An Unsupervised Prediction Model for Salmonella Detection with Hyperspectral Microscopy: A Multi-Year Validation


Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation and Collection
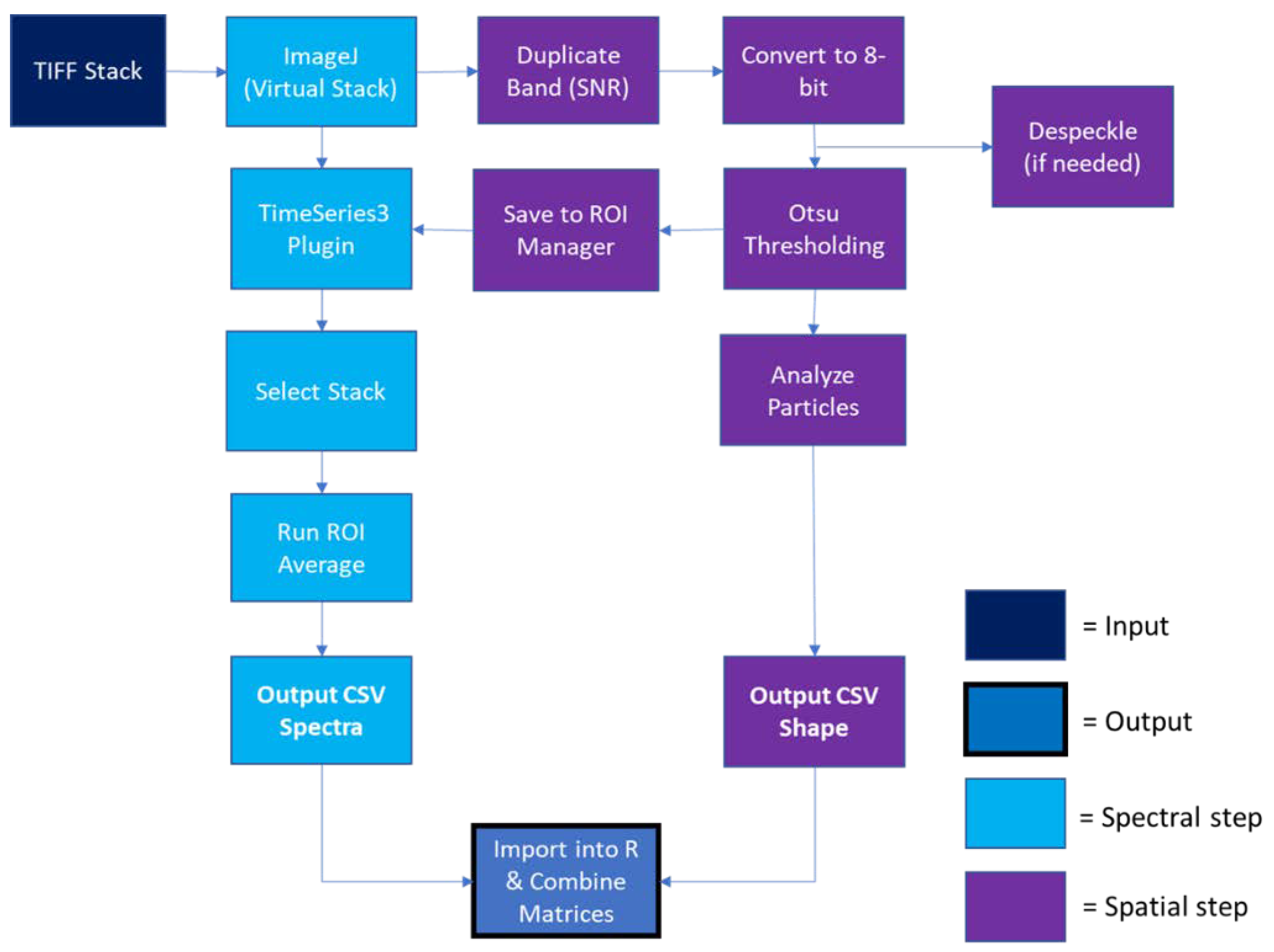
2.2. HMI Processing
2.3. Spectral Pre-Processing
2.4. SIMCA Classification Model
2.5. SIMCA Validation
3. Results and Discussion
