Optimising Hydrogel Release Profiles for Viro-Immunotherapy Using Oncolytic Adenovirus Expressing IL-12 and GM-CSF with Immature Dendritic Cells



Abstract
1. Introduction
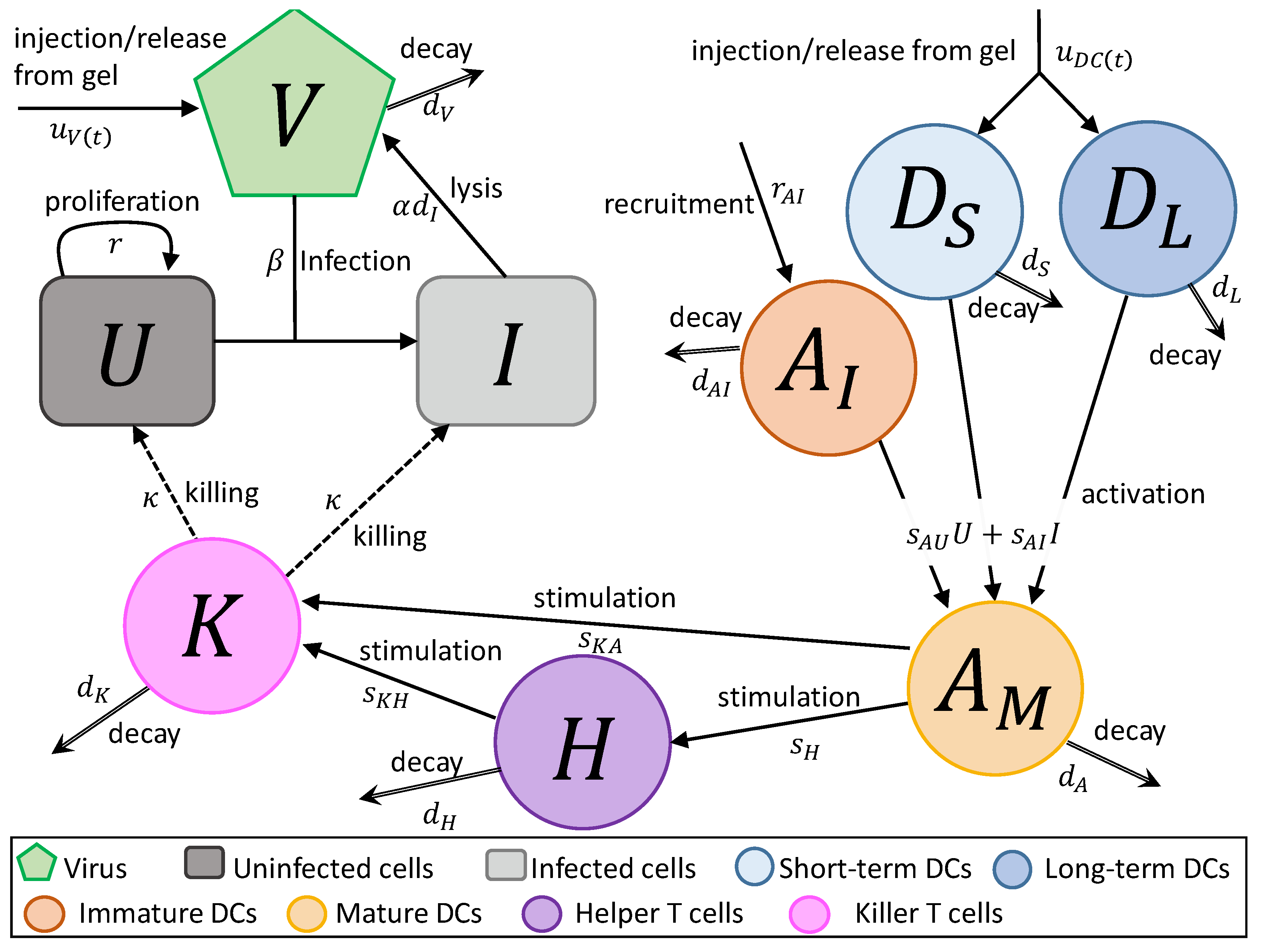
2. Mathematical Model
- In Equation (1), uninfected tumour cells, U, are growing at a rate described by a Gompertz function with proliferation rate r and carrying capacity L. Uninfected cells are infected by virus particles, V, at a frequency-dependent rate with rate constant . Killer T cells, K, induce apoptosis in uninfected cells at a frequency-dependent rate with rate constant .
