Defect Processes in Halogen Doped SnO2

 ,
, 

Abstract
1. Introduction
2. Methodology
3. Results
3.1. Structural Properties
3.2. Electrical Properties
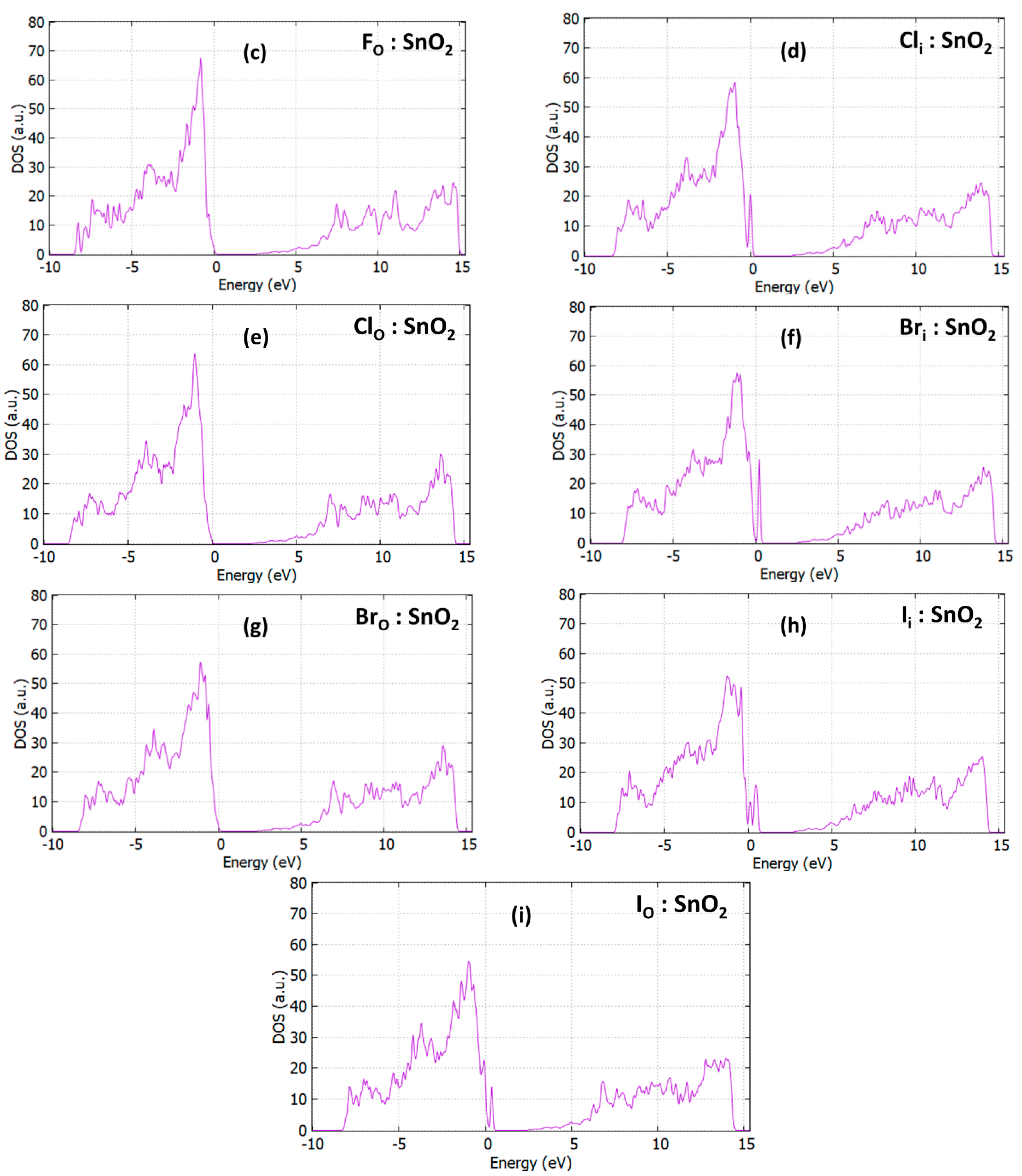
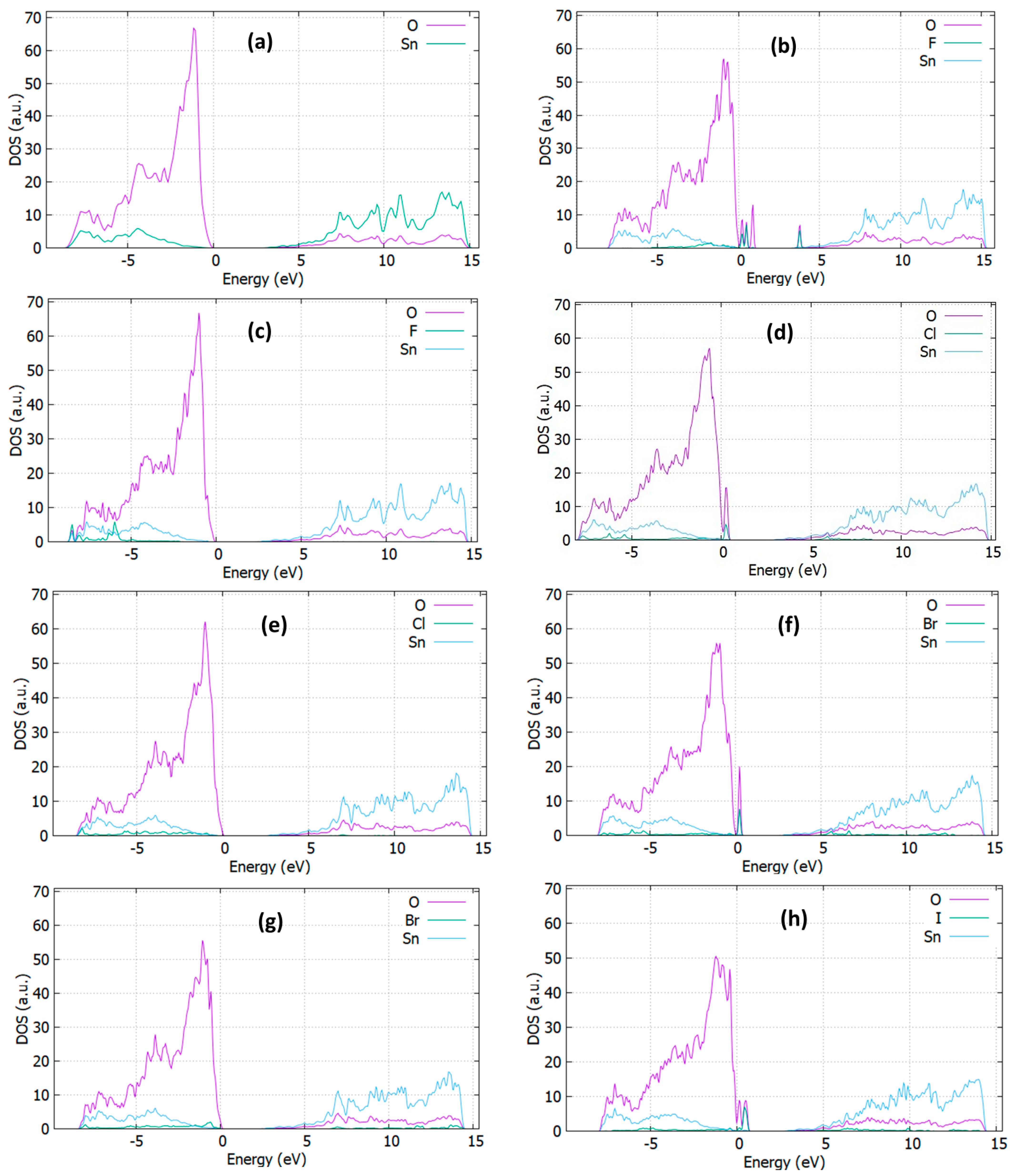
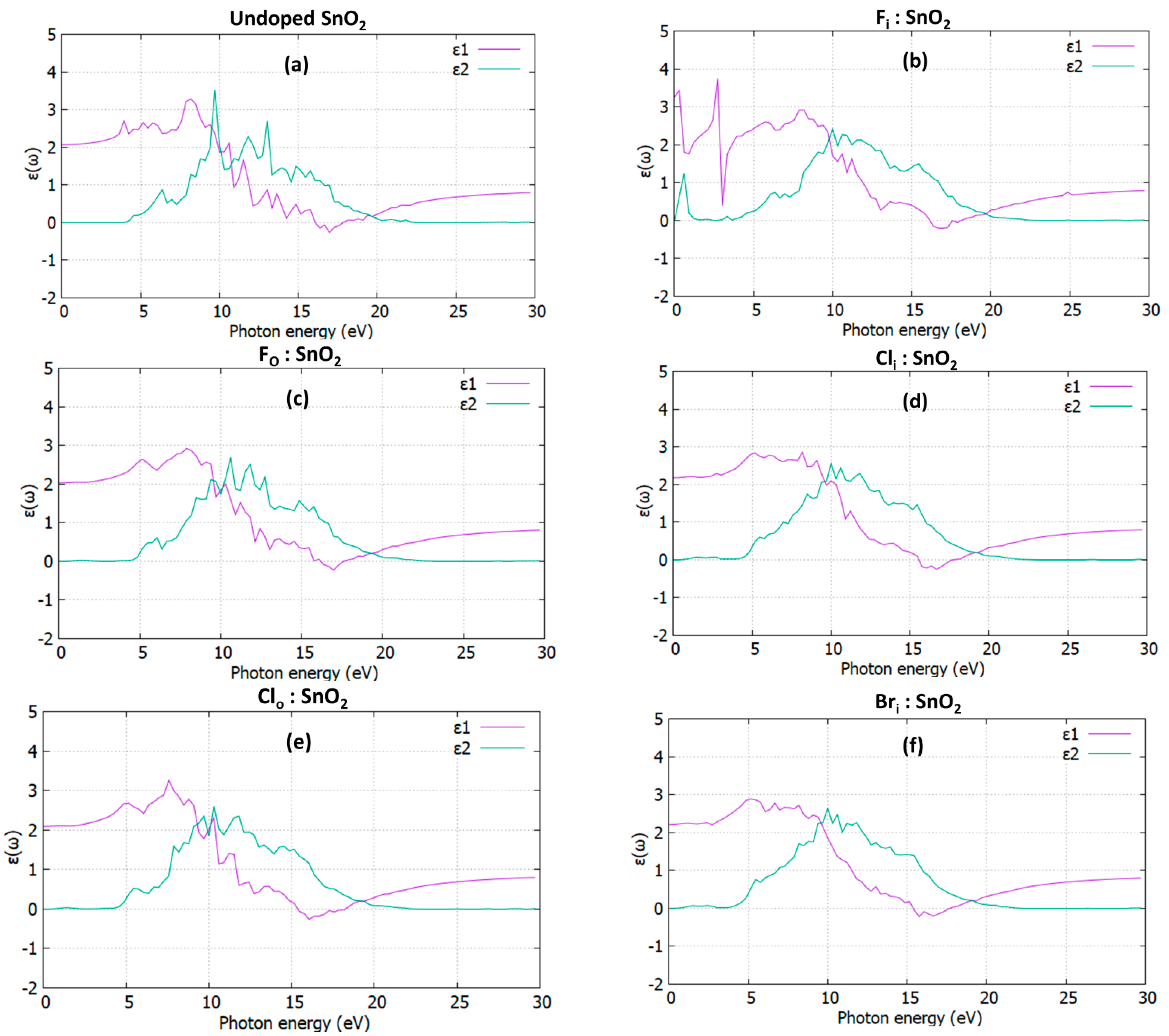
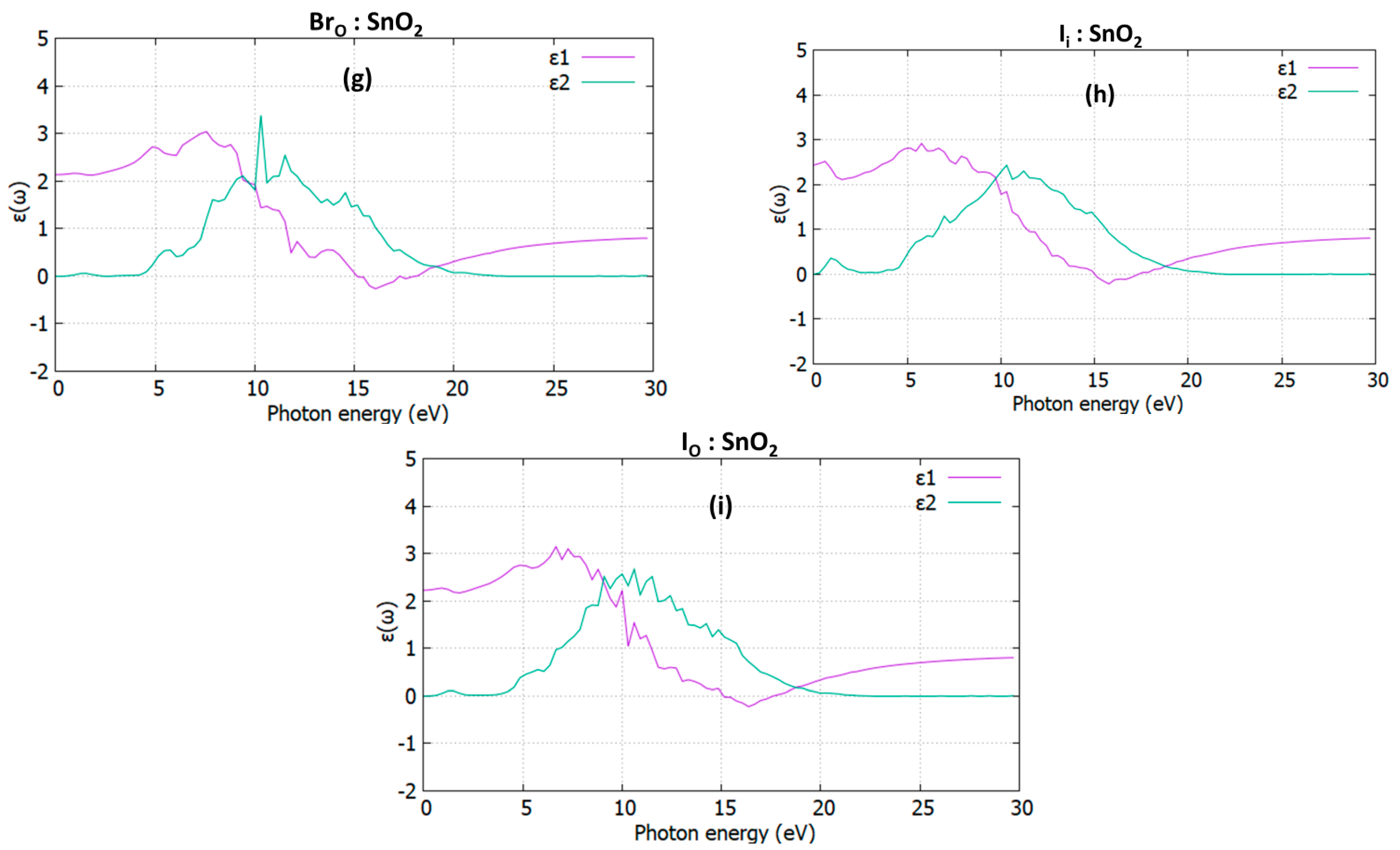
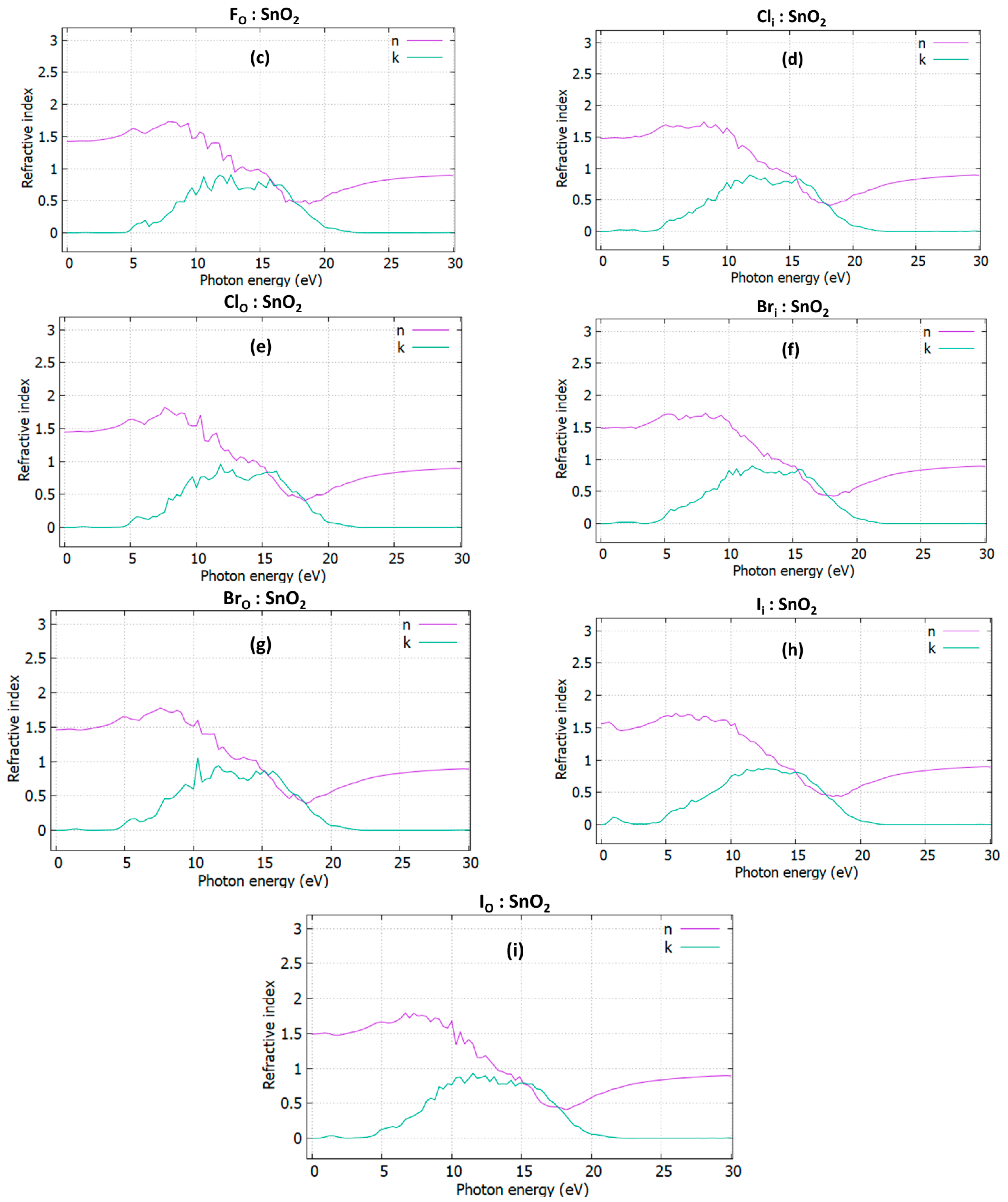
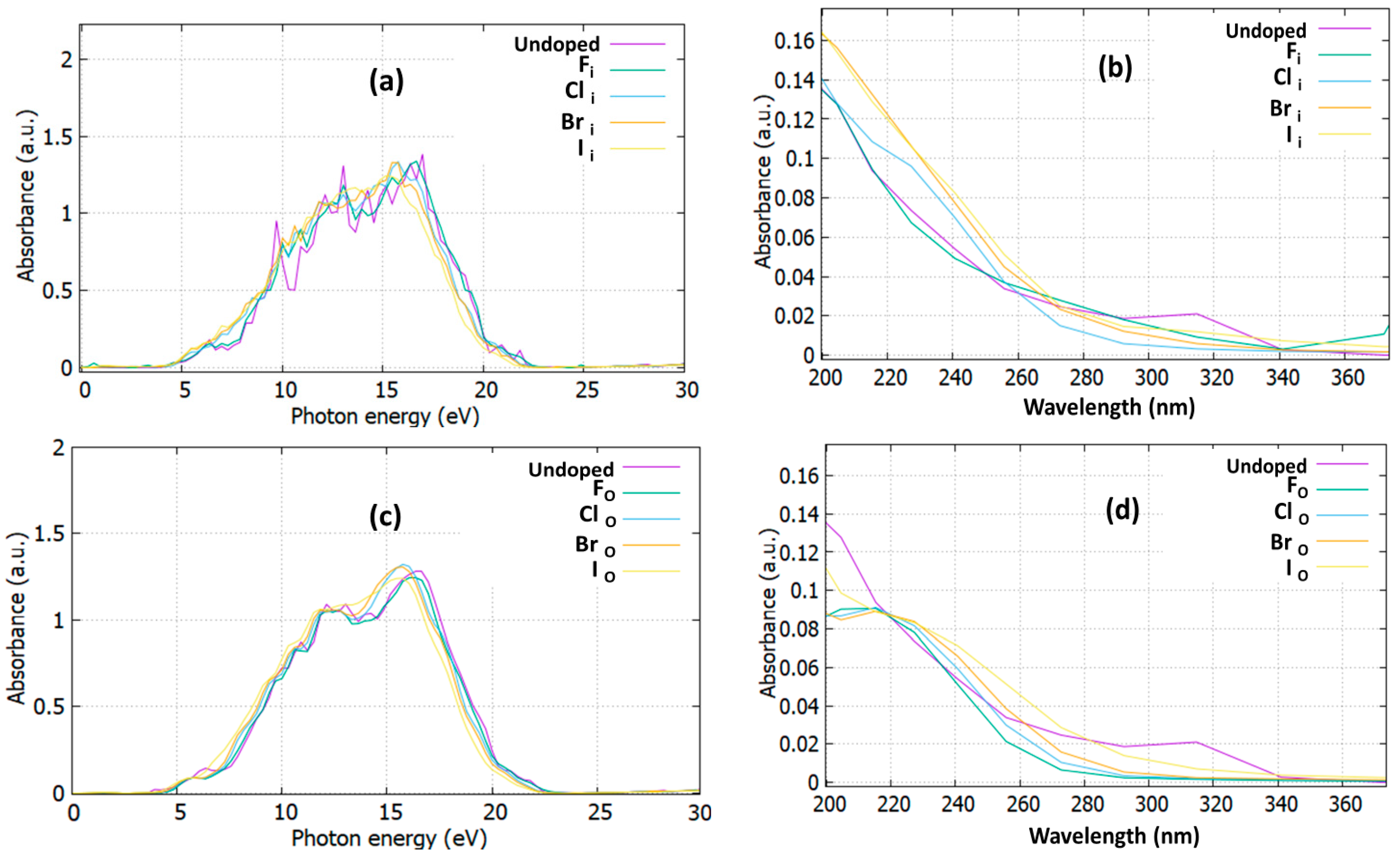
3.3. Optical Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Searle, A.B. The Glazer’s Book; The Technical Press: London, UK, 1935. [Google Scholar]
- Holleman, A.F.; Wiberg, E. Inorganic Chemistry; Academic Press: Cambridge, MA, USA, 2001. [Google Scholar]
- Greenwood, N.N.; Earnshaw, A. Chemistry of the Elements; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Tountas, M.; Topal, Y.; Kus, M.; Ersöz, M.; Fakis, M.; Argitis, P.; Vasilopoulou, M. Water-Soluble Lacunary Polyoxometalates with Excellent Electron Mobilities and Hole Blocking Capabilities for High Efficiency Fluorescent and Phosphorescent Organic Light Emitting Diodes. Adv. Funct. Mater. 2016, 26, 2655–2665. [Google Scholar] [CrossRef]
- Batzill, M.; Diebold, U. The Surface and Materials Science of Tin Oxide. Prog. Surf. Sci. 2005, 79, 47–154. [Google Scholar] [CrossRef]
- Fortunato, E.; Ginley, D.; Hosono, H.; Paine, D.C. Transparent conducting oxides for photovoltaics. MRS Bull. 2007, 32, 242–247. [Google Scholar] [CrossRef]
- Göpel, W.; Schierbaum, K.D. SnO2 sensors: Current status and future prospects. Sens. Actuators B Chem. 1995, 26, 1–12. [Google Scholar] [CrossRef]