3.1. Standard Normal Variant and Spectra
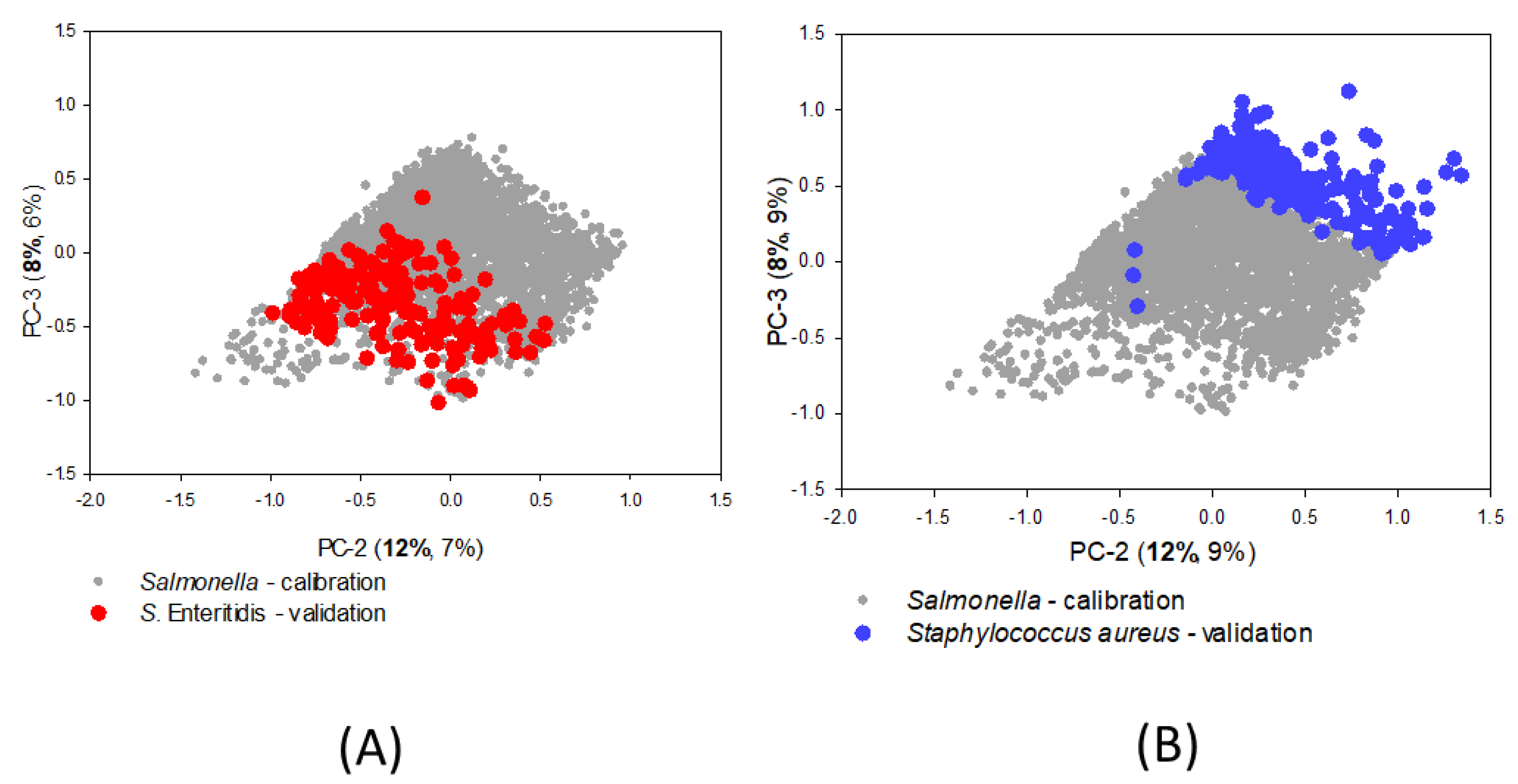
3.2. SIMCA Calibration Model
3.3. SIMCA Validation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Salmonella (Non-Typhoidal). 20 February 2018. Available online: https://www.who.int/news-room/fact-sheets/detail/salmonella-(non-typhoidal) (accessed on 13 August 2019).