- In Equation (2), tumour cells become infected cells, I, at rate . Infected cells lyse at rate , and, like uninfected cells, are killed at a frequency-dependent rate.
- In Equation (3), virus particles enter the system at rate , either by a single injection or release from the hydrogel. Virus particles decay at a rate , and new viruses are created through lysis.
- In Equation (4), short-term immature DCs, , enter the system at a rate , either by a single injection or release from a hydrogel. They are then stimulated to become mature or activated APCs, , at rate or due to the interaction with either uninfected or infected tumour cells. They decay at a fast rate , where .
- In Equation (5), long-term immature DCs, , enter the system at rate , where is the rate of injection or release from the hydrogel and f is the fraction of the initially loaded DCs that are long-term DCs, . Similarly to short-term DCs, they become activated APCs at rate and . These DCs decay slowly at rate .
- In Equation (6), immature APCs, , are recruited to the tumour site by infected cells at rate . Immature APCs are then stimulated at the same rate as immature short-term and long-term DCs. The immature APCs decay at rate .
- In Equation (7), mature APCs are generated through the maturation of introduced long-term and short-term DCs and immature DCs recruited by uninfected tumour cells at rate and infected tumour cells at rate . These cells decay at rate .
- In Equation (8), helper T cels, H, are stimulated by mature APCs at a rate . These cells decay at a rate . We assume the rate APCs stimulate helper cells and helper cells stimulate killer cells is independent of antigen.
- In Equation (9), killer T cells, K, are activated by helper T cells and mature APCs at rates of and , respectively. These cells either die or leave the tumour site at a rate .
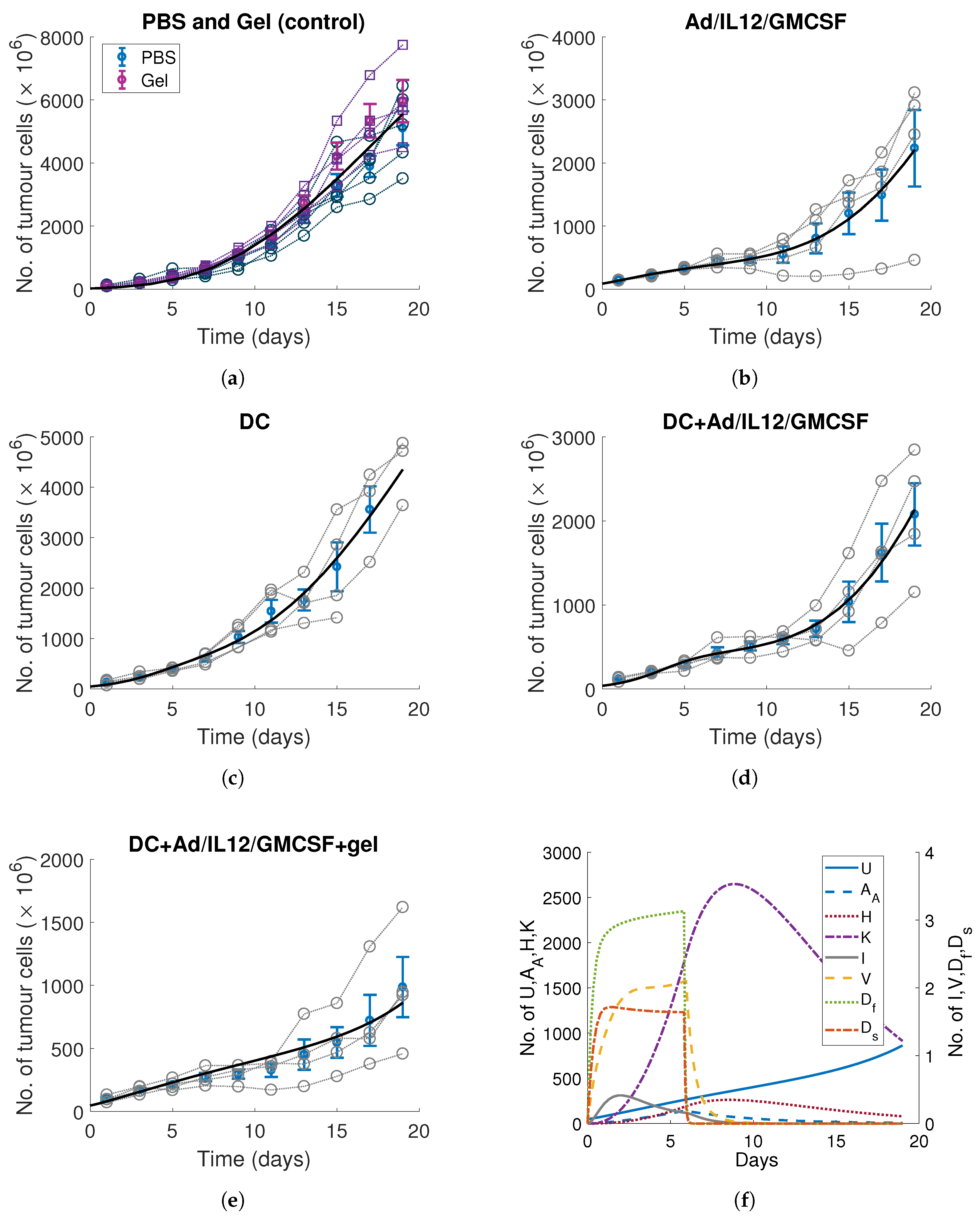
3. Calibrating Parameters to In Vitro and In Vivo Time-Series Measurements
3.1. DC Release-Profile and Decay Rates
3.2. Tumour, Immune and Viral Parameters