- Zhao, J.; Tan, R.; Shen, W.; Yang, Y.; Guo, Y.; Li, J.; Zhou, Z.; Jian, J.; Song, W. Highly selective Sn2O3-based sensors for NO detection. Mater. Lett. 2012, 84, 94–96. [Google Scholar] [CrossRef]
- Wang, C.; Du, G.; Ståhl, K.; Huang, H.; Zhong, Y.; Jiang, J.Z. Ultrathin SnO2 nanosheets: Oriented attachment mechanism, nonstoichiometric defects, and enhanced lithium-ion battery performances. J. Phys. Chem. C 2012, 116, 4000–4011. [Google Scholar] [CrossRef]
- Rakhshani, A.E.; Makdisi, Y.; Ramazaniyan, H.A. Electronic and optical properties of fluorine-doped tin oxide films. J. Appl. Phys. 1998, 83, 1049–1057. [Google Scholar] [CrossRef]
- Wu, S.; Yuan, S.; Shi, L.; Zhao, Y.; Fang, J. Preparation, characterization and electrical properties of fluorine-doped tin dioxide nanocrystals. J. Coll. Interface Sci. 2010, 346, 12–16. [Google Scholar] [CrossRef]
- Garces, F.A.; Budini, N.; Koropecki, R.R.; Arce, R.D. Structural mosaicity and electrical properties of pyrolytic SnO2:F thin films. Thin Solid Films 2013, 531, 172–178. [Google Scholar] [CrossRef]
- Elangovan, E.; Ramamurthi, K. A study on low cost-high conducting fluorine and antimony-doped tin oxide thin films. Appl. Surf. Sci. 2005, 249, 183–196. [Google Scholar] [CrossRef]
- Yang, J.; Liu, W.; Dong, L.; Li, Y.; Li, C.; Zhao, H. Studies on the structural and electrical properties of F-doped SnO2 film prepared by APCVD. Appl. Surf. Sci. 2011, 257, 10499–10502. [Google Scholar] [CrossRef]
