
A Coarse-Grained Molecular Model for Simulating Self-Healing of Bitumen

 ,
, 
 , ,
, , 


 , and
, and
Abstract
1. Introduction
2. Establishment of Molecular Model
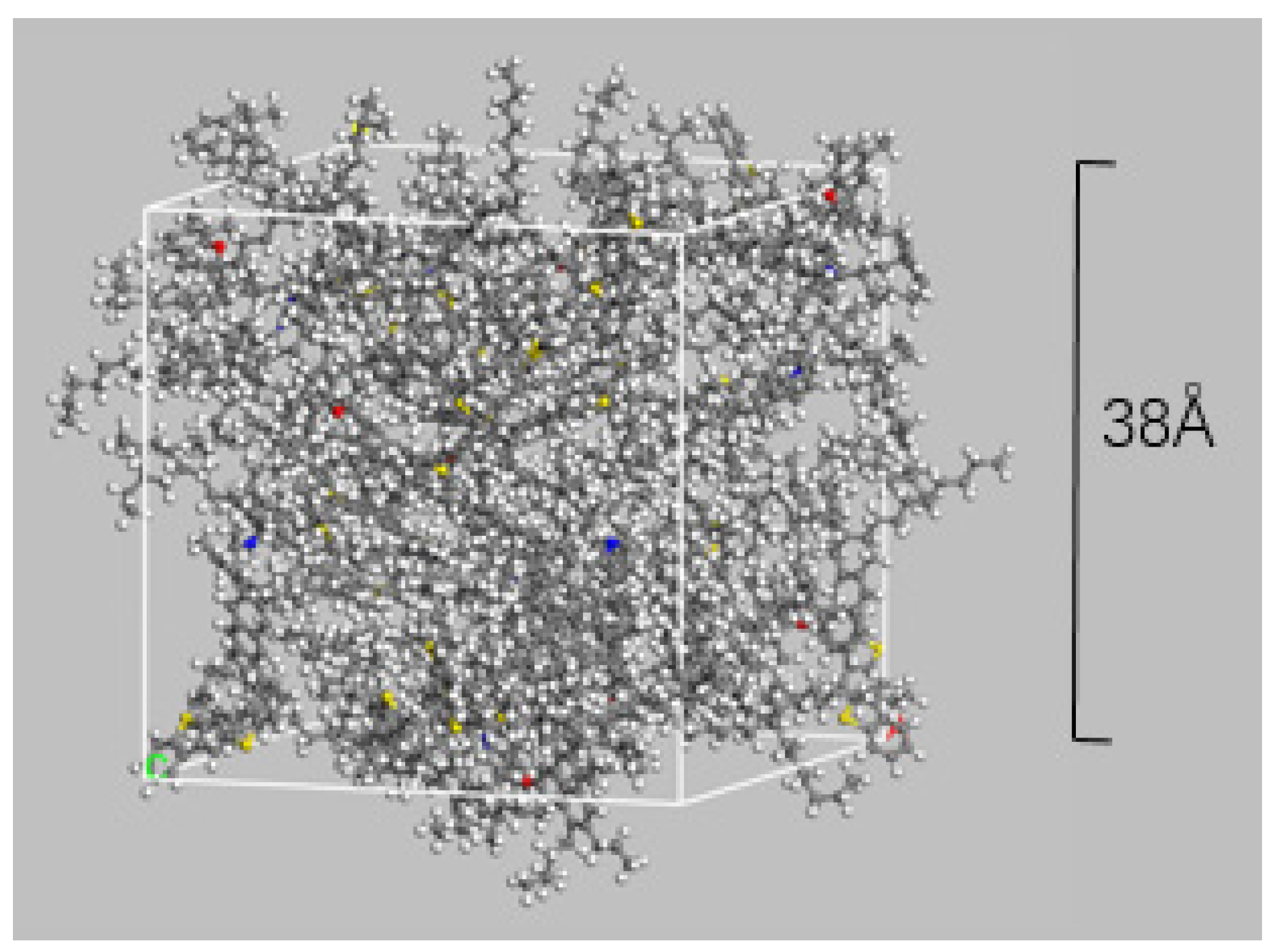
2.1. All-Atom Molecular Model (MD)
- (1)
- Twelve different bitumen molecules are drawn and geometrically optimized to eliminate errors in bond lengths, bond angles, etc.;
- (2)
- Molecules are placed into a computational box with periodic boundary conditions with an initial density of 0.1 g/cm3;
- (3)
- The system is relaxed for 300 ps in the NVT ensemble, the temperature is set to 298 K. The NVT ensemble refers to a constant temperature, constant volume ensemble where, through NVT relaxation, the bitumen molecules will fully move in the calculation box, thus mixing uniformly and reaching a relatively low energy position, with no change in density during this process;
- (4)
- It is annealed five times in the temperature range of 263–433 K, which corresponds to the working temperature of road bitumen. Annealing means that the temperature of the system is repeatedly raised and lowered, allowing some of the molecules to break through the energy barrier to reach a less energetic and more stable structure upon return to normal temperature;
- (5)
- It is simulated for 300 ps in the NPT ensemble, T = 298 K, P = 1 atm. The NPT ensemble refers to a constant temperature and pressure ensemble where the total energy and system volume can be freely varied, and the density of the bitumen system will stabilize through NPT relaxation.
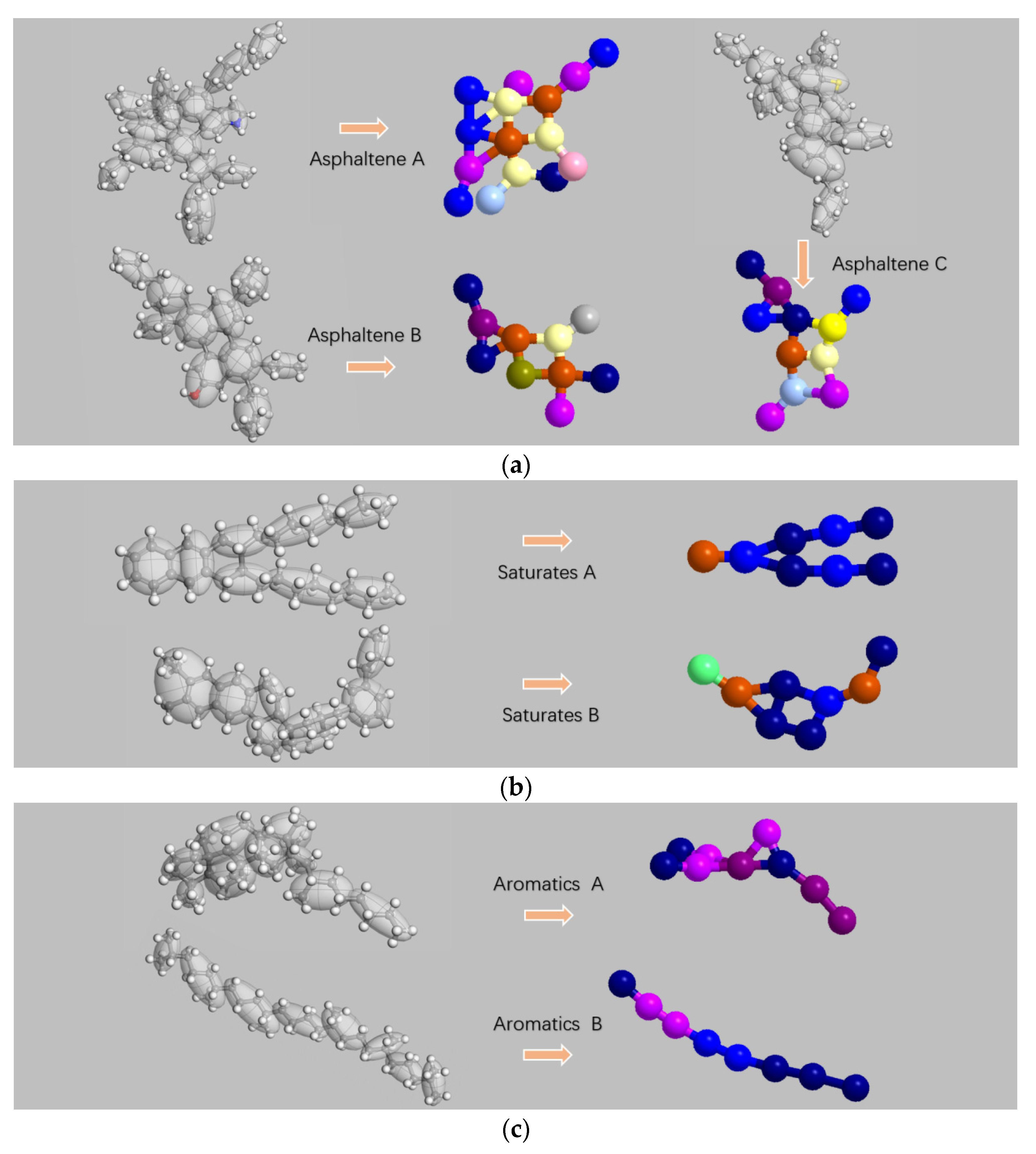
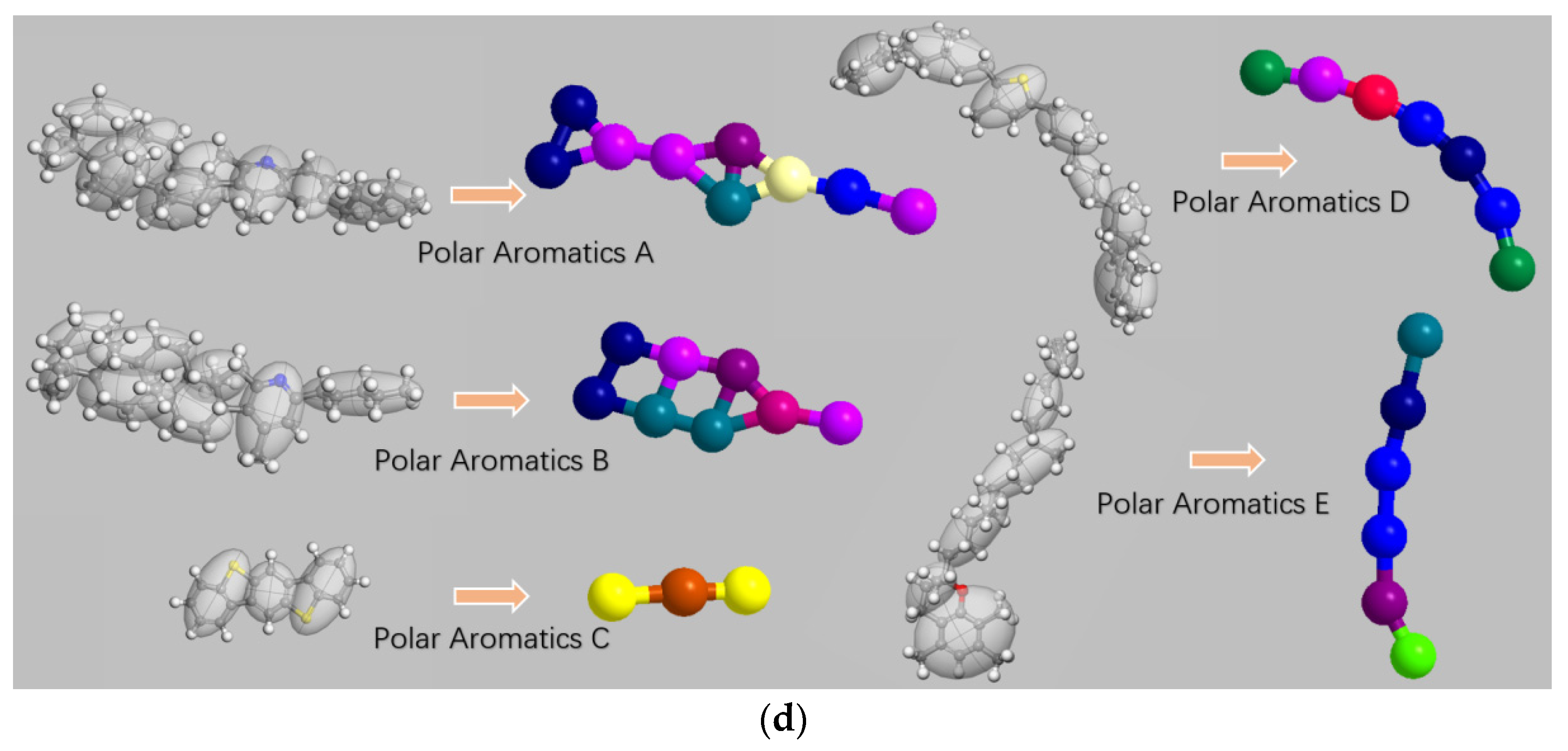
2.2. Coarse-Grained Model