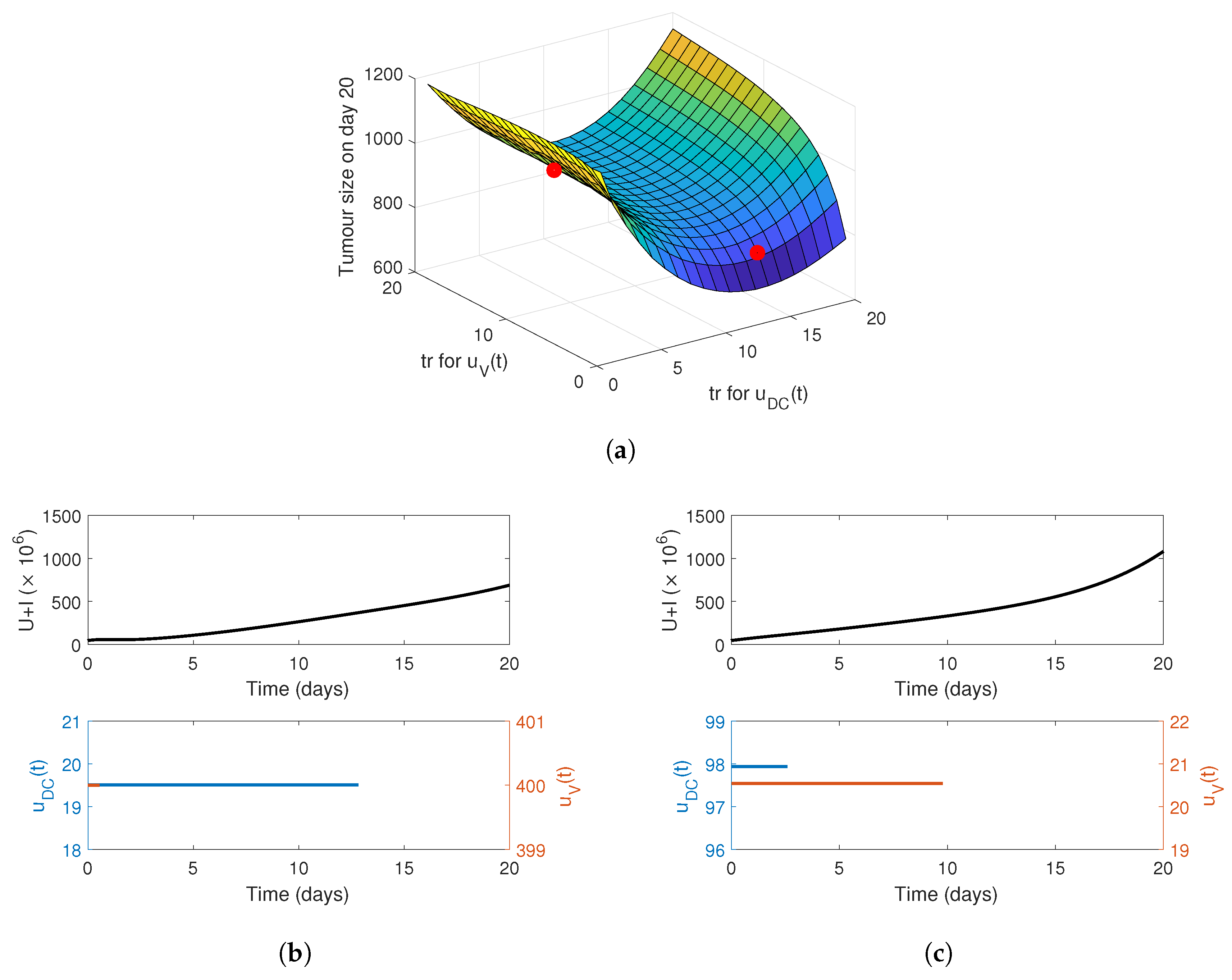
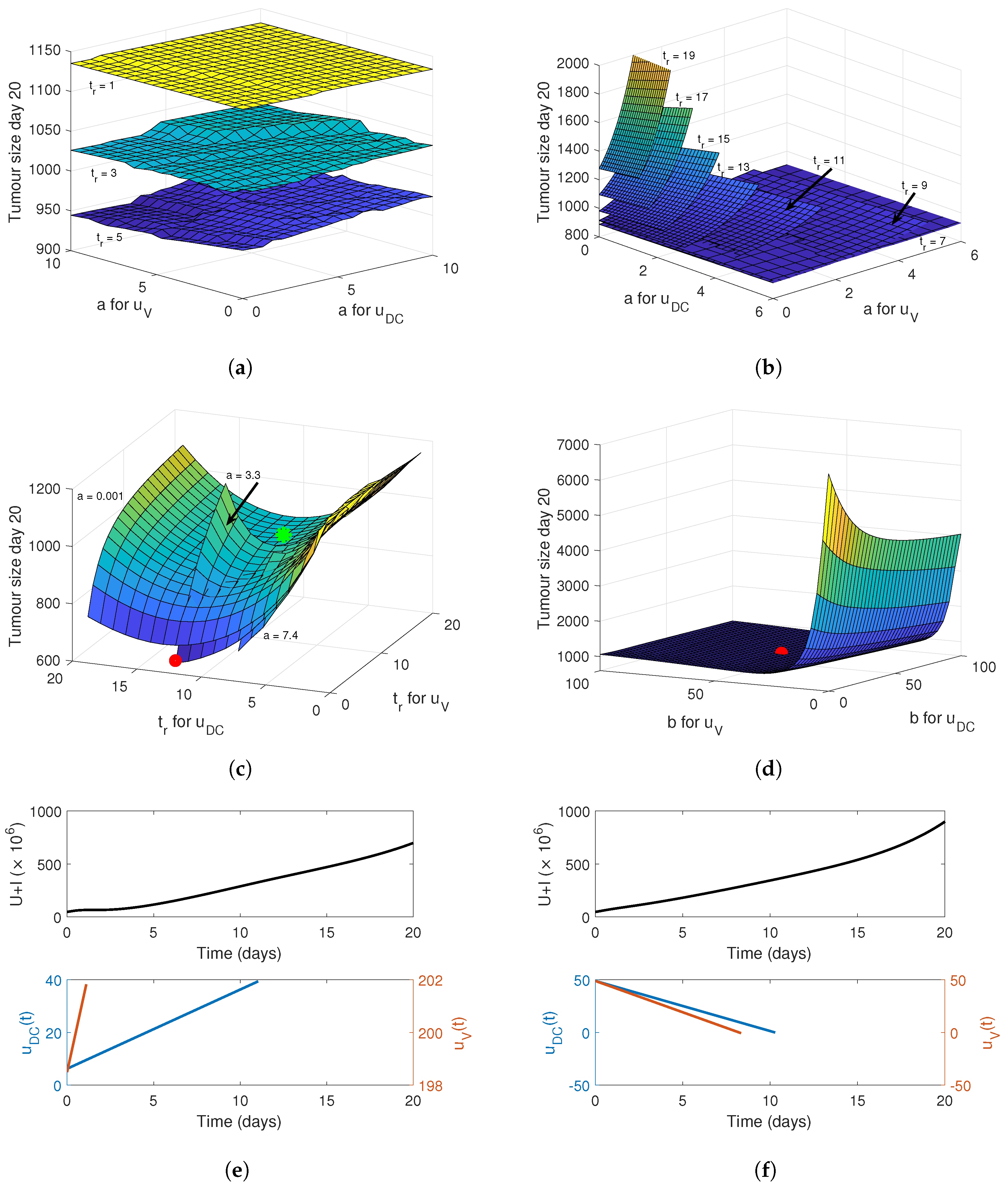
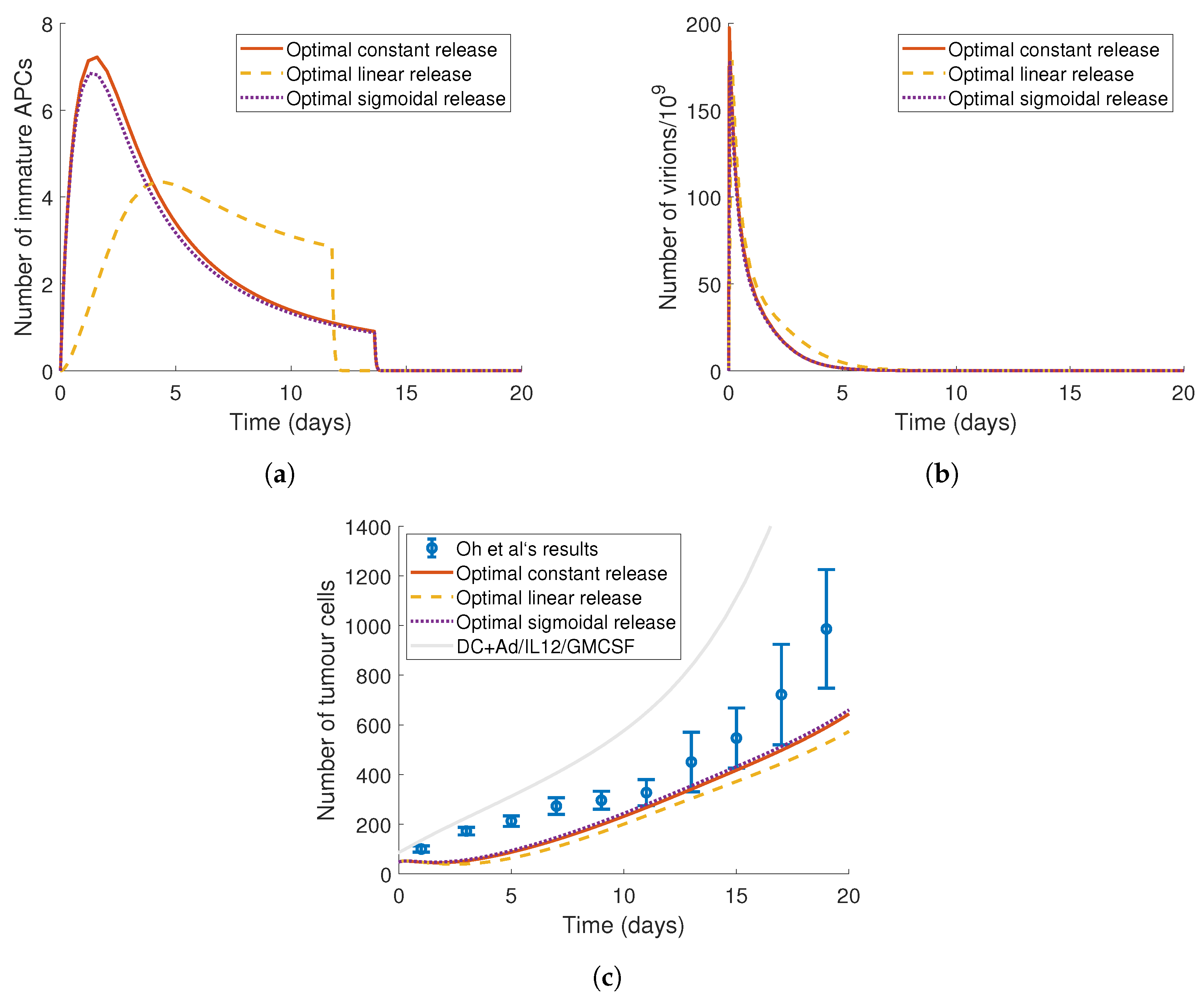
4. Optimising the Gel Release Profile
4.1. Constant Release Profile
4.2. Linear Release Profile
4.3. Sigmoidal Release Profile
4.4. Implementing a Genetic Algorithm to Determine the True Optimal Release Curves
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ta, H.T.; Dass, C.R.; Dunstan, D.E. Injectable chitosan hydrogels for localised cancer therapy. J. Control. Release 2008, 126, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Sahu, P.; Kashaw, S.K.; Sau, S.; Kushwah, V.; Jain, S.; Agrawal, R.K.; Iyer, A.K. pH responsive 5-fluorouracil loaded biocompatible nanogels for topical chemotherapy of aggressive melanoma. Colloids Surf. B Biointerfaces 2019, 174, 232–245. [Google Scholar] [CrossRef]
- Wade, S.J.; Zuzic, A.; Foroughi, J.; Talebian, S.; Aghmesheh, M.; Moulton, S.E.; Vine, K.L. Preparation and in vitro assessment of wet-spun gemcitabine-loaded polymeric fibers: Towards localized drug delivery for the treatment of pancreatic cancer. Pancreatology 2017, 17, 795–804. [Google Scholar] [CrossRef]
- Liu, X.; Li, Z.; Loh, X.J.; Chen, K.; Li, Z.; Wu, Y.L. Targeted and sustained corelease of chemotherapeutics and gene by injectable supramolecular hydrogel for drug-resistant cancer T=therapy. Macromol. Rapid Commun. 2019, 40, 1800117. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Kang, E.; Kwon, O.; Yun, T.; Park, H.; Kim, P.; Kim, S.; Kim, J.H.; Yun, C.O. Local sustained delivery of oncolytic adenovirus with injectable alginate gel for cancer virotherapy. Gene Ther. 2013, 20, 880–892. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.H.; Choi, J.W.; Yun, C.O.; Yhee, J.Y.; Price, R.; Kim, S.H.; Kwon, I.C.; Ghandehari, H. Sustained local delivery of oncolytic short hairpin RNA adenoviruses for treatment of head and neck cancer. J. Gene Med. 2014, 16, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.K.; Oh, E.; Hong, J.; Lee, Y.; Park, K.D.; Yun, C.O. A hydrogel matrix prolongs persistence and promotes specific localization of an oncolytic adenovirus in a tumor by restricting nonspecific shedding and an antiviral immune response. Biomaterials 2017, 147, 26–38. [Google Scholar] [CrossRef]
