The Neurochaperonopathies: Anomalies of the Chaperone System with Pathogenic Effects in Neurodegenerative and Neuromuscular Disorders


 , and
, and 
Abstract
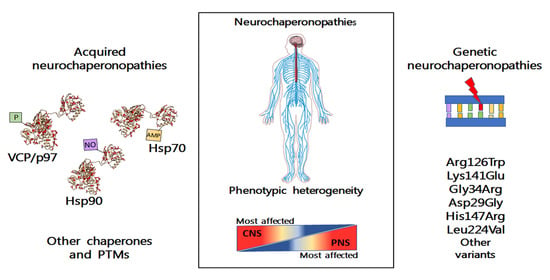
1. Introduction
2. Types of Chaperonopathies in NCPs
3. Genetic NCPs
3.1. Small Heat Shock Proteins
3.2. DnaJ(Hsp40)
3.3. Hsp70(DnaK)
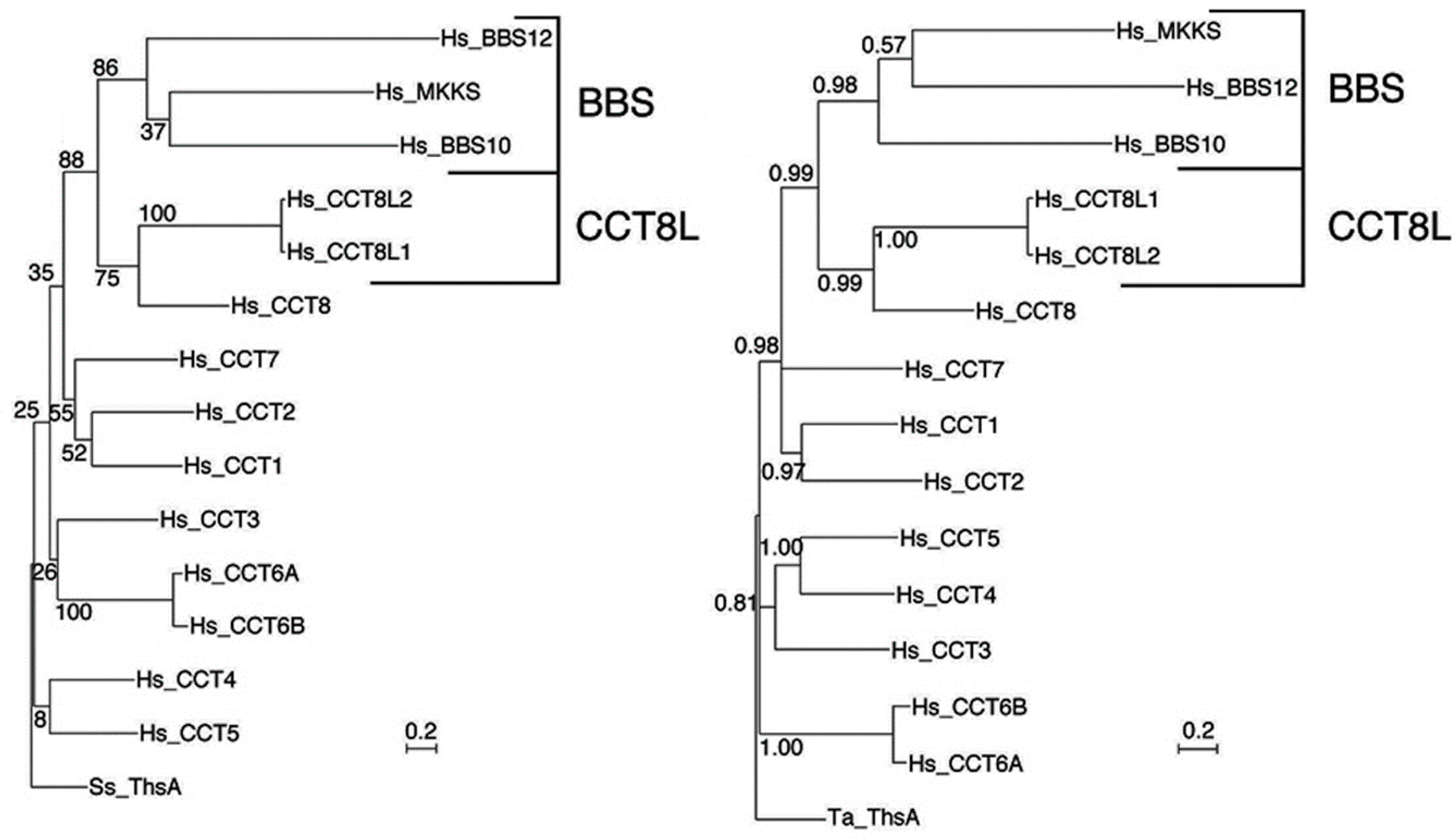
3.4. The Chaperonins
4. Acquired Neurochaperonopathies
4.1. Hsp70(DnaK)
4.2. Hsp90 (HSPC)
4.3. VCP/p97
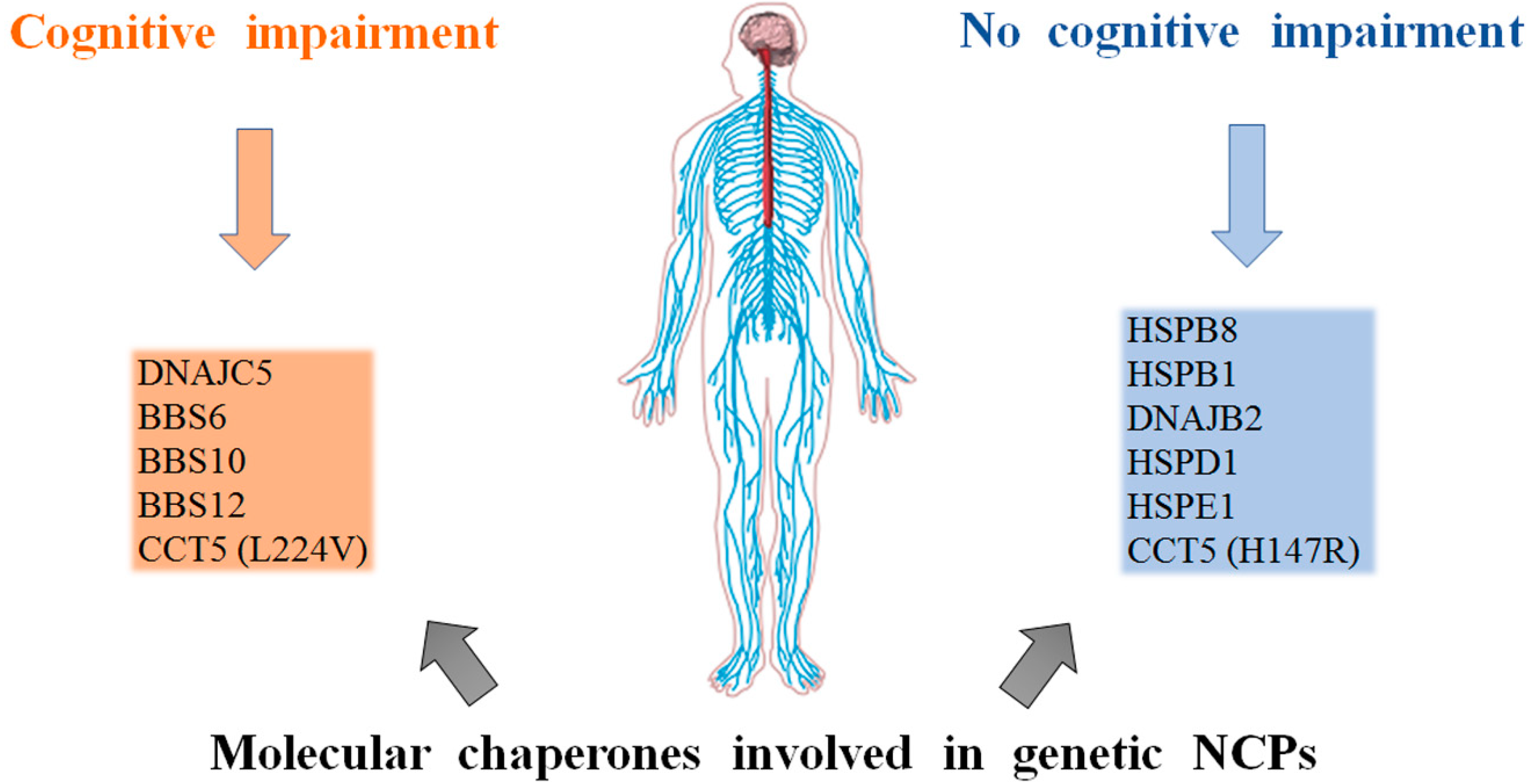
5. Discussion
6. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Briston, T.; Hicks, A.R. Mitochondrial dysfunction and neurodegenerative proteinopathies: Mechanisms and prospects for therapeutic intervention. Biochem. Soc. Trans. 2018, 46, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Dugger, B.N.; Dickson, D.W. Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, L.; Yan, T.; Perry, G.; Wang, X. TDP-43 proteinopathy and mitochondrial abnormalities in neurodegeneration. Mol. Cell. Neurosci. 2019, 100, 103396. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.L.; Lee, E.B.; Xie, S.X.; Rennert, L.; Suh, E.; Bredenberg, C.; Caswell, C.; Van Deerlin, V.M.; Yan, N.; Yousef, A.; et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 2018, 141, 2181–2193. [Google Scholar] [CrossRef] [PubMed]
- Carlisle, C.; Prill, K.; Pilgrim, D. Chaperones and the proteasome system: Regulating the construction and demolition of striated muscle. Int. J. Mol. Sci. 2018, 19, 32. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I. Proteasomal and autophagic degradation systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef]
- Nam, T.; Han, J.H.; Devkota, S.; Lee, H.W. Emerging paradigm of crosstalk between autophagy and the ubiquitin-proteasome system. Mol. Cells 2017, 40, 897–905. [Google Scholar]
- Catarino, S.; Pereira, P.; Girao, H. Molecular control of chaperone-mediated autophagy. Essays Biochem. 2017, 61, 663–674. [Google Scholar]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk between mammalian autophagy and the ubiquitin-proteasome system. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef]
- Horwich, A.L.; Fenton, W.A.; Chapman, E.; Farr, G.W. Two families of chaperonin: Physiology and mechanism. Annu. Rev. Cell Dev. Biol. 2007, 23, 115–145. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Ulrich Hartl, F. Molecular Chaperone Functions in Protein Folding and Proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef] [PubMed]
- Finka, A.; Sharma, S.K.; Goloubinoff, P. Multi-layered molecular mechanisms of polypeptide holding, unfolding and disaggregation by HSP70/HSP110 chaperones. Front. Mol. Biosci. 2015, 2, 29. [Google Scholar] [CrossRef] [PubMed]
- Mogk, A.; Bukau, B.; Kampinga, H.H. Cellular Handling of Protein Aggregates by Disaggregation Machines. Mol. Cell 2018, 69, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Willison, K.R. The structure and evolution of eukaryotic chaperonin-containing TCP-1 and its mechanism that folds actin into a protein spring. Biochem. J. 2018, 475, 3009–3034. [Google Scholar] [CrossRef]
- Adams, B.M.; Oster, M.E.; Hebert, D.N. Protein Quality Control in the Endoplasmic Reticulum. Protein J. 2019, 38, 317–329. [Google Scholar] [CrossRef]
- Dahiya, V.; Buchner, J. Functional principles and regulation of molecular chaperones. In Advances in Protein Chemistry and Structural Biology; Academic Press Inc.: Cambridge, MA, USA, 2019; Volume 114, pp. 1–60. ISBN 9780128155578. [Google Scholar]
- Macario, A.J.L.; Conway de Macario, E. Sick chaperones, cellular stress, and disease. N. Engl. J. Med. 2005, 353, 1489–1501. [Google Scholar] [CrossRef]
- Macario, A.J.L.; Conway de Macario, E. The chaperoning system: Physiology and pathology. In Experimental Medicine Reviews; Plumelia: Bagheria, Italy, 2008; Volumes 2–3, pp. 9–21. [Google Scholar]
- Macario, A.J.L.; Conway de Macario, E. Chaperone proteins and chaperonopathies. In Handbook of Stress; Elsevier; Academic Press: Cambridge, MS, USA, 2019. [Google Scholar]
- Macario, A.J.L.; Conway de Macario, E.; Cappello, F. The Chaperonopathies. Diseases with Defective Molecular Chaperones; Springer: Berlin, Germany, 2013; ISBN 978-94-007-4666-4. [Google Scholar]
- Lanfranco, M.; Vassallo, N.; Cauchi, R.J. Spinal muscular atrophy: From defective chaperoning of snRNP assembly to neuromuscular dysfunction. Front. Mol. Biosci. 2017, 4, 41. [Google Scholar] [CrossRef]
- Lupo, V.; Aguado, C.; Knecht, E.; Espinós, C. Chaperonopathies: Spotlight on hereditary motor neuropathies. Front. Mol. Biosci. 2016, 3, 81. [Google Scholar] [CrossRef]
- Álvarez-Satta, M.; Castro-Sánchez, S.; Valverde, D. Bardet-Biedl syndrome as a chaperonopathy: Dissecting the major role of chaperonin-like BBS proteins (BBS6-BBS10-BBS12). Front. Mol. Biosci. 2017, 4, 55. [Google Scholar] [CrossRef]
- Caruso Bavisotto, C.; Alberti, G.; Vitale, A.M.; Paladino, L.; Campanella, C.; Rappa, F.; Gorska, M.; Conway de Macario, E.; Cappello, F.; Macario, A.J.L.; et al. Hsp60 Post-translational Modifications: Functional and Pathological Consequences. Front. Mol. Biosci. 2020, 7, 95. [Google Scholar] [CrossRef] [PubMed]
- Royer-Bertrand, B.; Castillo-Taucher, S.; Moreno-Salinas, R.; Cho, T.J.; Chae, J.H.; Choi, M.; Kim, O.H.; Dikoglu, E.; Campos-Xavier, B.; Girardi, E.; et al. Mutations in the heat-shock protein A9 (HSPA9) gene cause the EVEN-PLUS syndrome of congenital malformations and skeletal dysplasia. Sci. Rep. 2015, 5, 17154. [Google Scholar] [CrossRef] [PubMed]
- Ghaoui, R.; Palmio, J.; Brewer, J.; Lek, M.; Needham, M.; Evilä, A.; Hackman, P.; Jonson, P.H.; Penttilä, S.; Vihola, A.; et al. Mutations in HSPB8 causing a new phenotype of distal myopathy and motor neuropathy. Neurology 2016, 86, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Irobi, J.; Van Impe, K.; Seeman, P.; Jordanova, A.; Dierick, I.; Verpoorten, N.; Michalik, A.; De Vriendt, E.; Jacobs, A.; Van Gerwen, V.; et al. Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat. Genet. 2004, 36, 597–601. [Google Scholar] [CrossRef]
- Timmerman, V.; Raeymaekers, P.; Nelis, E.; De Jonghe, P.; Muylle, L.; Ceuterick, C.; Martin, J.J.; Van Broeckhoven, C. Linkage analysis of distal hereditary motor neuropathy type II (distal HMN II) in a single pedigree. J. Neurol. Sci. 1992, 109, 41–48. [Google Scholar] [CrossRef]