- Moholkar, A.V.; Pawar, S.M.; Rajpure, K.Y.; Bhosale, C.H.; Kim, J.H. Effect of fluorine doping onhighly transparent conductive spray deposited nanocrystalline tin oxide thin flms. Appl. Surf. Sci. 2009, 255, 9358–9364. [Google Scholar] [CrossRef]
- Suffner, J.; Ágoston, P.; Kling, J.; Hahn, H. Chemical vapor synthesis of fluorine-doped SnO2 (FTO) nanoparticles. J. Nanopart. Res. 2010, 12, 2579–2588. [Google Scholar] [CrossRef]
- Abass, A.K. Electrical and optical properties of chlorine doped SnO2 coatings. Solid State Commun. 1987, 61, 507–510. [Google Scholar] [CrossRef]
- Abass, A.K.; Bakr, H.; Jassim, S.A.; Fahad, T.A. Electrical and optical properties of chemically deposited SnO2: I coatings. Sol. Energy Mater. 1988, 17, 425–431. [Google Scholar] [CrossRef]
- Abass, A.K.; Al-Liabi, N.A.; Taha, W.A. Optical properties of bromine-doped SnO2 coatings for solar applications. Phys. Status Solidi (a) 1988, 106, 613–618. [Google Scholar] [CrossRef]
- Agashe, C.; Major, S.S. Effect of F, Cl and Br doping on electrical properties of sprayed SnO2 films. J. Mater. Sci. Lett. 1996, 15, 497–499. [Google Scholar] [CrossRef]
- Peng-Fei, L.; Yue, S.; Zhong-Yuan, Y.; Long, Z.; Qiong-Yao, L.; Shi-Jia, L.M.; Li-Hong, H.; Yu-Min, L. Electronic structure and optical properties of antimony-doped SnO2 from first-principle study. Commun. Theor. Phys. 2012, 57, 145. [Google Scholar]