- Park, C.G.; Hartl, C.A.; Schmid, D.; Carmona, E.M.; Kim, H.J.; Goldberg, M.S. Extended release of perioperative immunotherapy prevents tumor recurrence and eliminates metastases. Sci. Transl. Med. 2018, 10, eaar1916. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Wang, C.; Yu, J.; Wang, J.; Lu, Y.; Zhang, Y.; Zhang, X.; Hu, Q.; Sun, W.; He, C.; et al. Injectable bioresponsive gel depot for enhanced immune checkpoint blockade. Adv. Mater. 2018, 30, 1801527. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.; Oh, J.E.; Hong, J.; Chung, Y.; Lee, Y.; Park, K.D.; Kim, S.; Yun, C.O. Optimized biodegradable polymeric reservoir-mediated local and sustained co-delivery of dendritic cells and oncolytic adenovirus co-expressing IL-12 and GM-CSF for cancer immunotherapy. J. Control. Release 2017, 259, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.; O’Leary, M.; Fong, Y.; Chen, N. From benchtop to bedside: A review of oncolytic virotherapy. Biomedicines 2016, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Rabkin, S.D. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol. Res. 2014, 2, 295–300. [Google Scholar] [CrossRef]
- Choi, K.J.; Zhang, S.N.; Choi, I.K.; Kim, J.S.; Yun, C.O. Strengthening of antitumor immune memory and prevention of thymic atrophy mediated by adenovirus expressing IL-12 and GM-CSF. Gene Ther. 2012, 19, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Tugues, S.; Burkhard, S.; Ohs, I.; Vrohlings, M.; Nussbaum, K.; Vom Berg, J.; Kulig, P.; Becher, B. New insights into IL-12-mediated tumor suppression. Cell Death Differ. 2015, 22, 237. [Google Scholar] [CrossRef]
- Dai, S.; Wei, D.; Wu, Z.; Zhou, X.; Wei, X.; Huang, H.; Li, G. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol. Ther. 2008, 16, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, L.H. Dendritic cells in cancer immunotherapy clinical trials: Are we making progress? Front. Immunol. 2013, 4, 454. [Google Scholar] [CrossRef] [PubMed]