- Tang, B.S.; Luo, W.; Xia, K.; Xiao, J.F.; Jiang, H.; Shen, L.; Tang, J.G.; Zhao, G.H.; Cai, F.; Pan, Q.; et al. A new locus for autosomal dominant Charcot-Marie-Tooth disease type 2 (CMT2L) maps to chromosome 12q. Hum. Genet. 2004, 114, 527–533. [Google Scholar]
- Tang, B.S.; Zhao, G.; Luo, W.; Xia, K.; Cai, F.; Pan, Q.; Zhang, R.; Zhang, F.; Liu, X.; Chen, B.; et al. Small heat-shock protein 22 mutated in autosomal dominant Charcot-Marie-Tooth disease type 2L. Hum. Genet. 2005, 116, 222–224. [Google Scholar] [CrossRef]
- Capponi, S.; Geroldi, A.; Fossa, P.; Grandis, M.; Ciotti, P.; Gulli, R.; Schenone, A.; Mandich, P.; Bellone, E. HSPB1 and HSPB8 in inherited neuropathies: Study of an Italian cohort of dHMN and CMT2 patients. J. Peripher. Nerv. Syst. 2011, 16, 287–294. [Google Scholar] [CrossRef]
- Houlden, H.; Laura, M.; Wavrant-De Vrièze, F.; Blake, J.; Wood, N.; Reilly, M.M. Mutations in the HSP27 (HSPB1) gene cause dominant, recessive, and sporadic distal HMN/CMT type. Neurology 2008, 71, 1660–1668. [Google Scholar] [CrossRef]
- James, P.A.; Rankin, J.; Talbot, K. Asymmetrical late onset motor neuropathy associated with a novel mutation in the small heat shock protein HSPB1 (HSP27). J. Neurol. Neurosurg. Psychiatry 2008, 79, 461–463. [Google Scholar] [CrossRef]
- Ho, C.C.; Tai, S.M.; Lee, E.C.N.; Mak, T.S.H.; Liu, T.K.T.; Tang, V.W.L.; Poon, W.T. Rapid identification of pathogenic variants in two cases of Charcot-Marie-tooth disease by gene-panel sequencing. Int. J. Mol. Sci. 2017, 18, 770. [Google Scholar] [CrossRef] [PubMed]
- Evgrafov, O.V.; Mersiyanova, I.; Irobi, J.; Van Den Bosch, L.V.; Dierick, I.; Leung, C.L.; Schagina, O.; Verpoorten, N.; Van Impe, K.; Fedotov, V.; et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat. Genet. 2004, 36, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Liu, X.; Zhao, G.; Luo, W.; Xia, K.; Pan, Q.; Cai, F.; Hu, Z.; Zhang, C.; Chen, B.; et al. Mutation analysis of the small heat shock protein 27 gene in Chinese patients with Charcot-Marie-Tooth disease. Arch. Neurol. 2005, 62, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, S.; Previtali, S.C.; Coviello, S.; Scarlato, M.; Cerri, F.; Di Pierri, E.; Piantoni, L.; Spiga, I.; Fazio, R.; Riva, N.; et al. Analyzing histopathological features of rare Charcot-Marie-Tooth neuropathies to unravel their pathogenesis. Arch. Neurol. 2010, 67, 1498–1505. [Google Scholar] [CrossRef] [PubMed]
- Oberstadt, M.; Mitter, D.; Classen, J.; Baum, P. Late onset dHMN II caused by c.404C>G mutation in HSPB1 gene. J. Peripher. Nerv. Syst. 2016, 21, 111–113. [Google Scholar] [CrossRef]
- Ismailov, S.M.; Fedotov, V.P.; Dadali, E.L.; Polyakov, A.V.; Van Broeckhoven, C.; Ivanov, V.I.; De Jonghe, P.; Timmerman, V.; Evgrafov, O.V. A new locus for autosomal dominant Charcot-Marie-Tooth disease type 2 (CMT2F) maps to chromosome 7q11-q. Eur. J. Hum. Genet. 2001, 9, 646–650. [Google Scholar] [CrossRef]
- Ikeda, Y.; Abe, A.; Ishida, C.; Takahashi, K.; Hayasaka, K.; Yamada, M. A clinical phenotype of distal hereditary motor neuronopathy type II with a novel HSPB1 mutation. J. Neurol. Sci. 2009, 277, 9–12. [Google Scholar] [CrossRef]
- Lin, K.-P.; Soong, B.-W.; Yang, C.-C.; Huang, L.-W.; Chang, M.-H.; Lee, I.-H.; Antonellis, A.; Lee, Y.-C. The Mutational Spectrum in a Cohort of Charcot-Marie-Tooth Disease Type 2 among the Han Chinese in Taiwan. PLoS ONE 2011, 6, e29393. [Google Scholar] [CrossRef]
- Luigetti, M.; Fabrizi, G.M.; Madia, F.; Ferrarini, M.; Conte, A.; Del Grande, A.; Tasca, G.; Tonali, P.A.; Sabatelli, M. A novel HSPB1 mutation in an Italian patient with CMT2/dHMN phenotype. J. Neurol. Sci. 2010, 298, 114–117. [Google Scholar] [CrossRef]
- Kijima, K.; Numakura, C.; Goto, T.; Takahashi, T.; Otagiri, T.; Umetsu, K.; Hayasaka, K. Small heat shock protein 27 mutation in a Japanese patient with distal hereditary motor neuropathy. J. Hum. Genet. 2005, 50, 473–476. [Google Scholar] [CrossRef]
- Bie, A.S.; Fernandez-Guerra, P.; Birkler, R.I.D.; Nisemblat, S.; Pelnena, D.; Lu, X.; Deignan, J.L.; Lee, H.; Dorrani, N.; Corydon, T.J.; et al. Effects of a mutation in the HSPE1 gene encoding the mitochondrial co-chaperonin HSP10 and its potential association with a neurological and developmental disorder. Front. Mol. Biosci. 2016, 3, 65. [Google Scholar] [CrossRef] [PubMed]
- Gess, B.; Auer-Grumbach, M.; Schirmacher, A.; Strom, T.; Zitzelsberger, M.; Rudnik-Schöneborn, S.; Röhr, D.; Halfter, H.; Young, P.; Senderek, J. HSJ1-related hereditary neuropathies Novel mutations and extended clinical spectrum. Neurology 2014, 83, 1726–1732. [Google Scholar] [CrossRef] [PubMed]
- Blumen, S.C.; Astord, S.; Robin, V.; Vignaud, L.; Toumi, N.; Cieslik, A.; Achiron, A.; Carasso, R.L.; Gurevich, M.; Braverman, I.; et al. A rare recessive distal hereditary motor neuropathy with HSJ1 chaperone mutation. Ann. Neurol. 2012, 71, 509–519. [Google Scholar]
- Benitez, B.A.; Alvarado, D.; Cai, Y.; Mayo, K.; Chakraverty, S.; Norton, J.; Morris, J.C.; Sands, M.S.; Goate, A.; Cruchaga, C. Exome-sequencing confirms DNAJC5 mutations as cause of adult neuronal ceroid-lipofuscinosis. PLoS ONE 2011, 6, 26741. [Google Scholar] [CrossRef] [PubMed]
- Nosková, L.; Stránecký, V.; Hartmannová, H.; Přistoupilová, A.; Barešová, V.; Ivánek, R.; Hlková, H.; Jahnová, H.; Van Der Zee, J.; Staropoli, J.F.; et al. Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis. Am. J. Hum. Genet. 2011, 89, 241–252. [Google Scholar] [CrossRef]
- Velinov, M.; Dolzhanskaya, N.; Gonzalez, M.; Powell, E.; Konidari, I.; Hulme, W.; Staropoli, J.F.; Xin, W.; Wen, G.Y.; Barone, R.; et al. Mutations in the Gene DNAJC5 Cause Autosomal Dominant Kufs Disease in a Proportion of Cases: Study of the Parry Family and 8 Other Families. PLoS ONE 2012, 7, e29729. [Google Scholar] [CrossRef]
- Cadieux-Dion, M.; Andermann, E.; Lachance-Touchette, P.; Ansorge, O.; Meloche, C.; Barnabé, A.; Kuzniecky, R.I.; Andermann, F.; Faught, E.; Leonberg, S.; et al. Recurrent mutations in DNAJC5 cause autosomal dominant Kufs disease. Clin. Genet. 2013, 83, 571–575. [Google Scholar] [CrossRef]
- Magen, D.; Georgopoulos, C.; Bross, P.; Ang, D.; Segev, Y.; Goldsher, D.; Nemirovski, A.; Shahar, E.; Ravid, S.; Luder, A.; et al. Mitochondrial Hsp60 Chaperonopathy Causes an Autosomal-Recessive Neurodegenerative Disorder Linked to Brain Hypomyelination and Leukodystrophy. Am. J. Hum. Genet. 2008, 83, 30–42. [Google Scholar] [CrossRef]