- Canestraro, C.D.; Roman, L.S.; Persson, C. Polarization dependence of the optical response in SnO2 and the effects from heavily F doping. Thin Solid Films 2009, 517, 6301–6304. [Google Scholar] [CrossRef]
- Rivera, R.; Marcillo, F.; Chamba, W.; Puchaicela, P.; Stashans, A. SnO2 physical and chemical properties due to the impurity doping. Lect. Notes Eng. Comp. 2013, 2, 814–818. [Google Scholar]
- Velikokhatnyi, O.I.; Kumta, P.N. Ab-initio study of fluorine-doped tin dioxide: A prospective catalyst support for water electrolysis. Physica B 2011, 406, 471–477. [Google Scholar] [CrossRef]
- Golovanov, V.; Golovanova, V.; Kuisma, M.; Rantala, T.T. Electron spin resonance parameters of cation vacancies in tin dioxide doped with fluorine and hydrogen. J. Appl. Phys. 2013, 114, 143907. [Google Scholar] [CrossRef]
- Oshima, M.; Yoshino, K. Structural and Electronic Structure of SnO2 by the First-Principle Study. Mater. Sci. Forum 2012, 725, 265–268. [Google Scholar] [CrossRef]
- Govaerts, K.; Partoens, B.; Lamoen, D. Extended homologous series of Sn–O layered systems: A first-principles study. Solid State Comm. 2016, 243, 36–43. [Google Scholar] [CrossRef]
- Marcillo, F.; Stashans, A. DFT calculations of tin dioxide crystals containing heavily-doped fluorine. J. Theor. Comput. Chem. 2014, 13, 1450069. [Google Scholar] [CrossRef]
- Perdew, J.P.; Levy, M. Physical content of the exact Kohn-Sham orbital energies: Band gaps and derivative discontinuities. Phys. Rev. Lett. 1983, 51, 1884. [Google Scholar] [CrossRef]