- Wang, W.; Peng, Y.; Huang, H.; Wu, J. Application of hyper-spectral imaging technique for the detection of total viable bacteria count in pork. Sens. Lett. 2011, 9, 1024–1030. [Google Scholar] [CrossRef]
- Feng, Y.Z.; Sun, D.W. Determination of total viable count (TVC) in chicken breast fillets by near-infrared hyperspectral imaging and spectroscopic transformations. Talanta 2013, 105, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Sun, D.W. Potential of time series-hyperspectral imaging (TS-HIS) for non-invasive determination of microbial spoilage of salmon flesh. Talanta 2013, 15, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.C.; Windham, W.R.; Ladley, S.R.; Heitschmidt, J.W.; Lawrence, K.C.; Park, B.; Narang, N.; Cray, W.C. Hyperspectral imaging for differentiating colonies of non-O157 Shiga-toxin producing Escherichia coli (STEC) serogroups on spread plates of pure cultures. J. Near Infrared Spectrosc. 2013, 21, 81–95. [Google Scholar] [CrossRef]
- Tang, Y.; Kim, H.; Singh, A.K.; Aroonnual, A.; Bae, E.; Rajwa, B.; Fratamico, P.M.; Bhunia, A. Light scattering sensor for direct identification of colonies of Escherichia coli serogroups O26, O45, O103, O111, O121, O145, and O157. PLoS ONE 2014, 9, e105272. [Google Scholar] [CrossRef] [PubMed]
- Foca, G.; Ferrari, C.; Ulrici, A.; Sciutto, G.; Prati, S.; Morandi, S.; Brasca, M.; Lavermicocca, P.; Lanteri, S.; Oliveri, P. The potential of spectral and hyperspectral-imaging techniques for bacteria detection in food: A case study on lactic acid bacteria. Talanta 2016, 153, 111–119. [Google Scholar] [CrossRef]
- Anderson, J.; Reynolds, C.; Ringelberg, D.; Edwards, J.; Foley, K. Differentiation of live-viable versus dead bacterial endospores by calibrated hyperspectral reflectance microscopy. J. Microsc. 2008, 232, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Eady, M.; Park, B.; Choi, S. Rapid and early detection of Salmonella serotypes with hyperspectral microscopy and multivariate data analysis. J. Food Prot. 2015, 78, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Esbensen, K.; Swarbrick, B. Multivariate Data Analysis, 6th ed.; Camo: Oslo, Norway, 2018. [Google Scholar]
- Karunathilaka, E.R.; Yakes, B.J.; He, K.; Chung, J.K.; Mossoba, M. Non-targeted NIR spectroscopy and SIMCA classification for commercial milk powder authentication: A study using eleven potential adulterants. Heliyon 2018, 4, e00806. [Google Scholar] [CrossRef] [PubMed]
- Zimbro, M.; Power, D. DIFCO and BBL Manual for Microbiological Culture Media, 2nd ed.; Dickinson and Co.: Sparks, MD, USA, 2009. [Google Scholar]
- Park, B.; Yoon, S.C.; Lee, S.; Sundahram, J.; Windham, W.R.; Hinton, A., Jr.; Lawrence, K.C. Acousto-optical tunable filter hyperspectral microscope imaging method for characterizing spectra from foodborne pathogens. Trans. ASABE 2012, 55, 1997–2006. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, S.; Saalfeld, C.; Schmid, S.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Meth. 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Haidekker, M. Advanced Biomedical Image Analysis; John Riley and Sons Inc.: Hoboken, NJ, USA, 2011. [Google Scholar]
- Balaji, J. Time Series Analyzer Version 3.0. 28 May 2014. Available online: https://imagej.nih.gov/ij/plugins/time-series.html (accessed on 20 October 2018).
