, Olmsted County, MN, USA, 1 January 1985; pp. 851–866. [Google Scholar]
- Knochel, J.P. The pathophysiology and clinical characteristics of severe hypophosphatemia. Arch. Intern. Med. 1977, 137, 203–220. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, J.E.; Velosa, J.A.; Kyle, R.A.; Wagoner, R.D.; Holley, K.E.; Salassa, R.M. Fanconi syndrome in adults: A manifestation of a latent form of myeloma. Am. J. Med. 1975, 58, 354–364. [Google Scholar] [CrossRef]
- Ruppe, M.D.; De Beur, S.J. Disorders of phosphate homeostasis. Primer Metab. Bone Dis. Disord. Miner. Metab. 2008, 7, 317–321. [Google Scholar]
- Khosravi, A.; Cutler, C.M.; Kelly, M.H.; Chang, R.; Royal, R.E.; Sherry, R.M.; Wodajo, F.M.; Fedarko, N.S.; Collins, M.T. Determination of the elimination half-life of fibroblast growth factor-23. J. Clin. Endocrinol. Metab. 2007, 92, 2374–2377. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Suzuki, H.; Ogura, S.; Imai, R.; Yamazaki, Y.; Yamashita, T.; Miyamoto, Y.; Okazaki, H.; Nakamura, K.; Nakahara, K. Venous sampling for fibroblast growth factor-23 confirms preoperative diagnosis of tumor-induced osteomalacia. J. Clin. Endocrinol. Metab. 2004, 89, 3979–3982. [Google Scholar] [CrossRef]
- Robertson, A.; Mansberg, R.; Mansberg, V.; Van der Wall, H.; Hooper, M. Tumor-induced osteomalacia: A case of diagnostic dilemma. Clin. Nucl. Med. 2007, 32, 631–634. [Google Scholar] [CrossRef]
- Roarke, M.C.; Nguyen, B.D. PET/CT localization of phosphaturic mesenchymal neoplasm causing tumor-induced osteomalacia. Clin. Nucl. Med. 2007, 32, 300–301. [Google Scholar] [CrossRef] [PubMed]
- Dupond, J.; Mahammedi, H.; Prie, D.; Collin, F.; Gil, H.; Blagosklonov, O.; Ricbourg, B.; Meaux-Ruault, N.; Kantelip, B. Oncogenic osteomalacia: Diagnostic importance of fibroblast growth factor 23 and F-18 fluorodeoxyglucose PET/CT scan for the diagnosis and follow-up in one case. Bone 2005, 36, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Rhee, Y.; Lee, J.D.; Shin, K.H.; Lee, H.C.; Huh, K.B.; Lim, S.K. Oncogenic osteomalacia associated with mesenchymal tumour detected by indium-111 octreotide scintigraphy. Clin. Endocrinol. 2001, 54, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Andreopoulou, P.; Dumitrescu, C.E.; Kelly, M.H.; Brillante, B.A.; Cutler Peck, C.M.; Wodajo, F.M.; Chang, R.; Collins, M.T. Selective venous catheterization for the localization of phosphaturic mesenchymal tumors. J. Bone Miner. Res. 2011, 26, 1295–1302. [Google Scholar] [CrossRef]
- Schapira, D.; Izhak, O.B.; Nachtigal, A.; Burstein, A.; Shalom, R.B.; Shagrawi, I.; Best, L.A. Tumor-induced osteomalacia. Semin. Arthritis Rheum. 1995, 25, 35–46. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).