- Anguille, S.; Smits, E.L.; Lion, E.; van Tendeloo, V.F.; Berneman, Z.N. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014, 15, e257–e267. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Travers, P.; Walport, M.; Shlomchik, M.J. Immunobiology: The Immune System in Health and Disease, 6th ed.; Garland Science Publishing: New York, NY, USA, 2005. [Google Scholar]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef]
- Chard, L.S.; Maniati, E.; Wang, P.; Zhang, Z.; Gao, D.; Wang, J.; Cao, F.; Ahmed, J.; El Khouri, M.; Hughes, J.; et al. A vaccinia virus armed with interleukin-10 is a promising therapeutic agent for treatment of murine pancreatic cancer. Clin. Cancer Res. 2015, 21, 405–416. [Google Scholar] [CrossRef]
- Bell, J.C.; Ilkow, C.S. A viro-immunotherapy triple play for the treatment of glioblastoma. Cancer Cell 2017, 32, 133–134. [Google Scholar] [CrossRef]
- Powathil, G.G.; Swat, M.; Chaplain, M.A. Systems oncology: Towards patient-specific treatment regimes informed by multiscale mathematical modelling. Semin. Cancer Biol. 2015, 30, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Cherruault, Y. Mathematical Modelling in Biomedicine: Optimal Control Of Biomedical Systems; Springer Science & Business Media: Berlin, Germany, 2012; Volume 23. [Google Scholar]
- Barbolosi, D.; Ciccolini, J.; Lacarelle, B.; Barlési, F.; André, N. Computational oncology—Mathematical modelling of drug regimens for precision medicine. Nat. Rev. Clin. Oncol. 2016, 13, 242. [Google Scholar] [CrossRef] [PubMed]
- Sbeity, H.; Younes, R. Review of optimization methods for cancer chemotherapy treatment planning. J. Comput. Sci. Syst. Biol. 2015, 8, 74. [Google Scholar] [CrossRef]