- Burger, J.; Geladi, P. Spectral pretreatments of hyperspectral near infrared images: Analysis of diffuse reflectance spectroscopy. J. Near Infrared Spectrosc. 2007, 15, 29–37. [Google Scholar] [CrossRef]
- Eady, M.; Park, B. Unsupervised classification of individual foodborne bacteria from a mixture of bacterial cultures with a hyperspectral microscope image. J. Spectr. Imaging 2018, 7, a6. [Google Scholar] [CrossRef]
- Mertens, B.; Thompson, M. Principal component outlier detection and SIMCA: A synthesis. Analyst 1994, 119, 2777–2784. [Google Scholar] [CrossRef]
- Vanden Branden, K.; Hubert, M. Robust classification in high dimension based on the SIMCA method. Chemom. Intell. Lab. Syst. 2005, 79, 10–21. [Google Scholar] [CrossRef]
- Wold, S.; Sjostrom, M. SIMCA: A method for analyzing chemical data in terms of similarity and analogy. Chemom. Theory Appl. 1977, 52, 243–282. [Google Scholar]
- Candolfi, A.; De Maesschalck, R.; Massart, D.L.; Hailey, P.A.; Harrington, A.C.E. Identification of pharmaceutical excipients using NIR spectroscopy and SIMCA. J. Pharm. Biomed. Anal. 1999, 19, 923–935. [Google Scholar] [CrossRef]
- De Maesschalk, R.; Candolfi, A.; Massart, D.L.; Heuerding, S. Decision criteria for soft independent modelling of class analogy applied to near infrared data. Chemom. Intell. Lab. Syst. 1999, 47, 65–77. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Salmonella Outbreaks. 1 October 2020. Available online: https://www.cdc.gov/salmonella/outbreaks.html (accessed on 27 December 2020).
- Borges, K.A.; Furian, T.Q.; de Souza, S.N.; Menezes, R.; Alves de Lima, D.; Bornancini Borges Fotes, F.; Tadeu Pippi Salle, C.; Luiz Souza Moraes, H.; Pinheiro Nascimento, V. Biofilm formation by Salmonella Enteritidis and Salmonella Typhimurium isolated from avian sources is partially related with their in vivo pathogenicity. Microb. Pathog. 2018, 118, 238–241. [Google Scholar] [CrossRef] [PubMed]
- Van Vuuren, H.J.J.; Kersters, K.; De Ley, J.; Toerien, D.F. The identification of Enterobacteriaceae from breweries: Combined use and comparison of API 20E system, gel electrophoresis of proteins and gas chromatography of volatile metabolites. J. Appl. Bacteriol. 1981, 51, 51–65. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microorganism | Microorganism |
|---|---|
| Campylobacter coli (Cc) | Salmonella Enteritidis (SE) |
| Campylobacter fetus (Cf) | Salmonella Heidelberg (SH) |
| Campylobacter jejuni (Cj) | Salmonella Infantis (SI) |
| Enterobacter cloacae (Ecl) | Salmonella Javiana (SJ) |
| Enterococcus faecalis (Ef) | Salmonella Kentucky (SKe) |
| Escherichia coli (Eco) | Salmonella Kiambu (SKi) |
| Klebsiella oxytoca (Ko) | Salmonella Mbandanka (SMb) |
| Listeria innocua (Li) | Salmonella Montevideo (SMo) |
| Listeria monocytogenes (Lm) | Salmonella Muenchen (SMu) |
| Macrococcus caseolyticus (Mc) | Salmonella Seftenberg (SSe) |
| Paenibacillus polymyxa (Ppo) | Salmonella Typhimurium (ST) |
| Pseudomonas putida (Ppu) | Salmonella Typhimurium–NAL (STN) |
| Staphylococcus aureus (Sa) | Salmonella Weltevreden (SW) |
| Staphylococcus simulans (Ss) |
| Calibration | Validation | ||||
|---|---|---|---|---|---|