2.3. Coarse-Grained Force Field
- (1)
- The non-bond potential is the energy that mainly affects the determination of macroscopic properties of the bitumen system, including van der Waals potential, hydrogen bond potential and charge energy. Among the non-bonded potential, van der Waals potential plays a major role in the bitumen system. Hydrogen bonds only exist between hydrogen atoms, oxygen atoms, fluorine atoms and nitrogen atoms and must conform to a certain structural general formula; the bitumen molecular model in this study has few oxygen atoms and nitrogen atoms and little influence of hydrogen bonds, so hydrogen bond potential is not considered in this study. The CG beads divided in this study are all neutral particles without charge, so charge energy is not considered in this study.
- (2)
- The bond potential energy function includes simple harmonic vibration potential energy function, plane bending potential energy function and out-of-plane rocking potential energy function. The bond potential energy plays a minor role in the determination of the macroscopic properties of the bitumen system. In this study, the out-of-plane rocking potential energy function with the least influence on the bitumen system is not considered.
2.3.1. Non-Bond Potential
2.3.2. Bond Potential
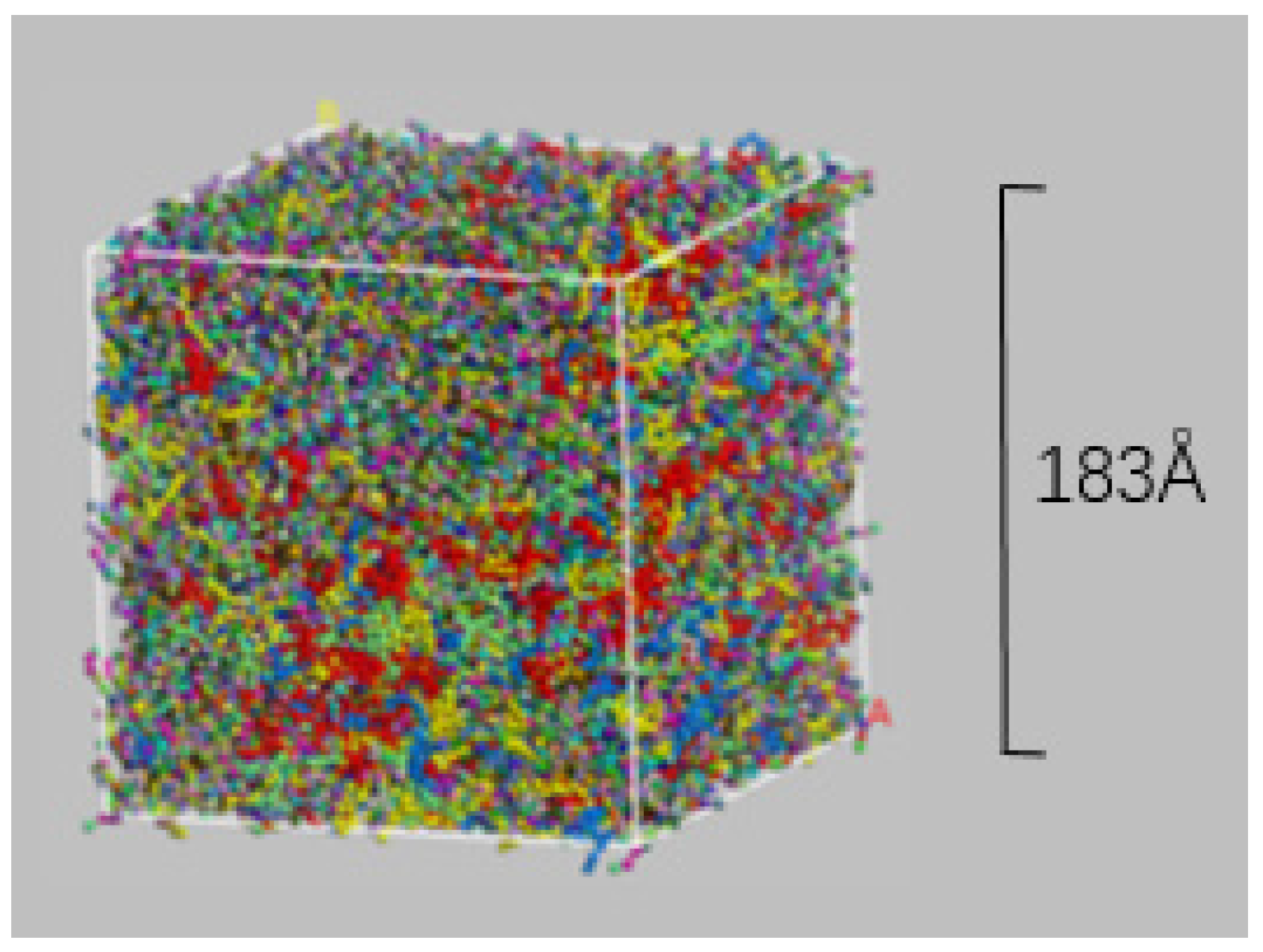
2.4. Coarse-Grained Molecular Dynamics
- (1)
- Color the CG molecule models of each component of the bitumen separately for subsequent studies on the effect of the components upon the properties of the bitumen system. The asphaltenes are colored red, the aromatics green, the polar aromatics yellow and the saturates blue.