- Carrere, C. Optimization of an in vitro chemotherapy to avoid resistant tumours. J. Theor. Biol. 2017, 413, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Engelhart, M.; Lebiedz, D.; Sager, S. Optimal control for selected cancer chemotherapy ODE models: A view on the potential of optimal schedules and choice of objective function. Math. Biosci. 2011, 229, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Piretto, E.; Delitala, M.; Kim, P.S.; Frascoli, F. Effects of mutations and immunogenicity on outcomes of anti-cancer therapies for secondary lesions. Math. Biosci. 2019, 315, 108238. [Google Scholar] [CrossRef]
- Cassidy, T.; Craig, M. Determinants of combination GM-CSF immunotherapy and oncolytic virotherapy success identified through in silico treatment personalization. PLoS Comput. Biol. 2019, 15, e1007495. [Google Scholar] [CrossRef]
- Frascoli, F.; Kim, P.S.; Hughes, B.D.; Landman, K.A. A dynamical model of tumour immunotherapy. Math. Biosci. 2014, 253, 50–62. [Google Scholar] [CrossRef]
- Zhu, J.; Liu, R.; Jiang, Z.; Wang, P.; Yao, Y.; Shen, Z. Optimization of drug regimen in chemotherapy based on semi-mechanistic model for myelosuppression. J. Biomed. Inform. 2015, 57, 20–27. [Google Scholar] [CrossRef][Green Version]
- De Pillis, L.G.; Gu, W.; Radunskaya, A.E. Mixed immunotherapy and chemotherapy of tumors: Modeling, applications and biological interpretations. J. Theor. Biol. 2006, 238, 841–862. [Google Scholar] [CrossRef]
- Tzafriri, A.R.; Lerner, E.I.; Flashner-Barak, M.; Hinchcliffe, M.; Ratner, E.; Parnas, H. Mathematical modeling and optimization of drug delivery from intratumorally injected microspheres. Clin. Cancer Res. 2005, 11, 826–834. [Google Scholar] [PubMed]
- El-Kareh, A.W.; Secomb, T.W. A mathematical model for comparison of bolus injection, continuous infusion, and liposomal delivery of doxorubicin to tumor cells. Neoplasia 2000, 2, 325. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.S.; Crivelli, J.J.; Choi, I.K.; Yun, C.O.; Wares, J.R. Quantitative impact of immunomodulation versus oncolysis with cytokine-expressing virus therapeutics. Math. Biosci. Eng. 2015, 12, 841–858. [Google Scholar] [CrossRef] [PubMed]