| Microorganism | Reps | Cells | Microorganism | Reps | Cells |
| Salmonella Enteritidis | 4 | 346 | Campylobacter coli | 2 | 27 |
| Salmonella Heidelberg | 4 | 388 | Campylobacter fetus | 2 | 26 |
| Salmonella Infantis | 3 | 282 | Campylobacter jejuni | 2 | 65 |
| Salmonella Javiana | 2 | 231 | Enterobacter cloacae | 1 | 142 |
| Salmonella Kentucky | 3 | 313 | Enterococcus faecalis | 3 | 157 |
| Salmonella Kiambu | 2 | 279 | Escherichia coli | 8 | 767 |
| Salmonella Mbandanka | 2 | 274 | Klebsiella oxytoca | 3 | 82 |
| Salmonella Montevideo | 2 | 156 | Listeria innocua | 3 | 79 |
| Salmonella Muenchen | 2 | 259 | Listeria monocytogenes | 2 | 116 |
| Salmonella Seftenberg | 3 | 165 | Macrococcus caseolyticus | 3 | 24 |
| Salmonella Typhimurium | 3 | 345 | Paenibacillus polymyxa | 2 | 66 |
| Salmonella Typhimurium-NAL | 3 | 140 | Pseudomonas putida | 3 | 151 |
| Salmonella Weltevreden | 2 | 137 | Staphylococcus aureus | 2 | 212 |
| Staphylococcus simulans | 2 | 190 | |||
| Salmonella Enteritdis | 8 | 350 | |||
| Salmonella Heidelberg | 6 | 149 | |||
| Salmonella Infantis | 5 | 284 | |||
| Salmonella Kentucky | 3 | 239 | |||
| Salmonella Typhimurium | 8 | 295 | |||
| Total | 35 | 3315 | Total | 68 | 3421 |
| Salmonella | ||||
|---|---|---|---|---|
| Microorganism | Cells | Yes | No | Accuracy (%) |
| Campylobacter coli | 27 | 6 | 21 | 77.8 |
| Campylobacter fetus | 26 | 3 | 23 | 88.5 |
| Campylobacter jejuni | 65 | 9 | 56 | 86.2 |
| Enterobacter cloacae | 142 | 4 | 138 | 97.2 |
| Enterococcus faecalis | 157 | 1 | 156 | 99.4 |
| Escherichia coli | 767 | 9 | 758 | 98.8 |
| Klebsiella oxytoca | 82 | 1 | 81 | 98.8 |
| Listeria innocua | 79 | 9 | 70 | 88.6 |
| Listeria monocytogenes | 116 | 1 | 115 | 99.1 |
| Macrococcus caseolyticus | 24 | 0 | 24 | 100 |
| Paenibacillus polymyxa | 66 | 6 | 60 | 90.9 |
| Pseudomonas putida | 151 | 55 | 96 | 63.6 |
| Staphylococcus aureus | 212 | 10 | 202 | 95.3 |
| Staphylococcus simulans | 190 | 5 | 185 | 97.4 |
| Salmonella Enteritdis | 350 | 343 | 7 | 98.0 |
| Salmonella Heidelberg | 149 | 141 | 8 | 94.6 |
| Salmonella Infantis | 284 | 277 | 7 | 97.5 |
| Salmonella Kentucky | 239 | 233 | 6 | 97.5 |
| Salmonella Typhimurium | 295 | 283 | 12 | 95.9 |
| Total | 3421 | 1277 | 1985 | 95.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eady, M.; Park, B. An Unsupervised Prediction Model for Salmonella Detection with Hyperspectral Microscopy: A Multi-Year Validation. Appl. Sci. 2021, 11, 895. https://doi.org/10.3390/app11030895
Eady M, Park B. An Unsupervised Prediction Model for Salmonella Detection with Hyperspectral Microscopy: A Multi-Year Validation. Applied Sciences. 2021; 11(3):895. https://doi.org/10.3390/app11030895
Chicago/Turabian StyleEady, Matthew, and Bosoon Park. 2021. "An Unsupervised Prediction Model for Salmonella Detection with Hyperspectral Microscopy: A Multi-Year Validation" Applied Sciences 11, no. 3: 895. https://doi.org/10.3390/app11030895
APA StyleEady, M., & Park, B. (2021). An Unsupervised Prediction Model for Salmonella Detection with Hyperspectral Microscopy: A Multi-Year Validation. Applied Sciences, 11(3), 895. https://doi.org/10.3390/app11030895