- (2)
- Construct a square calculation box with periodic boundary conditions, side lengths of the box are 30 nm and the initial density of the box is 0.15 g/cm3, then input the CG molecular model of each component into the box in terms of the mass ratio;
- (3)
- Conduct geometric optimization (energy minimization), which should be as full as possible since the CG molecular model is larger with more particles than the all-atom one, setting the number of iterations at 40,000;
- (4)
- Relaxing the model for 2000 ps at 533 K in the NVT ensemble to reach the energy minimum conformation;
- (5)
- Simulate the model for 3000 ps at 533 K and 10 atm in the NPT ensemble, then 1000 ps at 1 atm. The 10 atm run is for accelerating the compression of the model, and the 1atm run is to achieve the desired final pressure.
2.5. Validation of the Bitumen CG Molecular Model
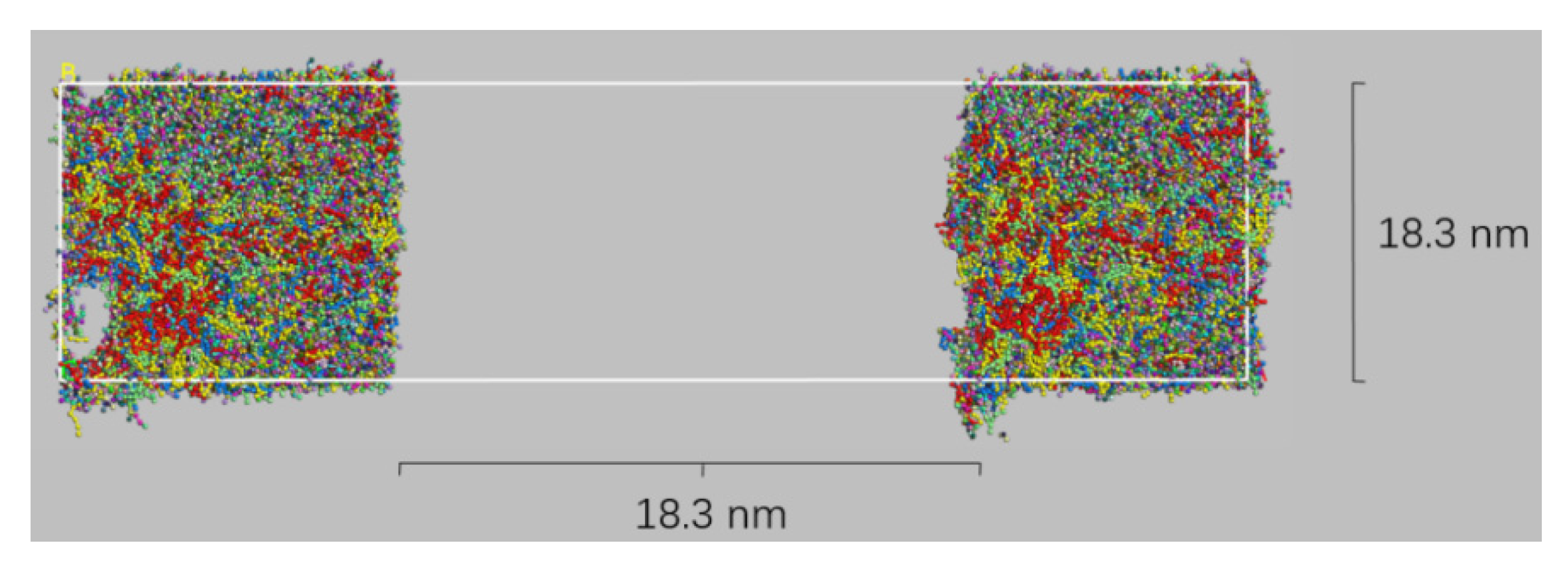
2.6. Bitumen Healing
3. Results and Discussion
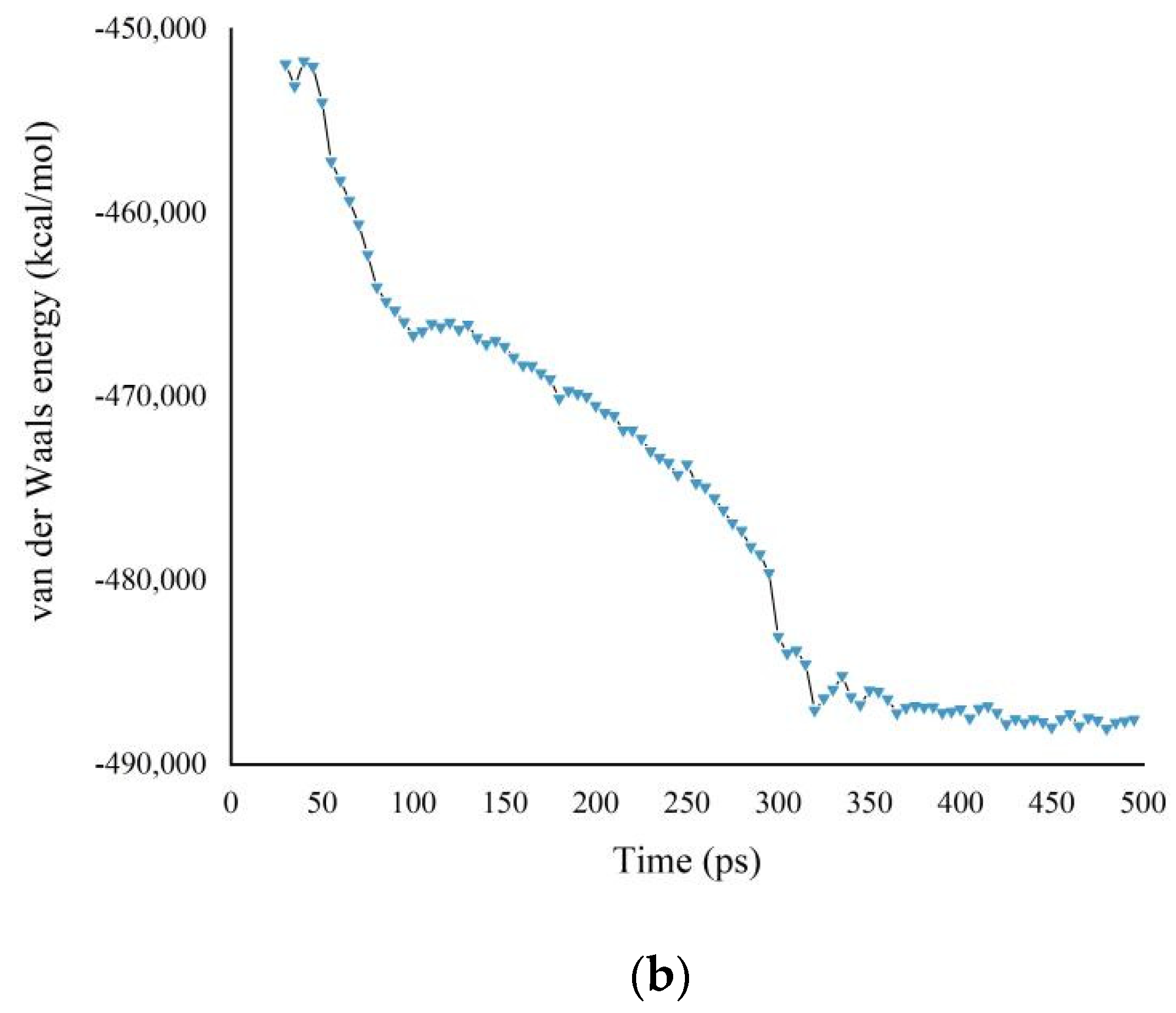
3.1. Potential Energy
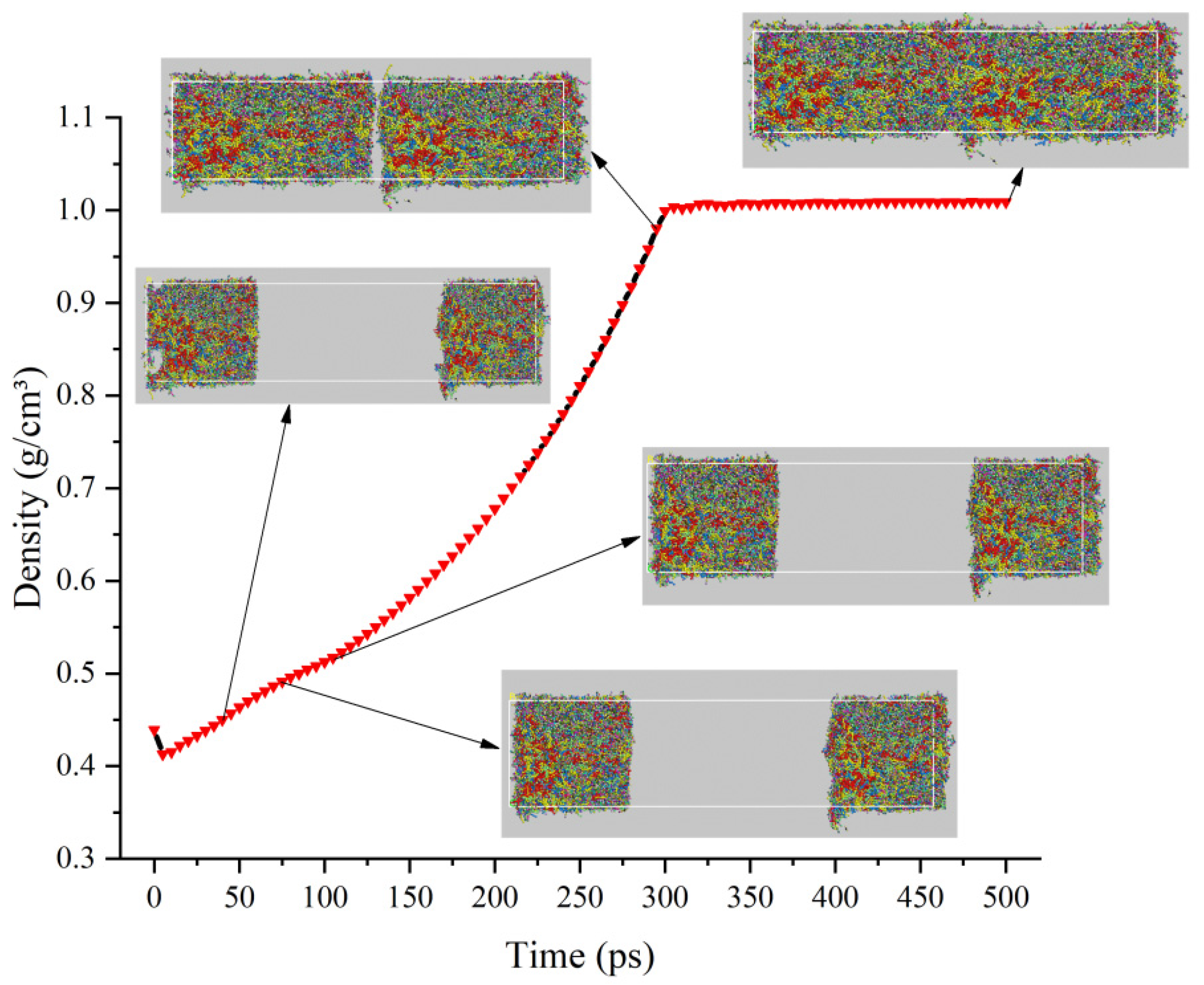
3.2. Density
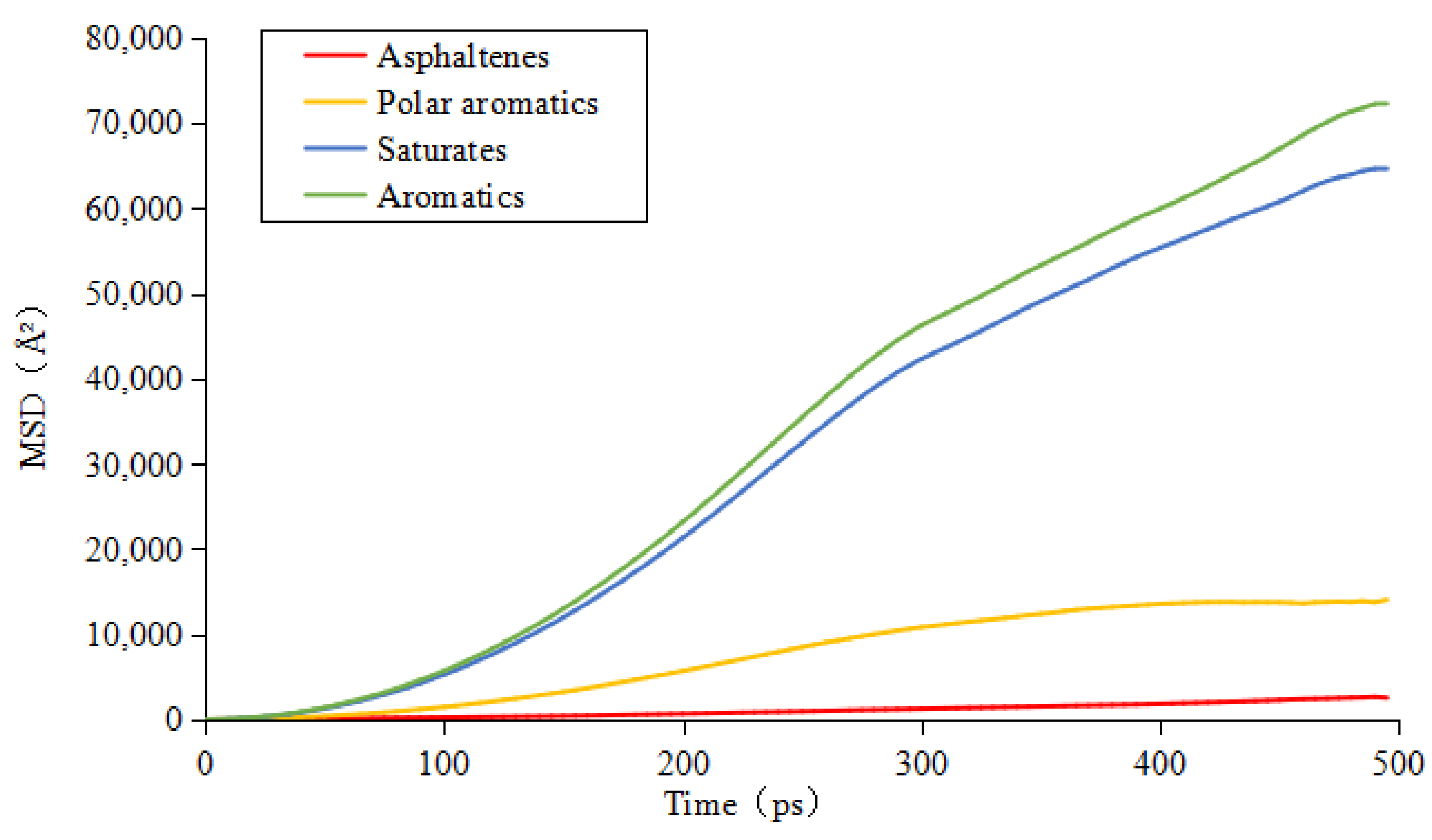
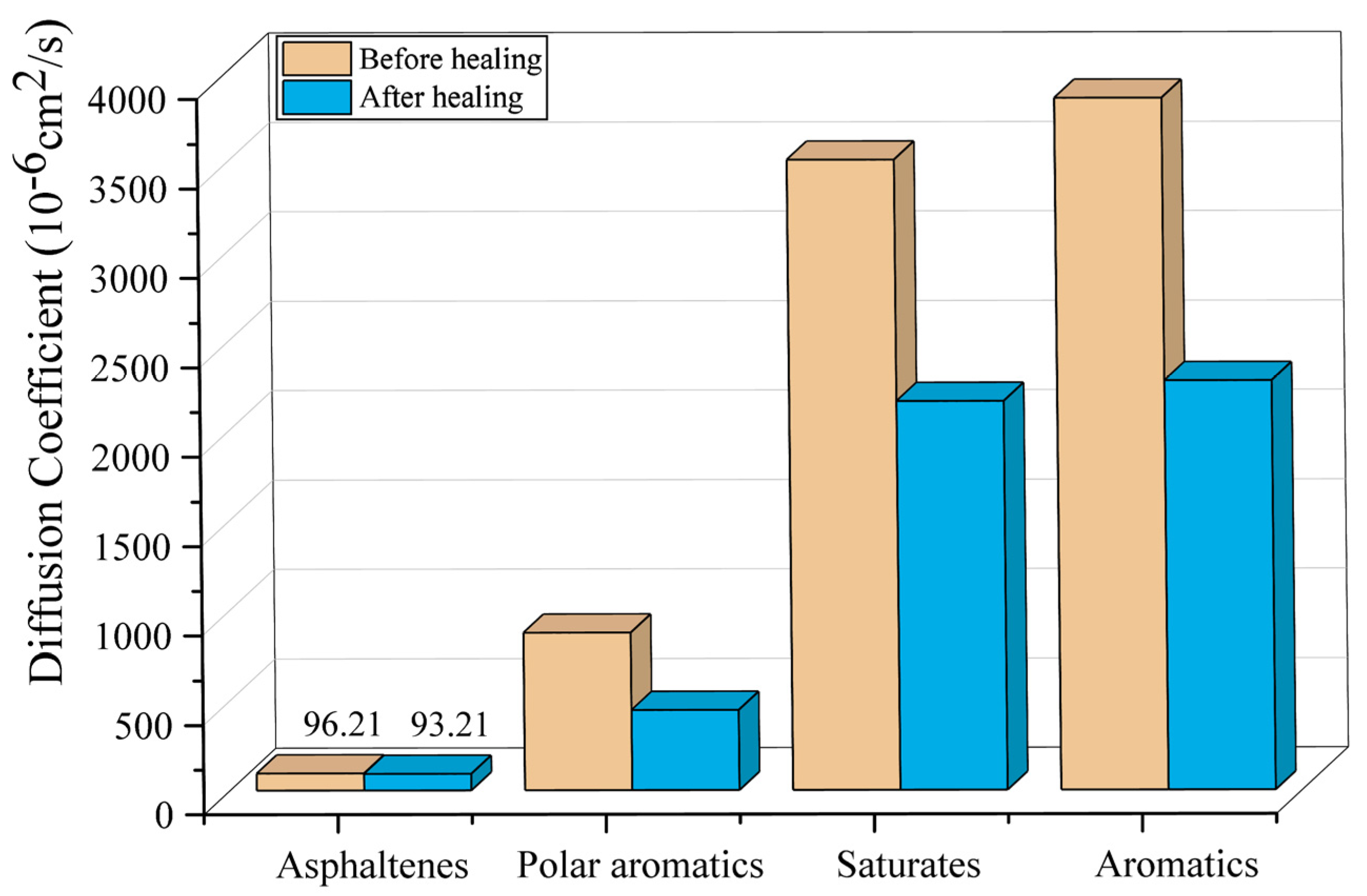
3.3. Diffusion Coefficient
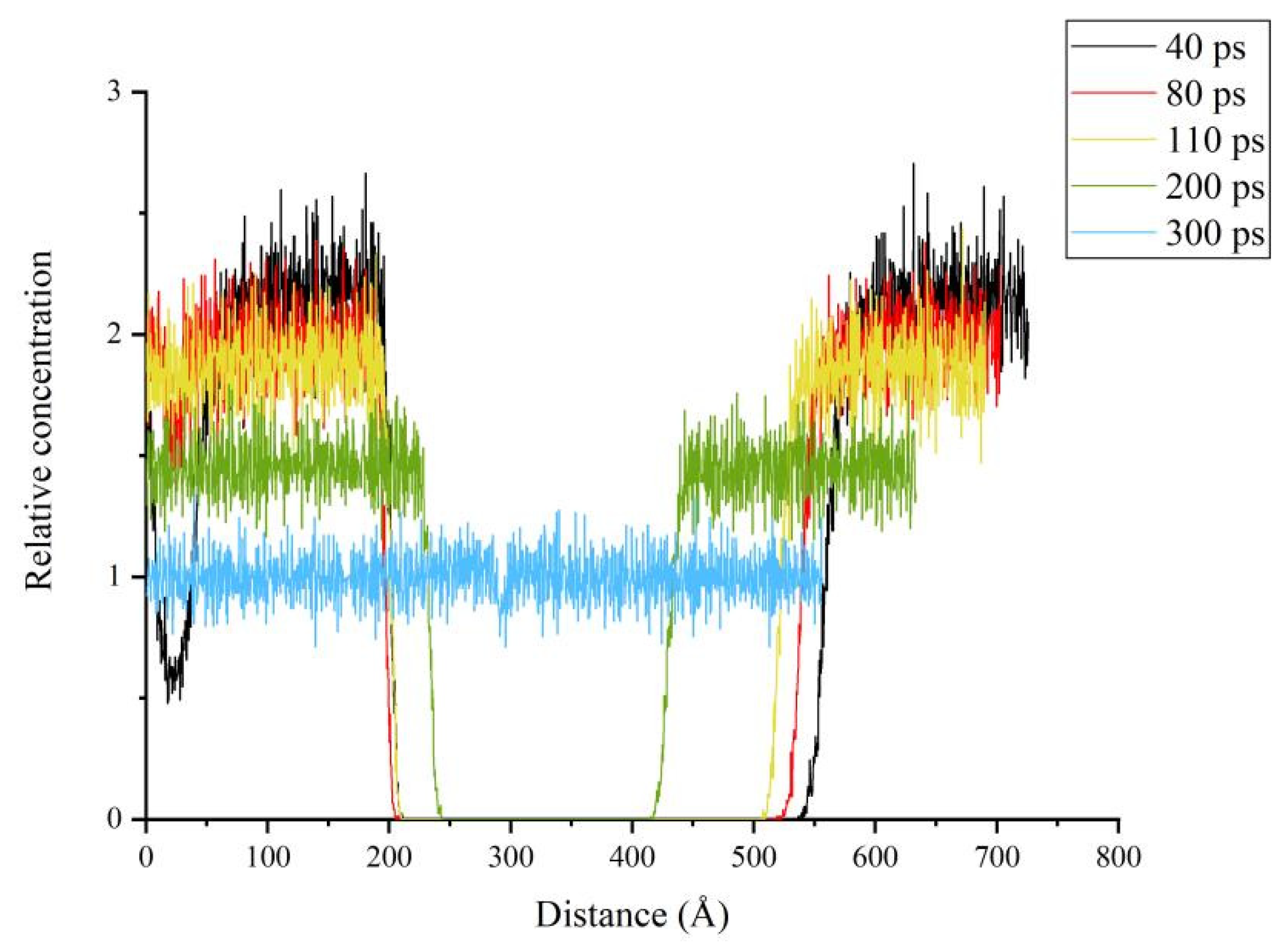
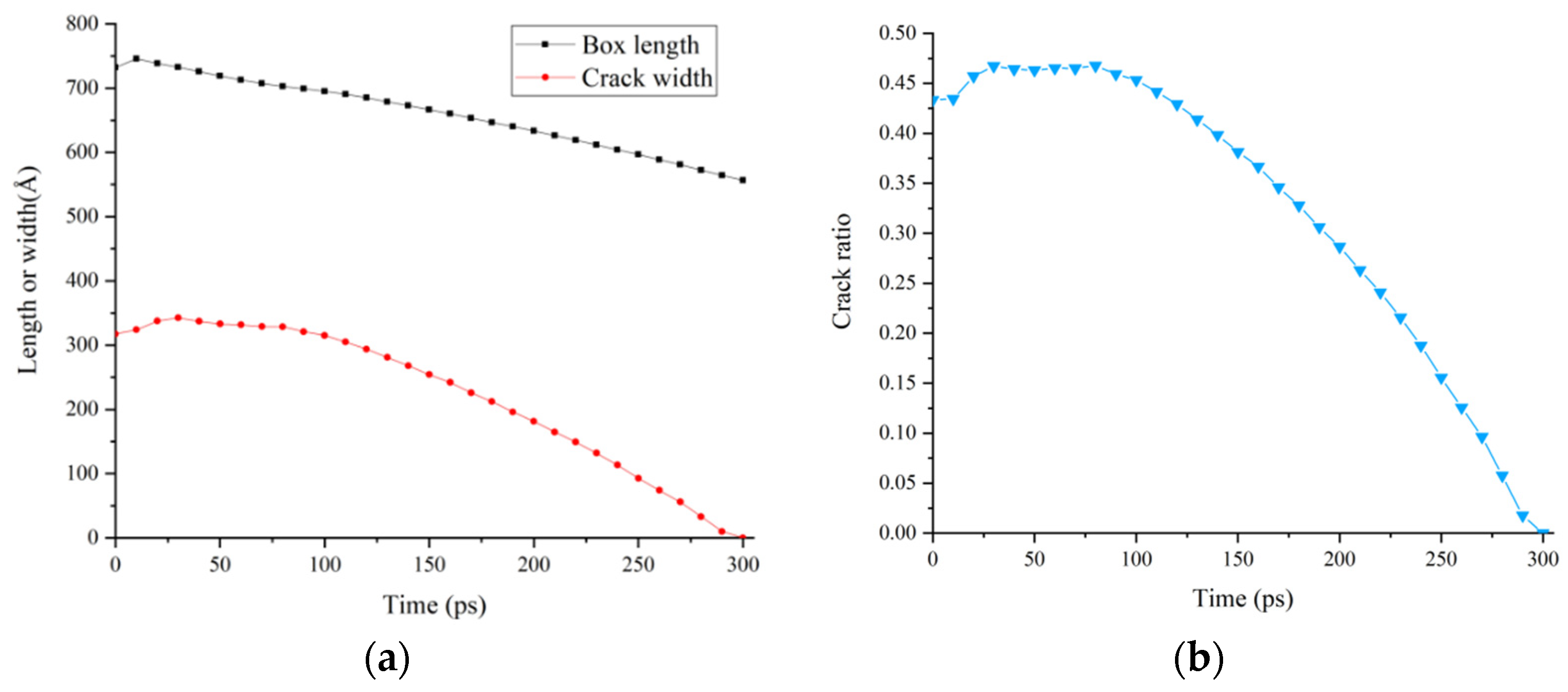
3.4. Relative Concentration and Crack Ratio
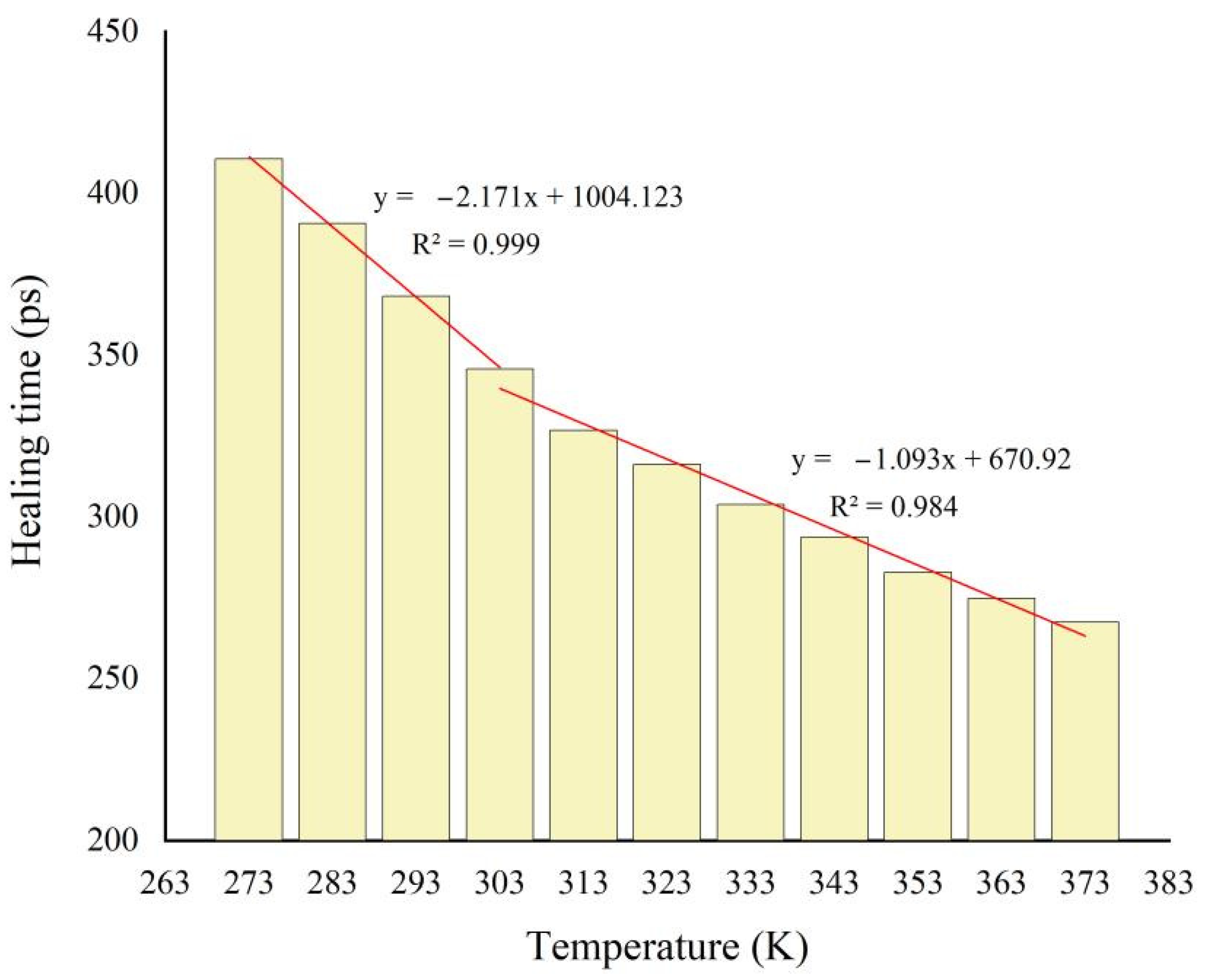
3.5. Temperature
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, L.; Greenfield, M.L. Molecular Orientation in Model Asphalts Using Molecular Simulation. Energy Fuels 2007, 21, 1102–1111. [Google Scholar] [CrossRef]
- Li, D.D.; Greenfield, M.L. Chemical compositions of improved model asphalt systems for molecular simulations. Fuel 2014, 115, 347–356. [Google Scholar] [CrossRef]
- Li, D.D.; Greenfield, M.L. Viscosity, relaxation time, and dynamics within a model asphalt of larger molecules. J. Chem. Phys. 2014, 140, 034507. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Lu, X.; Liu, L.; Zhang, C. Investigation of the influence of crack width on healing properties of asphalt binders at multi-scale levels. Constr. Build. Mater. 2016, 126, 197–205. [Google Scholar] [CrossRef]
- Sun, D.; Sun, G.; Zhu, X.; Ye, F.; Xu, J. Intrinsic temperature sensitive self-healing character of asphalt binders based on molecular dynamics simulations. Fuel 2018, 211, 609–620. [Google Scholar] [CrossRef]
- Qu, X.; Wang, D.; Hou, Y.; Liu, Q.; Oeser, M.; Wang, L. Investigation on Self-Healing Behavior of Asphalt Binder Using a Six-Fraction Molecular Model. J. Mater. Civ. Eng. 2019, 31, 04019046. [Google Scholar] [CrossRef]
- He, L.; Li, G.; Lv, S.; Gao, J.; Kowalski, K.J.; Valentin, J.; Alexiadis, A. Self-healing behavior of asphalt system based on molecular dynamics simulation. Constr. Build. Mater. 2020, 254, 119225. [Google Scholar] [CrossRef]