- Sham, L.J.; Schlüter, M. Density-functional theory of the energy gap. Phys. Rev. Lett. 1983, 51, 1888. [Google Scholar] [CrossRef]
- Thangaraju, B. Structural and electrical studies on highly conducting spray deposited fluorine and antimony doped SnO2 thin films from SnCl2 precursor. Thin Solid Films 2002, 402, 71–78. [Google Scholar] [CrossRef]
- Zervos, M.; Lathiotakis, N.; Kelaidis, N.; Othonos, A.; Tanasa, E.; Vasile, E. Epitaxial Highly Ordered Sb:SnO2 Nanowires Grown by the Vapor Liquid Solid Mechanism on m-, r- and a-Al2O3. Nanoscale Adv. 2019, 1, 1980–1990. [Google Scholar] [CrossRef]
- Deng, H.X.; Li, S.S.; Li, J. Quantum confinement effects and electronic properties of SnO2 quantum wires and dots. J. Phys. Chem. C 2010, 114, 4841–4845. [Google Scholar] [CrossRef]
- Payne, M.C.; Teter, M.P.; Allan, D.C.; Arias, T.A.; Joannopoulos, J.D. CASTEP 4.2 Academic version, licensed under the UKCP-MSI Agreement. Rev. Mod. Phys. 1992, 64, 1045–1097. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.; Probert, M.A.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Cond. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Ceperley, D.M.; Alder, B.J. Exchange-correlation potential and energy for density-functional calculation. Phys. Rev. Lett. 1980, 45, 567–581. [Google Scholar]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Paier, J.; Marsman, M.; Hummer, K.; Kresse, G.; Gerber, I.C.; Angyan, J.G. Screened hybrid density functionals applied to solids. J. Chem. Phys. 2006, 124, 154709. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Huang, S.; Wang, Z. First principle study on the electronic structure of fluorine-doped SnO2. Solid State Commun. 2009, 149, 527–531. [Google Scholar] [CrossRef]