- Jenner, A.L.; Yun, C.O.; Yoon, A.; Coster, A.C.; Kim, P.S. Modelling combined virotherapy and immunotherapy: Strengthening the antitumour immune response mediated by IL-12 and GM-CSF expression. Lett. Biomath. 2018, 5, S99–S116. [Google Scholar] [CrossRef]
- Wares, J.R.; Crivelli, J.J.; Yun, C.O.; Choi, I.K.; Gevertz, J.L.; Kim, P.S. Treatment strategies for combining immunostimulatory oncolytic virus therapeutics with dendritic cell injections. Math. Biosci. Eng. 2015, 12, 1237–1256. [Google Scholar] [CrossRef]
- Barish, S.; Ochs, M.F.; Sontag, E.D.; Gevertz, J.L. Evaluating optimal therapy robustness by virtual expansion of a sample population, with a case study in cancer immunotherapy. Proc. Natl. Acad. Sci. USA 2017, 114, E6277–E6286. [Google Scholar] [CrossRef]
- Sompayrac, L.M. How the Immune System Works; Wiley-Blackwell: Hoboken, NJ, USA, 2019. [Google Scholar]
- Jenner, A.L. Applications of Mathematical Modelling in Oncolytic Virotherapy and Immunotherapy. Ph.D. Thesis, University of Sydney, Sydney, NSW, Australia, 2019. [Google Scholar]
- Helft, J.; Böttcher, J.; Chakravarty, P.; Zelenay, S.; Huotari, J.; Schraml, B.U.; Goubau, D.; e Sousa, C.R. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c+ MHCII+ macrophages and dendritic cells. Immunity 2015, 41, 1197–1211. [Google Scholar] [CrossRef]
- Kim, S.E.; Hwang, J.H.; Kim, Y.K.; Lee, H.T. Heterogeneity of porcine bone marrow-derived dendritic cells induced by GM-CSF. PLoS ONE 2019, 14, e0223590. [Google Scholar] [CrossRef]
- Chow, K.V.; Lew, A.M.; Sutherland, R.M.; Zhan, Y. Monocyte-derived dendritic cells promote Th polarization, whereas conventional dendritic cells promote Th proliferation. J. Immunol. 2016, 196, 624–636. [Google Scholar] [CrossRef]
- Lee, Y.; Bae, J.W.; Lee, J.W.; Suh, W.; Park, K.D. Enzyme-catalyzed in situ forming gelatin hydrogels as bioactive wound dressings: Effects of fibroblast delivery on wound healing efficacy. J. Mater. Chem. B 2014, 2, 7712–7718. [Google Scholar] [CrossRef]