- He, L.; Zheng, Y.; Alexiadis, A.; Cannone Falchetto, A.; Li, G.; Valentin, J.; Van den bergh, W.; Emmanuilovich Vasiliev, Y.; Kowalski, K.J.; Grenfell, J. Research on the self-healing behavior of asphalt mixed with healing agents based on molecular dynamics method. Constr. Build. Mater. 2021, 295, 123430. [Google Scholar] [CrossRef]
- Sun, W.; Wang, H. Self-healing of asphalt binder with cohesive failure: Insights from molecular dynamics simulation. Constr. Build. Mater. 2020, 262, 120538. [Google Scholar] [CrossRef]
- Hu, D.; Pei, J.; Li, R.; Zhang, J.; Jia, Y.; Fan, Z. Using thermodynamic parameters to study self-healing and interface properties of crumb rubber modified asphalt based on molecular dynamics simulation. Front. Struct. Civ. Eng. 2020, 14, 109–122. [Google Scholar] [CrossRef]
- Gong, Y.; Xu, J.; Yan, E.; Cai, J. The Self-Healing Performance of Carbon-Based Nanomaterials Modified Asphalt Binders Based on Molecular Dynamics Simulations. Front. Mater. 2021, 7, 599551. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, X.; Zhang, F.; Ji, W.; Otto, F. Study of the self-healing properties of aged asphalt-binder regenerated using residual soybean-oil. J. Appl. Polym. Sci. 2022, 139, 51523. [Google Scholar] [CrossRef]
- Tripathy, M.; Agarwal, U.; Kumar, P.B.S. Toward Transferable Coarse-Grained Potentials for Poly-Aromatic Hydrocarbons: A Force Matching Approach. Macromol. Theory Simul. 2018, 28, 1800040. [Google Scholar] [CrossRef]
- Mohammadi, K.; Madadi, A.A.; Bajalan, Z.; Pishkenari, H.N. Analysis of mechanical and thermal properties of carbon and silicon nanomaterials using a coarse-grained molecular dynamics method. Int. J. Mech. Sci. 2020, 187, 106112. [Google Scholar] [CrossRef]
- Eichenberger, A.P.; Huang, W.; Riniker, S.; van Gunsteren, W.F. Supra-Atomic Coarse-Grained GROMOS Force Field for Aliphatic Hydrocarbons in the Liquid Phase. J. Chem. Theory Comput. 2015, 11, 2925–2937. [Google Scholar] [CrossRef] [PubMed]
- Yaxin, A.; Bejagam, K.K.; Deshmukh, S.A. Development of New Transferable Coarse-Grained Models of Hydrocarbons. J. Phys. Chem. B 2018, 122, 7143–7153. [Google Scholar]
- Garrido, J.M.; Cartes, M.; Mejía, A. Coarse-grained theoretical modeling and molecular simulations of nitrogen + n -alkanes: ( n -pentane, n -hexane, n -heptane, n -octane). J. Supercrit. Fluids 2017, 129, 83–90. [Google Scholar] [CrossRef]
- Jiménez-Serratos, G.; Totton, T.S.; Jackson, G.; Müller, E.A. Aggregation Behavior of Model Asphaltenes Revealed from Large-Scale Coarse-Grained Molecular Simulations. J. Phys. Chem. B 2019, 123, 2380–2396. [Google Scholar] [CrossRef]