- Kusk, M.S.; Damgaard, B.; Risom, L.; Hansen, B.; Ostergaard, E. Hypomyelinating Leukodystrophy due to HSPD1 Mutations: A New Patient. Neuropediatrics 2016, 47, 332–335. [Google Scholar] [CrossRef]
- Hansen, J.J.; Dürr, A.; Cournu-Rebeix, I.; Georgopoulos, C.; Ang, D.; Nielsen, M.N.; Davoine, C.S.; Brice, A.; Fontaine, B.; Gregersen, N.; et al. Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin Hsp60. Am. J. Hum. Genet. 2002, 70, 1328–1332. [Google Scholar] [CrossRef]
- Hansen, J.; Svenstrup, K.; Ang, D.; Nielsen, M.N.; Christensen, J.H.; Gregersen, N.; Nielsen, J.E.; Georgopoulos, C.; Bross, P. A novel mutation in the HSPD1 gene in a patient with hereditary spastic paraplegia. J. Neurol. 2007, 254, 897–900. [Google Scholar] [CrossRef] [PubMed]
- Bouhouche, A.; Benomar, A.; Bouslam, N.; Chkili, T.; Yahyaoui, M. Mutation in the epsilon subunit of the cytosolic chaperonin-containing t-complex peptide-1 (Cct5) gene causes autosomal recessive mutilating sensory neuropathy with spastic paraplegia. J. Med. Genet. 2006, 43, 441–443. [Google Scholar] [CrossRef] [PubMed]
- Bouhouche, A.; Benomar, A.; Bouslam, N.; Ouazzani, R.; Chkili, T.; Yahyaoui, M. Autosomal recessive mutilating sensory neuropathy with spastic paraplegia maps to chromosome 5p15.31-14. Eur. J. Hum. Genet. 2006, 14, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Antona, V.; Scalia, F.; Giorgio, E.; Radio, F.C.; Brusco, A.; Oliveri, M.; Corsello, G.; Lo Celso, F.; Vadalà, M.; Conway de Macario, E.; et al. A novel cct5 missense variant associated with early onset motor neuropathy. Int. J. Mol. Sci. 2020, 21, 7631. [Google Scholar] [CrossRef] [PubMed]
- Katsanis, N.; Beales, P.L.; Woods, M.O.; Lewis, R.A.; Green, J.S.; Parfrey, P.S.; Ansley, S.J.; Davidson, W.S.; Lupski, J.R. Mutations in MKKS cause obesity, retinal dystrophy and renal malformations associated with Bardet-Biedl syndrome. Nat. Genet. 2000, 26, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Katsanis, N.; Lupski, J.R.; Beales, P.L. Exploring the molecular basis of Bardet-Biedl syndrome. Hum. Mol. Genet. 2001, 10, 2293–2299. [Google Scholar] [CrossRef] [PubMed]
- Slavotinek, A.M.; Stone, E.M.; Mykytyn, K.; Heckenlively, J.R.; Green, J.S.; Heon, E.; Musarella, M.A.; Parfrey, P.S.; Sheffield, V.C.; Biesecker, L.G. Mutations in MKKS cause Bardet-Biedl syndrome. Nat. Genet. 2000, 26, 15–16. [Google Scholar] [CrossRef]
- Laurier, V.; Stoetzel, C.; Muller, J.; Thibault, C.; Corbani, S.; Jalkh, N.; Salem, N.; Chouery, E.; Poch, O.; Licaire, S.; et al. Pitfalls of homozygosity mapping: An extended consanguineous Bardet-Biedl syndrome family with two mutant genes (BBS2, BBS10), three mutations, but no triallelism. Eur. J. Hum. Genet. 2006, 14, 1195–1203. [Google Scholar] [CrossRef][Green Version]
- Stoetzel, C.; Laurier, V.; Davis, E.E.; Muller, J.; Rix, S.; Badano, J.L.; Leitch, C.C.; Salem, N.; Chouery, E.; Corbani, S.; et al. BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nat. Genet. 2006, 38, 521–524. [Google Scholar] [CrossRef]
- Putoux, A.; Mougou-Zerelli, S.; Thomas, S.; Elkhartoufi, N.; Audollent, S.; Le Merrer, M.; Lachmeijer, A.; Sigaudy, S.; Buenerd, A.; Fernandez, C.; et al. BBS10 mutations are common in ’Meckel’-type cystic kidneys. J. Med. Genet. 2010, 47, 848–852. [Google Scholar] [CrossRef]
- Stoetzel, C.; Muller, J.; Laurier, V.; Davis, E.E.; Zaghloul, N.A.; Vicaire, S.; Jacquelin, C.; Plewniak, F.; Leitch, C.C.; Sarda, P.; et al. Identification of a novel BBS gene (BBS12) highlights the major role of a vertebrate-specific branch of chaperonin-related proteins in Bardet-Biedl syndrome. Am. J. Hum. Genet. 2007, 80, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Paul, C.; Seigneuric, R.; Kampinga, H.H. The small heat shock proteins family: The long forgotten chaperones. Int. J. Biochem. Cell Biol. 2012, 44, 1588–1592. [Google Scholar] [CrossRef] [PubMed]
- Bruey, J.M.; Ducasse, C.; Bonniaud, P.; Ravagnan, L.; Susin, S.A.; Diaz-Latoud, C.; Gurbuxani, S.; Arrigo, A.P.; Kroemer, G.; Solary, E.; et al. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat. Cell Biol. 2000, 2, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Farber, R.; Nakazawa, A.; Kumar, S.; Bharti, A.; Nalin, C.; Weichselbaum, R.; Kufe, D.; Kharbanda, S. Hsp27 functions as a negative regulator of cytochrome c-dependent activation of procaspase. Oncogene 2000, 19, 1975–1981. [Google Scholar] [CrossRef] [PubMed]
- Chowdary, T.K.; Raman, B.; Ramakrishna, T.; Rao, C.M. Mammalian Hsp22 is a heat-inducible small heat-shock protein with chaperone-like activity. Biochem. J. 2004, 381, 379–387. [Google Scholar] [CrossRef]
- Vos, M.J.; Zijlstra, M.P.; Kanon, B.; van Waarde-Verhagen, M.A.W.H.; Brunt, E.R.P.; Oosterveld-Hut, H.M.J.; Carra, S.; Sibon, O.C.M.; Kampinga, H.H. HSPB7 is the most potent polyQ aggregation suppressor within the HSPB family of molecular chaperones. Hum. Mol. Genet. 2010, 19, 4677–4693. [Google Scholar] [CrossRef]
- Kawano, F.; Fujita, R.; Nakai, N.; Terada, M.; Ohira, T.; Ohira, Y. HSP25 can modulate myofibrillar desmin cytoskeleton following the phosphorylation at Ser15 in rat soleus muscle. J. Appl. Physiol. 2012, 112, 176–186. [Google Scholar] [CrossRef]
- Van Montfort, R.; Slingsby, C.; Vierling, E. Structure and function of the small heat shock protein/α-crystallin family of molecular chaperones. Adv. Protein Chem. 2001, 59, 105–156. [Google Scholar]
- Boncoraglio, A.; Minoia, M.; Carra, S. The family of mammalian small heat shock proteins (HSPBs): Implications in protein deposit diseases and motor neuropathies. Int. J. Biochem. Cell Biol. 2012, 44, 1657–1669. [Google Scholar] [CrossRef]
- Berger, P.; Young, P.; Suter, U. Molecular cell biology of Charcot-Marie-Tooth disease. Neurogenetics 2002, 4, 1–15. [Google Scholar] [CrossRef]
- Harding, A.E. Inherited Neuronal Atrophy and Degeneration Predominantly of Lower Motor Neurons. In Peripheral Neuropathy; Elsevier: Amsterdam, The Netherlands, 2005; Volume 2, pp. 1603–1621. ISBN 9780721694917. [Google Scholar]