- Yahla, H.; Boukra, A.; Belhakem, M.; Lippens, P.E. First-principles calculations of the electronic structure and Mössbauer parameters of Sb-doped SnO2. Solid State Commun. 2009, 149, 2202–2206. [Google Scholar] [CrossRef]
- Borges, P.D.; Scolfaro, L.M.; Alves, H.W.L.; da Silva, E.F. DFT study of the electronic, vibrational, and optical properties of SnO2. Theor. Chem. Acc. 2009, 126, 39–44. [Google Scholar] [CrossRef]
- Filippatos, P.P.; Kelaidis, N.; Vasilopoulou, M.; Davazoglou, D.; Lathiotakis, N.N.; Chroneos, A. Defect processes in F and Cl doped anatase TiO2. Sci. Rep. 2019, 9, 19970. [Google Scholar] [CrossRef] [PubMed]
- Ramaiah, K.S.; Raja, V.S. Structural and electrical properties of fluorine doped tin oxide films prepared by spray-pyrolysis technique. Appl. Surf. Sci. 2006, 253, 1451–1458. [Google Scholar] [CrossRef]
- Yadav, A.A.; Masumdar, E.U.; Moholkar, A.V.; Neumann-Spallart, M.; Rajpure, K.Y.; Bhosale, C.H. Electrical, structural and optical properties of SnO2:F thin films: Effect of the substrate temperature. J. Alloys Compd. 2009, 488, 350–355. [Google Scholar] [CrossRef]
- Elangovan, E.; Ramamurthi, K. Studies on micro-structural and electrical properties of spray-deposited fluorine-doped tin oxide thin films from low-cost precursor. Thin Solid Films 2005, 476, 231–236. [Google Scholar] [CrossRef]