- Li, G.; Han, M.; Tan, Y.; Meng, A.; Li, J.; Li, S. Research on bitumen molecule aggregation based on coarse-grained molecular dynamics. Constr. Build. Mater. 2020, 263, 120933. [Google Scholar] [CrossRef]
- Li, L.; Zhang, N.; Sun, D. Road Engineering Materials; China Communication Press: Beijing, China, 2011. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A Gen. Phys. 1985, 31, 1695. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.J. Characterization of Asphalt Properties and Asphalt-Aggregate Interaction Using Molecular Dynamics Simulation. Ph.D. Thesis, Rutgers University Community Repository, Rutgers, NJ, USA, 2017. [Google Scholar]
- Robertson, R.E.; Branthaver, J.F.; Harnsberger, P.M.; Peterson, J.C.; Dorrence, S.M.; Mckay, J.F.; Turner, T.F.; Pauli, A.T.; Huang, S.C.; Huh, J.D. Fundamental Properties of Asphalt and Modified Asphalts; Western Research Institute: Laramie, WY, USA, 2001; Volume I. [Google Scholar]
- Xu, G.; Wang, H. Study of cohesion and adhesion properties of asphalt concrete with molecular dynamics simulation. Comput. Mater. Sci. 2016, 112, 161–169. [Google Scholar] [CrossRef]
- Voth, G.A. On the Development of Coarse-Grained Protein Models. In Coarse-Graining of Condensed Phase and Biomolecular Systems; CRC Press: Boca Raton, FL, USA, 2008; pp. 141–156. [Google Scholar]
- Youngs, T.G.A.; Del Pópolo, M.G.; Kohanoff, J. Development of complex classical force fields through force matching to ab initio data: Application to a room-temperature ionic liquid. J. Phys. Chem. B 2006, 110, 5697–5707. [Google Scholar] [CrossRef] [PubMed]
- Voth, G.A. Coarse-Graining of Condensed Phase and Biomolecular Systems; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar]
- Shinoda, W.; Delvane, R.; Klein, M.L. Multi-property fitting and parameterization of a coarse grained model for aqueous surfactants. Mol. Simul. 2007, 33, 27–36. [Google Scholar] [CrossRef]
- Lyubartsev, A.P.; York, R.; Heald, E.F. Modeling a Boltzmann Distribution: Simbo (Simulated Boltzmann). J. Chem. Educ. 2003, 80, 109. [Google Scholar] [CrossRef]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef]
- Andrews, A.; Guerra, R.; Mullins, O.; Sen, P. Diffusivity of Asphaltene Molecules by Fluorescence Correlation Spectroscopy. J. Phys. Chem. A 2006, 110, 8093–8097. [Google Scholar] [CrossRef]
- Mullins, O.C. The Modified Yen Model. Energy Fuels 2010, 24, 2179–2207. [Google Scholar] [CrossRef]
- Wu, G.; He, L.; Chen, D. Sorption and Distribution of Asphaltene, Resin, Aromatic and Saturate Fractions of Heavy Crude Oil on Quartz Surface: Molecular Dynamic Simulation. Chemosphere 2013, 92, 1465–1471. [Google Scholar] [CrossRef]