- Rossor, A.M.; Kalmar, B.; Greensmith, L.; Reilly, M.M. The distal hereditary motor neuropathies. J. Neurol. Neurosurg. Psychiatry 2012, 83, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Solla, P.; Vannelli, A.; Bolino, A.; Marrosu, G.; Coviello, S.; Murru, M.R.; Tranquilli, S.; Corongiu, D.; Benedetti, S.; Marrosu, M.G. Heat shock protein 27 R127W mutation: Evidence of a continuum between axonal Charcot-Marie-Tooth and distal hereditary motor neuropathy. J. Neurol. Neurosurg. Psychiatry 2010, 81, 958–962. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Weeks, S.D.; Muranova, L.K.; Heirbaut, M.; Beelen, S.; Strelkov, S.V.; Gusev, N.B. Characterization of human small heat shock protein HSPB1 α-crystallin domain localized mutants associated with hereditary motor neuron diseases. Sci. Rep. 2018, 8, 688. [Google Scholar] [CrossRef] [PubMed]
- Irobi, J.; Almeida-Souza, L.; Asselbergh, B.; de Winter, V.; Goethals, S.; Dierick, I.; Krishnan, J.; Timmermans, J.P.; Robberecht, W.; de Jonghe, P.; et al. Mutant HSPB8 causes motor neuron-specific neurite degeneration. Hum. Mol. Genet. 2010, 19, 3254–3265. [Google Scholar] [CrossRef] [PubMed]
- Ackerley, S.; James, P.A.; Kalli, A.; French, S.; Davies, K.E.; Talbot, K. A mutation in the small heat-shock protein HSPB1 leading to distal hereditary motor neuronopathy disrupts neurofilament assembly and the axonal transport of specific cellular cargoes. Hum. Mol. Genet. 2006, 15, 347–354. [Google Scholar] [CrossRef]
- Zhai, J.; Lin, H.; Julien, J.P.; Schlaepfer, W.W. Disruption of neurofilament network with aggregation of light neurofilament protein: A common pathway leading to motor neuron degeneration due to Charcot-Marie-Tooth disease-linked mutations in NFL and HSPB. Hum. Mol. Genet. 2007, 16, 3103–3116. [Google Scholar] [CrossRef]
- Almeida-Souza, L.; Asselbergh, B.; d’Ydewalle, C.; Moonens, K.; Goethals, S.; de Winter, V.; Azmi, A.; Irobi, J.; Timmermans, J.P.; Gevaert, K.; et al. Small heat-shock protein HSPB1 mutants stabilize microtubules in Charcot-Marie-Tooth neuropathy. J. Neurosci. 2011, 31, 15320–15328. [Google Scholar] [CrossRef]
- Kalmar, B.; Innes, A.; Wanisch, K.; Kolaszynska, A.K.; Pandraud, A.; Kelly, G.; Abramov, A.Y.; Reilly, M.M.; Schiavo, G.; Greensmith, L. Mitochondrial deficits and abnormal mitochondrial retrograde axonal transport play a role in the pathogenesis of mutant Hsp27-induced charcot marie tooth disease. Hum. Mol. Genet. 2017, 26, 3313–3326. [Google Scholar] [CrossRef]
- Tanabe, H.; Higuchi, Y.; Yuan, J.H.; Hashiguchi, A.; Yoshimura, A.; Ishihara, S.; Nozuma, S.; Okamoto, Y.; Matsuura, E.; Ishiura, H.; et al. Clinical and genetic features of Charcot-Marie-Tooth disease 2F and hereditary motor neuropathy 2B in Japan. J. Peripher. Nerv. Syst. 2018, 23, 40–48. [Google Scholar] [CrossRef]
- Lee, J.; Jung, S.-C.; Joo, J.; Choi, Y.-R.; Moon, H.W.; Kwak, G.; Yeo, H.K.; Lee, J.-S.; Ahn, H.-J.; Jung, N.; et al. Overexpression of mutant HSP27 causes axonal neuropathy in mice. J. Biomed. Sci. 2015, 22, 43. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Renusch, S.R.; Naiman, N.E.; Gu, S.; Sneh, A.; Arnold, W.D.; Sahenk, Z.; Kolb, S.J. Mutant HSPB1 overexpression in neurons is sufficient to cause age-related motor neuronopathy in mice. Neurobiol. Dis. 2012, 47, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Muchowski, P.J.; Schaffar, G.; Sittler, A.; Wanker, E.E.; Hayer-Hartl, M.K.; Hartl, F.U. Hsp70 and Hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 2000, 97, 7841–7846. [Google Scholar] [CrossRef] [PubMed]
- Jana, N.R.; Tanaka, M.; Wang, G.H.; Nukina, N. Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: Their role in suppression of aggregation and cellular toxicity. Hum. Mol. Genet. 2000, 9, 2009–2018. [Google Scholar] [CrossRef] [PubMed]
- Howarth, J.L.; Kelly, S.; Keasey, M.P.; Glover, C.P.J.; Lee, Y.B.; Mitrophanous, K.; Chapple, J.P.; Gallo, J.M.; Cheetham, M.E.; Uney, J.B. Hsp40 molecules that target to the ubiquitin-proteasome system decrease inclusion formation in models of polyglutamine disease. Mol. Ther. 2007, 15, 1100–1105. [Google Scholar] [CrossRef] [PubMed]
- Frasquet, M.; Chumillas, M.J.; Vílchez, J.J.; Márquez-Infante, C.; Palau, F.; Vázquez-Costa, J.F.; Lupo, V.; Espinós, C.; Sevilla, T. Phenotype and natural history of inherited neuropathies caused by HSJ1 c.352+1G>A mutation. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1265–1268. [Google Scholar] [CrossRef]
- Greaves, J.; Chamberlain, L.H. Dual role of the cysteine-string domain in membrane binding and palmitoylation-dependent sorting of the molecular chaperone cysteine-string protein. Mol. Biol. Cell 2006, 17, 4748–4759. [Google Scholar] [CrossRef]
- Martin, J.J. Adult type of neuronal ceroid-lipofuscinosis. J. Inherit. Metab. Dis. 1993, 16, 237–240. [Google Scholar] [CrossRef]
- Haltia, M. The neuronal ceroid-lipofuscinoses. J. Neuropathol. Exp. Neurol. 2003, 62, 1–13. [Google Scholar] [CrossRef]
- Xu, F.; Proft, J.; Gibbs, S.; Winkfein, B.; Johnson, J.N.; Syed, N.; Braun, J.E.A. Quercetin targets cysteine string protein (CSPα) and impairs synaptic transmission. PLoS ONE 2010, 5, e11045. [Google Scholar] [CrossRef]
- Brocchieri, L.; Conway de Macario, E.; Macario, A.J.L. hsp70 genes in the human genome: Conservation and differentiation patterns predict a wide array of overlapping and specialized functions. BMC EVolume Biol. 2008, 8, 19. [Google Scholar] [CrossRef]
- Cheng, M.Y.; Hartl, F.U.; Martin, J.; Pollock, R.A.; Kalousek, F.; Neuper, W.; Hallberg, E.M.; Hallberg, R.L.; Horwich, A.L. Mitochondrial heat-shock protein hsp60 is essential for assembly of proteins imported into yeast mitochondria. Nature 1989, 337, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Bross, P.; Fernandez-Guerra, P. Disease-associated mutations in the HSPD1 gene encoding the large subunit of the mitochondrial HSP60/HSP10 chaperonin complex. Front. Mol. Biosci. 2016, 3, 49. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.H.; Nielsen, M.N.; Hansen, J.; Füchtbauer, A.; Füchtbauer, E.M.; West, M.; Corydon, T.J.; Gregersen, N.; Bross, P. Inactivation of the hereditary spastic paraplegia-associated Hspd1 gene encoding the Hsp60 chaperone results in early embryonic lethality in mice. Cell Stress Chaperones 2010, 15, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, B.; Davoine, C.S.; Dürr, A.; Paternotte, C.; Feki, I.; Weissenbach, J.; Hazan, J.; Brice, A. A new locus for autosomal dominant pure spastic paraplegia, on chromosome 2q24-q. Am. J. Hum. Genet. 2000, 66, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Bross, P.; Naundrup, S.; Hansen, J.; Nielsen, M.N.; Christensen, J.H.; Kruhøffer, M.; Palmfeldt, J.; Corydon, T.J.; Gregersen, N.; Ang, D.; et al. The Hsp60-(p.V98I) mutation associated with hereditary spastic paraplegia SPG13 compromises chaperonin function both in vitro and in vivo. J. Biol. Chem. 2008, 283, 15694–15700. [Google Scholar] [CrossRef] [PubMed]