- Chinnappa, L.; Ravichandran, K.; Saravanakumar, K.; Muruganantham, G.; Sakthivel, B. The combined effects of molar concentration of the precursor solution and fluorine doping on the structural and electrical properties of tin oxide films. J. Mater. Sci. Mater. Electron. 2001, 22, 1827–1834. [Google Scholar] [CrossRef]
- Bakr, N.A.; Salman, S.A.; Ali, M.N. Effect of Fluorine Doping on Structural and Optical Properties of SnO2 Thin Films Prepared by Chemical Spray Pyrolysis Method. Adv. Mater. 2006, 5, 23. [Google Scholar] [CrossRef]
- Tingting, S.; Fuchun, Z.; Weihu, Z. Density functional theory study on the electronic structure and optical properties of SnO2. Rare Met. Mat. Eng. 2015, 44, 2409–2414. [Google Scholar] [CrossRef]
- Khan, A.F.; Mehmood, M.; Aslam, M.; Ashraf, M. Characteristics of electron beam evaporated nanocrystalline SnO2 thin films annealed in air. Appl. Surf. Sci. 2010, 256, 2252–2258. [Google Scholar] [CrossRef]
- Afify, H.H.; Momtaz, R.S.; Badawy, W.A.; Nasser, S.A. Some physical properties of fluorine-doped SnO2 films prepared by spray pyrolysis. J. Mater. Sci. Mater. Electron. 1991, 2, 40–45. [Google Scholar] [CrossRef]
- Doyan, A.; Susilawati; Imawanti, Y.D. Synthesis and Characterization of SnO2 thin layer with a doping Aluminum is deposited on Quartz Substrates. AIP Conf. Proc. 2017, 1801, 020005. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a (Å) | b (Å) | c (Å) | Vol (Å3) | |