- Einstein, A. On the Movement of Small Particles Suspended in Stationary Liquids Required by the Molecular-Kinetic Theory of Heat. Ann. Phys. 1905, 17, 549–560. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Name | Chemical Formula | Number of Molecules Contained in the System |
|---|---|---|
| Asphaltene A | C66H81N | 3 |
| Asphaltene B | C42H54O | 2 |
| Asphaltene C | C51H62S | 3 |
| Saturate A | C30H62 | 4 |
| Saturate B | C35H62 | 4 |
| Aromatic A | C35H44 | 11 |
| Aromatic B | C30H46 | 13 |
| Polar aromatic A | C40H59N | 4 |
| Polar aromatic B | C36H57N | 4 |
| Polar aromatic C | C18H10S2 | 15 |
| Polar aromatic D | C40H60S | 4 |
| Polar aromatic E | C29H50O | 5 |
| Properties | Calculation | Reference Value |
|---|---|---|
| Density (g/cm3) (at 288 K) | 0.996 | 1.025 [8] |
| Diffusion coefficient(cm2/s) (at 298 K) | 0.4 × 10−6 | 0.092 × 10−6 [23] |
| Glass transition temperature (K) | 262.9 | 261.7 [24] |
| Solubility parameter (J/cm3)0.5 | 17.6–18.2 | 13.3–22.5 [25] |
| Bead Name | Structural Formula | Chemical Formula | Mapping Ratio | Legend | Bead Name | Structural Formula | Chemical Formula | Mapping Ratio | Legend |
|---|---|---|---|---|---|---|---|---|---|
| C3 |  | C3Hn | 3 |  | S1 |  | C5HnS | 6 |  |
| C4a |  | C4Hn | 4 |  | B1 |  | C6Hn | 6 |  |
| C4b |  | C4Hn | 4 |  | B2 |  | C4Hn | 4 |  |
| C5a |  | C5Hn | 5 |  | B3 |  | C7Hn | 7 |  |
| C5b |  | C5Hn | 5 |  | B4 |  | C9Hn | 9 |  |
| C6 |  | C6Hn | 6 |  | B5 |  | C9HnO | 10 |  |
| N1 |  | C2HnN | 3 |  | B6 |  | C6HnS | 7 |  |
| N2 |  | C6HnN | 7 |  | B7 |  | C4HnO | 5 |  |
| ε | C3 | C4a | C4b | C5a | C5b | C6 | B1 | B2 | B3 | B4 | B5 | B6 | B7 | N1 | N2 | S1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C3 | 0.805 | 0.806 | 0.806 | 0.807 | 0.806 | 0.810 | 0.814 | 0.806 | 0.813 | 0.868 | 0.882 | 0.829 | 0.809 | 0.608 | 0.843 | 0.849 |
| C4a | 0.806 | 0.806 | 0.807 | 0.807 | 0.810 | 0.815 | 0.808 | 0.849 | 0.869 | 0.885 | 0.834 | 0.811 | 0.696 | 0.847 | 0.855 | |
| C4b | 0.806 | 0.807 | 0.807 | 0.810 | 0.815 | 0.807 | 0.846 | 0.870 | 0.889 | 0.836 | 0.812 | 0.657 | 0.850 | 0.856 | ||
| C5a | 0.811 | 0.811 | 0.811 | 0.817 | 0.809 | 0.873 | 0.883 | 0.907 | 0.840 | 0.814 | 0.762 | 0.856 | 0.862 | |||
| C5b | 0.811 | 0.811 | 0.817 | 0.809 | 0.873 | 0.885 | 0.908 | 0.840 | 0.815 | 0.738 | 0.859 | 0.861 | ||||
| C6 | 0.814 | 0.843 | 0.812 | 0.890 | 0.913 | 0.948 | 0.871 | 0.822 | 0.781 | 0.894 | 0.906 | |||||
| B1 | 0.962 | 0.816 | 0.903 | 1.014 | 1.017 | 0.993 | 0.838 | 0.842 | 0.998 | 1.007 | ||||||
| B2 | 0.808 | 0.855 | 0.842 | 0.866 | 0.827 | 0.817 | 0.713 | 0.839 | 0.840 | |||||||
| B3 | 1.002 | 0.905 | 0.929 | 0.904 | 0.830 | 0.746 | 0.906 | 0.919 | ||||||||