- Parnas, A.; Nadler, M.; Nisemblat, S.; Horovitz, A.; Mandel, H.; Azem, A. The MitCHAP-60 disease is due to entropic destabilization of the human mitochondrial Hsp60 oligomer. J. Biol. Chem. 2009, 284, 28198–28203. [Google Scholar] [CrossRef]
- Vitale, A.M.; Conway de Macario, E.; Alessandro, R.; Cappello, F.; Macario, A.J.L.; Marino Gammazza, A. Missense Mutations of Human Hsp60: A Computational Analysis to Unveil Their Pathological Significance. Front. Genet. 2020, 11, 969. [Google Scholar] [CrossRef]
- Bross, P.; Magnoni, R.; Sigaard Bie, A. Molecular Chaperone Disorders: Defective Hsp60 in Neurodegeneration. Curr. Top. Med. Chem. 2013, 12, 2491–2503. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Eguchi, T.; Kawahara, K.; Hasegawa, N.; Nakamura, K.; Funakoshi-Tago, M.; Tanoue, A.; Tamura, H.; Yamauchi, J. Hypomyelinating leukodystrophy-associated missense mutation in HSPD1 blunts mitochondrial dynamics. Biochem. Biophys. Res. Commun. 2015, 462, 275–281. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Megumi, F.T.; Hasegawa, N.; Eguchi, T.; Tanoue, A.; Tamura, H.; Yamauchi, J. Data supporting mitochondrial morphological changes by SPG13-associated HSPD1 mutants. Data Br. 2016, 6, 482–488. [Google Scholar] [CrossRef]
- Mukherjee, K.; Conway de Macario, E.; Macario, A.J.L.; Brocchieri, L. Chaperonin genes on the rise: New divergent classes and intense duplication in human and other vertebrate genomes. BMC EVolume Biol. 2010, 10, 64. [Google Scholar] [CrossRef] [PubMed]
- Min, W.; Angileri, F.; Luo, H.; Lauria, A.; Shanmugasundaram, M.; Almerico, A.M.; Cappello, F.; Conway de Macario, E.; Lednev, I.K.; Macario, A.J.L.; et al. A human CCT5 gene mutation causing distal neuropathy impairs hexadecamer assembly in an archaeal model. Sci. Rep. 2014, 4, 1–9. [Google Scholar] [CrossRef]
- Sergeeva, O.A.; Tran, M.T.; Haase-Pettingell, C.; King, J.A. Biochemical characterization of mutants in chaperonin proteins CCT4 and CCT5 associated with hereditary sensory neuropathy. J. Biol. Chem. 2014, 289, 27470–27480. [Google Scholar] [CrossRef] [PubMed]
- Spigolon, D.; Gallagher, D.T.; Velazquez-Campoy, A.; Bulone, D.; Narang, J.; San Biagio, P.L.; Cappello, F.; Macario, A.J.L.; Conway de Macario, E.; Robb, F.T. Quantitative analysis of the impact of a human pathogenic mutation on the CCT5 chaperonin subunit using a proxy archaeal ortholog. Biochem. Biophys. Rep. 2017, 12, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Nachury, M.V.; Loktev, A.V.; Zhang, Q.; Westlake, C.J.; Peränen, J.; Merdes, A.; Slusarski, D.C.; Scheller, R.H.; Bazan, J.F.; Sheffield, V.C.; et al. A Core Complex of BBS Proteins Cooperates with the GTPase Rab8 to Promote Ciliary Membrane Biogenesis. Cell 2007, 129, 1201–1213. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [CrossRef] [PubMed]
- Priya, S.; Nampoothiri, S.; Sen, P.; Sripriya, S. Bardet-Biedl syndrome: Genetics, molecular pathophysiology, and disease management. Indian J. Ophthalmol. 2016, 64, 620–627. [Google Scholar] [CrossRef] [PubMed]
- M’Hamdi, O.; Ouertani, I.; Chaabouni-Bouhamed, H. Update on the genetics of bardet-biedl syndrome. Mol. Syndromol. 2014, 5, 51–56. [Google Scholar] [CrossRef]
- Kim, J.C.; Ou, Y.Y.; Badano, J.L.; Esmail, M.A.; Leitch, C.C.; Fiedrich, E.; Beales, P.L.; Archibald, J.M.; Katsanis, N.; Rattner, J.B.; et al. MKKS/BBS6, a divergent chaperonin-like protein linked to the obesity disorder Bardet-Biedl syndrome, is a novel centrosomal component required for cytokinesis. J. Cell Sci. 2005, 118, 1007–1020. [Google Scholar] [CrossRef]
- Muller, J.; Stoetzel, C.; Vincent, M.C.; Leitch, C.C.; Laurier, V.; Danse, J.M.; Hellé, S.; Marion, V.; Bennouna-Greene, V.; Vicaire, S.; et al. Identification of 28 novel mutations in the Bardet-Biedl syndrome genes: The burden of private mutations in an extensively heterogeneous disease. Hum. Genet. 2010, 127, 583–593. [Google Scholar] [CrossRef]
- Billingsley, G.; Bin, J.; Fieggen, K.J.; Duncan, J.L.; Gerth, C.; Ogata, K.; Wodak, S.S.; Traboulsi, E.I.; Fishman, G.A.; Paterson, A.; et al. Mutations in chaperonin-like BBS genes are a major contributor to disease development in a multiethnic Bardet-Biedl syndrome patient population. J. Med. Genet. 2010, 47, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Baye, L.M.; Schulz, N.P.; Beck, J.S.; Zhang, Q.; Slusarski, D.C.; Sheffield, V.C. BBS6, BBS10, and BBS12 form a complex with CCT/TRiC family chaperonins and mediate BBSome assembly. Proc. Natl. Acad. Sci. USA 2010, 107, 1488–1493. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.E.; Swiderski, R.E.; Rahmouni, K.; Nishimura, D.Y.; Mullins, R.F.; Agassandian, K.; Philp, A.R.; Searby, C.C.; Andrews, M.P.; Thompson, S.; et al. A knockin mouse model of the Bardet-Biedl syndrome 1 M390R mutation has cilia defects, ventriculomegaly, retinopathy, and obesity. Proc. Natl. Acad. Sci. USA 2007, 104, 19422–19427. [Google Scholar] [CrossRef]
- Swiderski, R.E.; Agassandian, K.; Ross, J.L.; Bugge, K.; Cassell, M.D.; Yeaman, C. Structural defects in cilia of the choroid plexus, subfornical organ and ventricular ependyma are associated with ventriculomegaly. Fluids Barriers CNS 2012, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Badano, J.L.; Kim, J.C.; Hoskins, B.E.; Lewis, R.A.; Ansley, S.J.; Cutler, D.J.; Castellan, C.; Beales, P.L.; Leroux, M.R.; Katsanis, N. Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet-Beidl patients with two mutations at a second BBS locus. Hum. Mol. Genet. 2003, 12, 1651–1659. [Google Scholar] [CrossRef]
- Stone, D.L.; Slavotinek, A.; Bouffard, G.G.; Banerjee-Basu, S.; Baxevanis, A.D.; Barr, M.; Biesecker, L.G. Mutation of a gene encoding a putative chaperonin causes McKusick- Kaufman syndrome. Nat. Genet. 2000, 25, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, E.; Durand, M.; Stoetzel, C.; Doray, B.; Viville, B.; Hellé, S.; Danse, J.M.; Hamel, C.; Bitoun, P.; Goldenberg, A.; et al. Molecular diagnosis reveals genetic heterogeneity for the overlapping MKKS and BBS phenotypes. Eur. J. Med. Genet. 2011, 54, 157–160. [Google Scholar] [CrossRef]