|---|---|---|---|---|
| SnO2 | 4.717 | 4.717 | 3.189 | 70.956 |
| Fi: SnO2 | 4.739 | 4.715 | 3.195 | 71.390 |
| Fo: SnO2 | 4.735 | 4.735 | 3.205 | 71.857 |
| Cli: SnO2 | 4.789 | 4.790 | 3.201 | 73.429 |
| CloSnO2 | 4.778 | 4.778 | 3.213 | 73.350 |
| Bri: SnO2 | 4.795 | 4.845 | 3.194 | 74.202 |
| Bro: SnO2 | 4.795 | 4.795 | 3.218 | 73.988 |
| Ii: SnO2 | 5.055 | 4.830 | 3.165 | 77.276 |
| Io: SnO2 | 4.840 | 4.840 | 3.215 | 75.313 |
| Halogen Dopants | Band Gap | |
|---|---|---|
| Interstitial Doping | Substitutional Doping | |
| F | 3.12 | 2.90 |
| Cl | 2.85 | 2.70 |
| Br | 2.90 | 2.71 |
| I | 2.62 | 2.61 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filippatos, P.-P.; Kelaidis, N.; Vasilopoulou, M.; Davazoglou, D.; Chroneos, A. Defect Processes in Halogen Doped SnO2. Appl. Sci. 2021, 11, 551. https://doi.org/10.3390/app11020551
Filippatos P-P, Kelaidis N, Vasilopoulou M, Davazoglou D, Chroneos A. Defect Processes in Halogen Doped SnO2. Applied Sciences. 2021; 11(2):551. https://doi.org/10.3390/app11020551
Chicago/Turabian StyleFilippatos, Petros-Panagis, Nikolaos Kelaidis, Maria Vasilopoulou, Dimitris Davazoglou, and Alexander Chroneos. 2021. "Defect Processes in Halogen Doped SnO2" Applied Sciences 11, no. 2: 551. https://doi.org/10.3390/app11020551
APA StyleFilippatos, P.-P., Kelaidis, N., Vasilopoulou, M., Davazoglou, D., & Chroneos, A. (2021). Defect Processes in Halogen Doped SnO2. Applied Sciences, 11(2), 551. https://doi.org/10.3390/app11020551