| B4 | 1.036 | 1.051 | 0.985 | 0.856 | 0.861 | 1.006 | 1.028 | |||||||||
| B5 | 1.065 | 1.002 | 0.882 | 0.868 | 1.035 | 1.042 | ||||||||||
| B6 | 1.026 | 0.847 | 0.849 | 1.031 | 1.036 | |||||||||||
| B7 | 0.823 | 0.736 | 1.002 | 1.014 | ||||||||||||
| N1 | 0.634 | 0.854 | 0.857 | |||||||||||||
| N2 | 1.029 | 1.030 | ||||||||||||||
| S1 | 1.034 |
| σ | C3 | C4a | C4b | C5a | C5b | C6 | B1 | B2 | B3 | B4 | B5 | B6 | B7 | N1 | N2 | S1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C3 | 4.80 | 5.35 | 5.41 | 5.47 | 5.14 | 5.32 | 5.39 | 5.36 | 5.35 | 5.68 | 5.78 | 5.46 | 5.377 | 5.17 | 5.52 | 5.55 |
| C4a | 5.68 | 5.88 | 5.99 | 5.99 | 5.73 | 5.80 | 5.71 | 5.69 | 5.83 | 5.91 | 5.82 | 5.74 | 5.44 | 5.83 | 5.84 | |
| C4b | 5.79 | 6.05 | 6.11 | 5.85 | 5.82 | 5.74 | 5.81 | 5.83 | 5.91 | 5.82 | 5.77 | 5.400 | 5.83 | 5.855 | ||
| C5a | 5.95 | 6.15 | 5.966 | 6.03 | 5.82 | 5.95 | 6.05 | 6.11 | 6.04 | 5.87 | 5.67 | 6.06 | 6.08 | |||
| C5b | 6.15 | 5.97 | 6.03 | 5.86 | 5.94 | 6.055 | 6.11 | 6.055 | 5.888 | 5.62 | 6.044 | 6.08 | ||||
| C6 | 5.666 | 5.77 | 5.944 | 5.72 | 5.84 | 5.888 | 5.799 | 5.91 | 5.877 | 5.80 | 5.81 | |||||
| B1 | 5.83 | 5.78 | 5.78 | 6.02 | 6.13 | 5.88 | 5.80 | 5.89 | 5.966 | 6.011 | ||||||
| B2 | 5.70 | 5.744 | 5.76 | 5.79 | 5.77 | 5.74 | 5.51 | 5.77 | 5.79 | |||||||
| B3 | 5.78 | 5.85 | 5.877 | 5.811 | 5.85 | 5.69 | 5.83 | 5.83 | ||||||||
| B4 | 6.11 | 6.25 | 6.02 | 5.89 | 5.93 | 6.05 | 6.088 | |||||||||
| B5 | 6.30 | 6.08 | 5.94 | 5.95 | 6.12 | 6.16 | ||||||||||
| B6 | 5.97 | 5.88 | 5.90 | 5.99 | 6.02 | |||||||||||
| B7 | 5.81 | 5.56 | 5.90 | 5.93 | ||||||||||||
| N1 | 5.28 | 5.78 | 5.85 | |||||||||||||
| N2 | 6.03 | 6.044 | ||||||||||||||
| S1 | 6.08 |
| r0 | C3 | C4a | C4b | C5a | B1 | B2 | B3 | B4 | B5 | B6 | B7 | N1 | N2 | S1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C3 | 4.574 | 4.629 | 4.589 | 4.618 | 4.683 | 4.564 | ||||||||
| C4a | 4.686 | 4.684 | 4.65 | 4.593 | 4.688 | 4.689 | ||||||||
| C4b | 4.686 | 4.642 | 4.711 | 4.648 | 4.651 | 4.757 | 4.677 | |||||||
| C5a | 4.516 | 4.621 | 4.606 | 4.659 | 4.757 | |||||||||
| C5b | 4.589 | |||||||||||||
| C6 | 4.573 | 4.688 | 4.677 | |||||||||||
| B1 | 4.525 | 4.611 | 4.689 | 4.539 | ||||||||||
| B2 | 4.657 | 4.619 | 4.66 |
| Bead 1 | Middle Bead | Bead 2 | θ0 (°) | Bead 1 | Middle Bead | Bead 2 | θ0 (°) |
|---|---|---|---|---|---|---|---|
| C3 | C3 | C3 | 81.6 | C3 | B1 | C4a | 154 |
| C3 | C3 | C4a | 84.7 | C3 | B1 | C4b | 163.3 |
| C3 | C3 | C5a | 86 | C3 | B1 | C5b | 161.5 |
| C3 | C3 | B1 | 96.8 | C3 | B1 | B2 | 165.6 |
| C3 | C3 | B2 | 89.3 | C3 | B1 | B3 | 156.3 |
| C4a | C3 | C4a | 118.3 | C5a | B1 | B2 | 165 |
| C4a | C3 | C4b | 105.6 | C5b | B1 | B7 | 153.1 |