- Nitika; Porter, C.M.; Truman, A.W.; Truttmann, M.C. Post-translational modifications of Hsp70 family proteins: Expanding the chaperone code. J. Biol. Chem. 2020, 295, 10689–10708. [Google Scholar] [CrossRef]
- Engel, P.; Goepfert, A.; Stanger, F.V.; Harms, A.; Schmidt, A.; Schirmer, T.; Dehio, C. Adenylylation control by intra-or intermolecular active-site obstruction in Fic proteins. Nature 2012, 482, 107–110. [Google Scholar] [CrossRef]
- Truttmann, M.C.; Zheng, X.; Hanke, L.; Damon, J.R.; Grootveld, M.; Krakowiak, J.; Pincus, D.; Ploegh, H.L. Unrestrained AMPylation targets cytosolic chaperones and activates the heat shock response. Proc. Natl. Acad. Sci. USA 2017, 114, E152–E160. [Google Scholar] [CrossRef]
- Kielkowski, P.; Buchsbaum, I.Y.; Kirsch, V.C.; Bach, N.C.; Drukker, M.; Cappello, S.; Sieber, S.A. FICD activity and AMPylation remodelling modulate human neurogenesis. Nat. Commun. 2020, 11, 517. [Google Scholar] [CrossRef] [PubMed]
- Truttmann, M.C.; Pincus, D.; Ploegh, H.L. Chaperone AMPylation modulates aggregation and toxicity of neurodegenerative disease-associated polypeptides. Proc. Natl. Acad. Sci. USA 2018, 115, E5008–E5017. [Google Scholar] [CrossRef] [PubMed]
- Backe, S.J.; Sager, R.A.; Woodford, M.R.; Makedon, A.M.; Mollapour, M. Post-translational modifications of Hsp90 and translating the chaperone code. J. Biol. Chem. 2020, 295, 11099–11117. [Google Scholar] [CrossRef] [PubMed]
- Franco, M.C.; Ye, Y.; Refakis, C.A.; Feldman, J.L.; Stokes, A.L.; Basso, M.; De Mera, R.M.M.F.; Sparrow, N.A.; Calingasan, N.Y.; Kiaei, M.; et al. Nitration of Hsp90 induces cell death. Proc. Natl. Acad. Sci. USA 2013, 110, E1102–E1111. [Google Scholar] [CrossRef] [PubMed]
- Koike, M.; Fukushi, J.; Ichinohe, Y.; Higashimae, N.; Fujishiro, M.; Sasaki, C.; Yamaguchi, M.; Uchihara, T.; Yagishita, S.; Ohizumi, H.; et al. Valosin-containing protein (VCP) in novel feedback machinery between abnormal protein accumulation and transcriptional suppression. J. Biol. Chem. 2010, 285, 21736–21749. [Google Scholar] [CrossRef]
- Franco, M.C.; Ricart, K.C.; Gonzalez, A.S.; Dennys, C.N.; Nelson, P.A.; Janes, M.S.; Mehl, R.A.; Landar, A.; Estévez, A.G. Nitration of Hsp90 on Tyrosine 33 regulates mitochondrial metabolism. J. Biol. Chem. 2015, 290, 19055–19066. [Google Scholar] [CrossRef]
- Verba, K.A.; Agard, D.A. How Hsp90 and Cdc37 Lubricate Kinase Molecular Switches. Trends Biochem. Sci. 2017, 42, 799–811. [Google Scholar] [CrossRef]
- Gracia, L.; Lora, G.; Blair, L.J.; Jinwal, U.K. Therapeutic Potential of the Hsp90/Cdc37 Interaction in Neurodegenerative Diseases. Front. Neurosci. 2019, 13, 1263. [Google Scholar] [CrossRef]
- Miyata, Y.; Nishida, E. CK2 Controls Multiple Protein Kinases by Phosphorylating a Kinase-Targeting Molecular Chaperone, Cdc37. Mol. Cell. Biol. 2004, 24, 4065–4074. [Google Scholar] [CrossRef]
- Perez, D.I.; Gil, C.; Martinez, A. Protein kinases CK1 and CK2 as new targets for neurodegenerative diseases. Med. Res. Rev. 2011, 31, 924–954. [Google Scholar] [CrossRef]
- Zhang, S.H.; Liu, J.; Kobayashi, R.; Tonks, N.K. Identification of the cell cycle regulator VCP (p97/CDC48) as a substrate of the band 4.1-related protein-tyrosine phosphatase PTPH. J. Biol. Chem. 1999, 274, 17806–17812. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.S.; Fuentealba, R.A.; Miller, S.E.; Jackson, E.; Piwnica-Worms, D.; Baloh, R.H.; Weihl, C.C. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol. 2009, 187, 875–888. [Google Scholar] [CrossRef] [PubMed]
- Torrecilla, I.; Oehler, J.; Ramadan, K. The role of ubiquitin-dependent segregase p97 (VCP or Cdc48) in chromatin dynamics after DNA double strand breaks. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, P.; Coulombe, B. Regulation of molecular chaperones through post-translational modifications: Decrypting the chaperone code. Biochim. Biophys. Acta Gene Regul. Mech. 2013, 1829, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Astakhova, L.N.; Zatsepina, O.G.; Funikov, S.Y.; Zelentsova, E.S.; Schostak, N.G.; Orishchenko, K.E.; Evgen’Ev, M.B.; Garbuz, D.G. Activity of heat shock genes’ promoters in thermally contrasting animal species. PLoS ONE 2015, 10, 115536. [Google Scholar] [CrossRef]
- Murphy, M.E. The HSP70 family and cancer. Carcinogenesis 2013, 34, 1181–1188. [Google Scholar] [CrossRef]
- Sun, Y.; Zheng, J.; Xu, Y.; Zhang, X. Paraquat-induced inflammatory response of microglia through HSP60/TLR4 signaling. Hum. Exp. Toxicol. 2018, 37, 1161–1168. [Google Scholar] [CrossRef]
- Nguyen, P.; Kdeiss, B.; Ord, S.; Hess, K.; Oliverio, R.; Nikolaidis, N. Evolution and natural variation of HSPA1A, the major stress inducible gene, in humans. FASEB J. 2018, 31, 604–618. [Google Scholar]
- Hess, K.; Oliverio, R.; Nguyen, P.; Le, D.; Ellis, J.; Kdeiss, B.; Ord, S.; Chalkia, D.; Nikolaidis, N. Concurrent action of purifying selection and gene conversion results in extreme conservation of the major stress-inducible Hsp70 genes in mammals. Sci. Rep. 2018, 8, 5082. [Google Scholar] [CrossRef]
- Singh, R.; Kølvraa, S.; Bross, P.; Jensen, U.B.; Gregersen, N.; Tan, Q.; Knudsen, C.; Rattan, S.I.S. Reduced heat shock response in human mononuclear cells during aging and its association with polymorphisms in HSP70 genes. Cell Stress Chaperones 2006, 11, 208–215. [Google Scholar] [CrossRef]
- He, M.; Guo, H.; Yang, X.; Zhang, X.; Zhou, L.; Cheng, L.; Zeng, H.; Hu, F.B.; Tanguay, R.M.; Wu, T. Functional SNPs in HSPA1A Gene Predict Risk of Coronary Heart Disease. PLoS ONE 2009, 4, e4851. [Google Scholar] [CrossRef]
- Konings, A.; Van Laer, L.; Michel, S.; Pawelczyk, M.; Carlsson, P.I.; Bondeson, M.L.; Rajkowska, E.; Dudarewicz, A.; Vandevelde, A.; Fransen, E.; et al. Variations in HSP70 genes associated with noise-induced hearing loss in two independent populations. Eur. J. Hum. Genet. 2009, 17, 329–335. [Google Scholar] [CrossRef]
- Maugeri, N.; Radhakrishnan, J.; Knight, J.C. Genetic determinants of HSP70 gene expression following heat shock. Hum. Mol. Genet. 2010, 19, 4939–4947. [Google Scholar] [CrossRef] [PubMed]