| C4a | C3 | B1 | 107.6 | B2 | B1 | B2 | 163.4 |
| C4a | C3 | B2 | 100.6 | B2 | B1 | B3 | 160.3 |
| C4b | C3 | C4b | 88.5 | B2 | B1 | B7 | 169.7 |
| C4b | C3 | B1 | 91.3 | B6 | B1 | B6 | 174.3 |
| C3 | C4a | C3 | 86.3 | C3 | B2 | C3 | 89.6 |
| C3 | C4a | C4a | 95.7 | C3 | B2 | C4b | 81.6 |
| C3 | C4a | B1 | 86.9 | C3 | B2 | B1 | 80.9 |
| C3 | C4a | B4 | 86.4 | C3 | B2 | B6 | 91.6 |
| C3 | C4a | N3 | 83.7 | C4a | B2 | C5a | 89.1 |
| C4a | C4a | C5a | 95.3 | C4a | B2 | B2 | 89.6 |
| C5a | C4a | B5 | 162 | C4b | B2 | B1 | 74.4 |
| B2 | C4a | B2 | 169.5 | C4b | B2 | B6 | 84.8 |
| C3 | C4b | C3 | 167.6 | C5a | B2 | B1 | 80 |
| C3 | C4b | C4b | 170.2 | C5a | B2 | B6 | 93.8 |
| C3 | C4b | B1 | 157.3 | C5a | B2 | N2 | 85.6 |
| C4b | C4b | C5a | 165.5 | C6 | B2 | B1 | 166.4 |
| C5a | C4b | C5a | 144.2 | B1 | B2 | B1 | 80.5 |
| C5a | C4b | N2 | 159.5 | B1 | B2 | B2 | 75.4 |
| C3 | C5a | C4b | 82.5 | B1 | B2 | B6 | 85.4 |
| C3 | C5a | C5a | 75.5 | B1 | B2 | N1 | 84.6 |
| C4a | C5a | C5a | 77.7 | C5a | B3 | B1 | 89.9 |
| C4a | C5a | B1 | 104.9 | C4b | B6 | B2 | 152.5 |
| C4b | C5a | C5a | 104 | B2 | B6 | B2 | 153.6 |
| C4b | C5a | B1 | 97.5 | B1 | B7 | B1 | 91 |
| C4b | C5a | N2 | 97.4 | C4b | N2 | C5a | 164 |
| C5a | C5a | C5a | 107.6 | C4b | N2 | B2 | 162.7 |
| C5a | C5a | N2 | 108.4 | C5a | N2 | C5a | 161.3 |
| B1 | C5a | B2 | 101.2 | C5a | N2 | B2 | 158.2 |
| B4 | C6 | S1 | 89.6 | C4a | S1 | C6 | 152.7 |
| C3 | B1 | C3 | 158.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, L.; Zhou, Z.; Ling, F.; Alexiadis, A.; Van den Bergh, W.; Cannone Falchetto, A.; Balieu, R.; Zhu, J.; Valentin, J.; Kowalski, K.J.; et al. A Coarse-Grained Molecular Model for Simulating Self-Healing of Bitumen. Appl. Sci. 2022, 12, 10360. https://doi.org/10.3390/app122010360
He L, Zhou Z, Ling F, Alexiadis A, Van den Bergh W, Cannone Falchetto A, Balieu R, Zhu J, Valentin J, Kowalski KJ, et al. A Coarse-Grained Molecular Model for Simulating Self-Healing of Bitumen. Applied Sciences. 2022; 12(20):10360. https://doi.org/10.3390/app122010360
Chicago/Turabian StyleHe, Liang, Zhiguang Zhou, Fei Ling, Alessio Alexiadis, Wim Van den Bergh, Augusto Cannone Falchetto, Romain Balieu, Jiqing Zhu, Jan Valentin, Karol J. Kowalski, and et al. 2022. "A Coarse-Grained Molecular Model for Simulating Self-Healing of Bitumen" Applied Sciences 12, no. 20: 10360. https://doi.org/10.3390/app122010360
APA StyleHe, L., Zhou, Z., Ling, F., Alexiadis, A., Van den Bergh, W., Cannone Falchetto, A., Balieu, R., Zhu, J., Valentin, J., Kowalski, K. J., & Zhang, L. (2022). A Coarse-Grained Molecular Model for Simulating Self-Healing of Bitumen. Applied Sciences, 12(20), 10360. https://doi.org/10.3390/app122010360