- Oliverio, R.; Nguyen, P.; Kdeiss, B.; Ord, S.; Daniels, A.J.; Nikolaidis, N. Functional characterization of natural variants found on the major stress inducible 70-kDa heat shock gene, HSPA1A, in humans. Biochem. Biophys. Res. Commun. 2018, 506, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.G.; Wisén, S.; Gestwicki, J.E. Heat shock proteins 70 and 90 inhibit early stages of amyloid β-(1-42) aggregation in vitro. J. Biol. Chem. 2006, 281, 33182–33191. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Sun, W.; Taldone, T.; Rodina, A.; Chiosis, G. Heat shock protein 90 in neurodegenerative diseases. Mol. Neurodegener. 2010, 5, 24. [Google Scholar] [CrossRef]
- Chen, B.; Piel, W.H.; Gui, L.; Bruford, E.; Monteiro, A. The HSP90 family of genes in the human genome: Insights into their divergence and evolution. Genomics 2005, 86, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Barazi, H.O.; Zhou, L.; Templeton, N.S.; Krutzsch, H.C.; Roberts, D.D. Identification of heat shock protein 60 as a molecular mediator of alpha 3 beta 1 integrin activation. Cancer Res. 2002, 62, 1541–1548. [Google Scholar]
- Gorska, M.; Marino Gammazza, A.; Zmijewski, M.A.; Campanella, C.; Cappello, F.; Wasiewicz, T.; Kuban-Jankowska, A.; Daca, A.; Sielicka, A.; Popowska, U.; et al. Geldanamycin-Induced Osteosarcoma Cell Death Is Associated with Hyperacetylation and Loss of Mitochondrial Pool of Heat Shock Protein 60 (Hsp60). PLoS ONE 2013, 8, e71135. [Google Scholar] [CrossRef]
- Lin, C.Y.; Hu, C.T.; Cheng, C.C.; Lee, M.C.; Pan, S.M.; Lin, T.Y.; Wu, W.S. Oxidation of heat shock protein 60 and protein disulfide isomerase activates ERK and migration of human hepatocellular carcinoma HepG. Oncotarget 2016, 7, 11067–11082. [Google Scholar] [CrossRef] [PubMed]
- Marino Gammazza, A.; Campanella, C.; Barone, R.; Caruso Bavisotto, C.; Gorska, M.; Wozniak, M.; Carini, F.; Cappello, F.; D’Anneo, A.; Lauricella, M.; et al. Doxorubicin anti-tumor mechanisms include Hsp60 post-translational modifications leading to the Hsp60/p53 complex dissociation and instauration of replicative senescence. Cancer Lett. 2017, 385, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Kim, E.M.; Lee, J.; Yang, W.H.; Park, T.Y.; Kim, Y.M.; Cho, J.W. Heat shock protein 60 modified with O-linked N-acetylglucosamine is involved in pancreatic β-cell death under hyperglycemic conditions. FEBS Lett. 2006, 580, 2311–2316. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.; Mukherjee, A.; Patra, U.; Bhowmick, R.; Basak, T.; Sengupta, S.; Chawla-Sarkar, M. Tyrosine phosphorylation modulates mitochondrial chaperonin Hsp60 and delays rotavirus NSP4-mediated apoptotic signaling in host cells. Cell. Microbiol. 2017, 19. [Google Scholar] [CrossRef] [PubMed]
- Hayoun, D.; Kapp, T.; Edri-Brami, M.; Ventura, T.; Cohen, M.; Avidan, A.; Lichtenstein, R.G. HSP60 is transported through the secretory pathway of 3-MCA-induced fibrosarcoma tumour cells and undergoes N-glycosylation. FEBS J. 2012, 279, 2083–2095. [Google Scholar] [CrossRef]
- Baron, B. Role of the Post-Translational Modifications of HSP60 in Disease; Springer: Cham, Switzerland, 2019; Volume 18, pp. 69–94. [Google Scholar]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Macario, A.J.L.; Conway de Macario, E. Chaperonopathies and chaperonotherapy. FEBS Lett. 2007, 581, 3681–3688. [Google Scholar] [CrossRef]
- Macario, A.J.L.; Conway de Macario, E. Molecular mechanisms in chaperonopathies: Clues to understanding the histopathological abnormalities and developing novel therapies. J. Pathol. 2020, 250, 9–18. [Google Scholar] [CrossRef]
- Cappello, F.; Marino Gammazza, A.; Palumbo Piccionello, A.; Campanella, C.; Pace, A.; Conway de Macario, E.; Macario, A.J.L. Hsp60 chaperonopathies and chaperonotherapy: Targets and agents. Expert Opin. Ther. Targets 2014, 18, 185–208. [Google Scholar] [CrossRef]
- Meng, Q.; Li, B.X.; Xiao, X. Toward developing chemical modulators of Hsp60 as potential therapeutics. Front. Mol. Biosci. 2018, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Macario, A.J.L.; Conway de Macario, E. Hidden chaperonopathies: Alerting physicians and pathologists on the possibility that uncharacteristic, baffling clinical features in otherwise known diseases may be due to failure of the chaperoning system. Life Saf. Secur. 2020. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
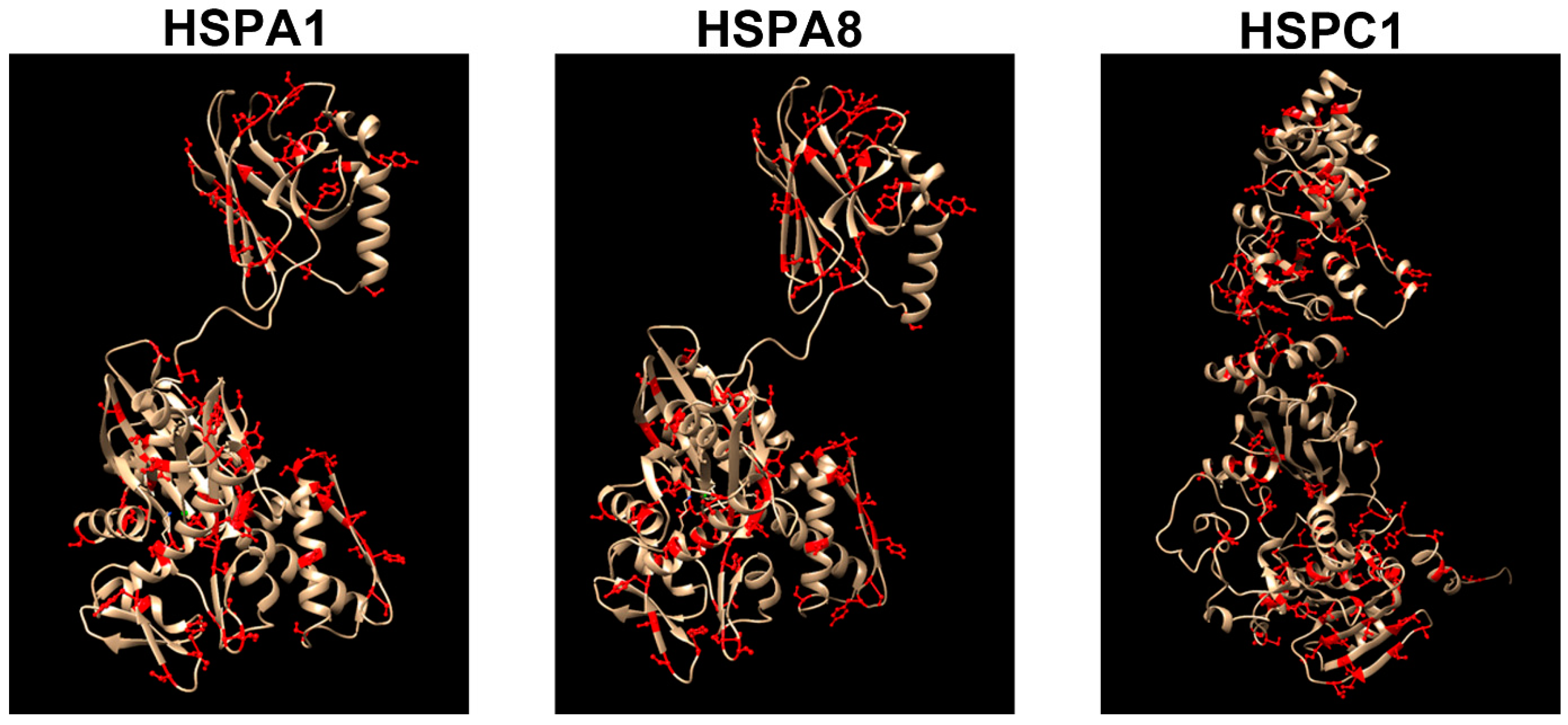{kind=link}
| Mol. Chap. | Mutation | Disease | M. I. | Ref. |
|---|---|---|---|---|
| HSPA9 | Homozygous c.376C-T transition in exon 4 resulting in p.Arg126Trp substitution | EVEN-plus syndrome | AR | [26] |
| Compound heterozygous for a c.383A-G transition in exon 4 resulting in p.Tyr128Cys substitution, and a 2-bp deletion in exon 8 (c.882_883delAG) causing a frameshift and resulting in a premature termination codon at amino acid 296 | EVEN-plus syndrome | AR | [26] | |
| HSPB8 | Heterozygous c.421A-G transition in exon 1 resulting in p.Lys141Glu substitution | dHMN2A | ADo | [27,28] |
| Heterozygous c.423G-C transversion in exon 1 resulting in p.Lys141Asn substitution | dHMN2A | ADo | [28,29] | |
| c.423G-T transversion in exon 1 resulting in p.Lys141Asn substitution | CMT2L | ADo | [30,31] | |
| HSPB1 | c.100G-A transition in exon 1 resulting in p.Gly34Arg substitution | dHMN2B | Sporadic | [32] |
| c.116C-T transition in exon 1 resulting in p.Pro39Leu substitution | dHMN2B/CMT2F | ADo | [32,33] | |
| c.121G-A transition in exon 1 resulting in p.Glu41Lys substitution | dHMN2B | ADo | [32] | |
| c.250G-C transversion in exon 1 resulting in p.Gly84Arg substitution | dHMN2B/CMT2F | ADo | [33,34,35] | |
| Homozygous c.295C-A transversion in exon 1 resulting in p.Leu99Met substitution | dHMN2B/CMT2F | AR | [33] | |
| c.379C-T transition in exon 1 resulting in p.Arg127Trp substitution | dHMN2B/CMT2F | ADo | [36,37] | |
| c.404C-G transversion in exon 1 resulting in p.Ser135Cys substitution | dHMN2B/CMT2F | ADo | [38,39] | |
| c.404C-T transition in exon 1 resulting in p.Ser135Phe substitution | dHMN2B/CMT2F | ADo | [33,36,40] | |
| c.406C-T transition in exon 2 resulting in p.Arg136Trp substitution | CMT2F | ADo | [36] | |
| c.407G-T transversion in exon 2 resulting in p.Arg136Leu substitution | dHMN2B/CMT2F | Sporadic | [32] | |
| c.418C-G transversion in exon 2 resulting in p.Arg140Gly substitution | dHMN2B/CMT2F | ADo or sporadic | [33] | |
| c.421A-C transversion in exon 2 resulting in p.Lys141Gln substitution | dHMN2B | ADo | [41] | |
| c.452C-T transition in exon 2 resulting in p.Thr151Ile substitution | dHMN2B | ADo | [36] | |
| c.490A-G transition in exon resulting in exon 3 resulting in p.Thr164Ala substitution | CMT2F | ADo | [42] | |
| c.539C-T transition in exon 3 resulting in p.Thr180Ile substitution | dHMN2B/CMT2F | ADo | [32,43] | |
| c.544C-T transition in exon 3 resulting in p.Pro182Ser substitution | dHMN2B | ADo | [44] | |
| c.545C-T transition in exon 3 resulting in p.Pro182Leu substitution | dHMN2B | ADo | [36] | |
| c.562C-T transition in exon 3 resulting in p.Arg188Trp substitution | CMT2F | Sporadic | [32] | |
| HSPE1 | Heterozygous c.217C-T transition in exon 2 resulting in p.Leu73Phe substitution | Undefined neurologic disorder | Sporadic | [45] |
| DNAJB2 | Homozygous c.14A-G transition in exon 1 resulting in p.Tyr5Cys substitution | CMT2F/DSMA5 | AR | [46] |
| Homozygous G-A transition in intron 4 (c.229+1G-A) | dHMN/DSMA5 | AR recessive | [46] | |
| Homozygous G-A transition in the donor splice site of exon 5 (c.352+1G-A) | DSMA5 | AR | [47] | |
| DNAJC5 | Heterozygous c.344T-G transversion in exon 3 resulting in p.Leu115Arg substitution | CLN4B | ADo | [48,49,50,51] |
| Heterozygous 3-bp deletion in exon 3 (c.346_348del) resulting in p.Leu116del | CLN4B | ADo | [48,49,50,51] | |
| HSPD1 | Homozygous c.86A-G transition in exon 2 resulting in p.Asp29Gly substitution | HLD4 or MitCHAP-60 disease | AR | [52,53] |
| Heterozygous c.292G-A transition in exon 3 resulting in p.Val98Ile substitution | SPG13 | ADo | [54] | |
| Heterozygous c.1381C-G transversion in exon 10 resulting in p.Gln461Glu substitution | SPG13 | ADo | [55] | |
| CCT5 | Homozygous c.440A-G transition in exon 4 resulting in a His147Arg substitution | Hereditary sensory neuropathy with spastic paraplegia | AR | [56,57] |
| Homozigous c.670C>G transversion in exon 5 resulting in Leu224Val substitution | Demyelinating neuropathy with severe motor disability. | AR | [58] | |
| BBS6 | c.110A-G transition in exon 1 resulting in p.Tyr37Cys substitution | BBS | AR | [59,60] |
| c.155G-A in exon 1 transition resulting in p.Gly52Asp substitution | BBS | AR | [61] | |
| c.169A-G transition in exon 1 resulting in Thr57Ala substitution | BBS | AR | [59] | |
| Homozygous 1-bp deletion (c.281del) in exon 2 resulting in a frameshift after amino acid Phe94 (p.Phe94fs), terminating the protein at amino acid 103 | BBS | AR | [59,61] | |
| Homozygosity for a complex 2-bp deletion (c.429_430del and c.433_434del) in exon 2 resulting in a frameshift and a premature termination of the protein at amino acid 157 | BBS | AR | [59,61] | |
| Nonsense mutation leading to premature termination (c.442C-T transition in exon 2 resulting in p.Gln148Ter) | BBS | AR | [60] | |
| c.792T-A transversion in exon 3 resulting in a premature termination (p.Tyr264Ter) | BBS | AR | [61] | |
| c.830C-T transition in exon 3 resulting in p.Leu277Pro substitution | BBS | AR | [59] | |
| c.1496G-C transversion in exon 3 resulting in p.Cys499Ser substitution | BBS | AR | [60] | |
| BBS10 | c.32T-G transversion in exon 1 resulting in p.Val11Gly substitution | BBS | AR | [62] |
| c.101G-C transversion in exon 1 resulting in p.Arg34Pro substitution | BBS | AR | [63] | |
| 1-bp insertion in exon 2 (c.271dupT) leading to premature termination (p.Cys91fsTer95) | BBS | AR | [63,64] | |
| c.273C-G transversion in exon 2 resulting in p.Cys91Trp substitution | BBS | AR | [64] | |
| 4-bp deletion in exon 2 (c.909_912del) resulting in premature termination (p.S303fsTer305) | BBS | AR | [63] | |
| c.931T-G transversion in exon 2 resulting in p.Ser311Ala substitution | BBS | AR | [62,63] | |
| 2-bp deletion in exon 2 (c.1044_1045del) resulting in a frameshift and premature termination (p.Pro350fs) | BBS | AR | [64] | |
| BBS12 | Homozygous 3-bp deletion in exon 2 (c.337_339del) resulting in p.Val113del | BBS | AR | [65] |
| Homozygous c.865G-C transversion in exon 3 resulting in p.Ala289Pro substitution | BBS | AR | [65] | |
| c.1063C-T transition in exon 3 resulting in a nonsense mutation (p.Arg355Ter) | BBS | AR | [65] | |
| 2-bp deletion in exon 3 (c.1115_1116del) resulting in frameshift and premature termination of the protein (p.Phe372fsTer373) | BBS | AR | [65] | |
| 2-bp deletion in exon 3 (c.1483_1484del) resulting in frameshift and premature termination (p.Glu495fsTer498) | BBS | AR | [65] |
| Model | Status | Molecular Chaperone | Human Ortholog | PTM | Effect | Ref. |
|---|---|---|---|---|---|---|
| Drosophila | Wt | Bip | Grp78 (HSPA5) | AMPylation | Blindness | [123] |
| Saccha-romyces cerevisiae | Wt | Hsp70 | HSPA | AMPylation | Decreased cell growth; protein misfolding; toxic protein aggregation | [125] |
| Caeno-rhabditis elegans | AD; PD | HSP-1; HSP-3; HSP-4 | HSPA | AMPylation | Large cytoprotective protein aggregates | [127] |
| PC12 cells | Wt | Hsp90 | HSPC | Nitration | ALS | [128,129] |
| PC12 cells | Wt | VCP | p97 | Phosphorylation and acetylation | Neurite retraction and shrinkage | [130] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scalia, F.; Vitale, A.M.; Santonocito, R.; Conway de Macario, E.; Macario, A.J.L.; Cappello, F. The Neurochaperonopathies: Anomalies of the Chaperone System with Pathogenic Effects in Neurodegenerative and Neuromuscular Disorders. Appl. Sci. 2021, 11, 898. https://doi.org/10.3390/app11030898
Scalia F, Vitale AM, Santonocito R, Conway de Macario E, Macario AJL, Cappello F. The Neurochaperonopathies: Anomalies of the Chaperone System with Pathogenic Effects in Neurodegenerative and Neuromuscular Disorders. Applied Sciences. 2021; 11(3):898. https://doi.org/10.3390/app11030898
Chicago/Turabian StyleScalia, Federica, Alessandra Maria Vitale, Radha Santonocito, Everly Conway de Macario, Alberto J. L. Macario, and Francesco Cappello. 2021. "The Neurochaperonopathies: Anomalies of the Chaperone System with Pathogenic Effects in Neurodegenerative and Neuromuscular Disorders" Applied Sciences 11, no. 3: 898. https://doi.org/10.3390/app11030898
APA StyleScalia, F., Vitale, A. M., Santonocito, R., Conway de Macario, E., Macario, A. J. L., & Cappello, F. (2021). The Neurochaperonopathies: Anomalies of the Chaperone System with Pathogenic Effects in Neurodegenerative and Neuromuscular Disorders. Applied Sciences, 11(3), 898. https://doi.org/10.3390/app11030898