- Narayani, R.; Rao, K.P. Gelatin microsphere cocktails of different sizes for the controlled release of anticancer drugs. Int. J. Pharm. 1996, 143, 255–258. [Google Scholar] [CrossRef]
- Li, Z.; Li, L.; Liu, Y.; Zhang, H.; Li, X.; Luo, F.; Mei, X. Development of interferon alpha-2b microspheres with constant release. Int. J. Pharm. 2011, 410, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Tabata, Y.; Langer, R. Polyanhydride mierospheres that display near-constant release of water-soluble model drug compounds. Pharm. Res. 1993, 10, 391–399. [Google Scholar] [CrossRef]
- Tabata, Y.; Gutta, S.; Langer, R. Controlled delivery systems for proteins using polyanhydride microspheres. Pharm. Res. 1993, 10, 487–496. [Google Scholar] [CrossRef]
- Yu, D.G.; Li, X.Y.; Wang, X.; Chian, W.; Liao, Y.Z.; Li, Y. Zero-order drug release cellulose acetate nanofibers prepared using coaxial electrospinning. Cellulose 2013, 20, 379–389. [Google Scholar] [CrossRef]
- Berkland, C.; King, M.; Cox, A.; Kim, K.K.; Pack, D.W. Precise control of PLG microsphere size provides enhanced control of drug release rate. J. Control. Release 2002, 82, 137–147. [Google Scholar] [CrossRef]
- Conte, U.; Maggi, L.; Colombo, P.; La Manna, A. Multi-layered hydrophilic matrices as constant release devices (GeomatrixTM Systems). J. Control. Release 1993, 26, 39–47. [Google Scholar] [CrossRef]
- Cheng, X.; Kuhn, L. Chemotherapy drug delivery from calcium phosphate nanoparticles. Int. J. Nanomed. 2007, 2, 667. [Google Scholar]
- McCall, J. Genetic algorithms for modelling and optimisation. J. Comput. Appl. Math. 2005, 184, 205–222. [Google Scholar] [CrossRef]
- Holland, J.H. Adaptation in Natural and Artificial Systems: An Introductory Analysis with Applications to Biology, Control, and Artificial Intelligence; MIT Press: Cambridge, MA, USA, 1992. [Google Scholar]
- Kiran, K.L.; Lakshminarayanan, S. Optimization of chemotherapy and immunotherapy: In silico analysis using pharmacokinetic–pharmacodynamic and tumor growth models. J. Process Control 2013, 23, 396–403. [Google Scholar] [CrossRef]
- Mahasa, K.J.; Eladdadi, A.; De Pillis, L.; Ouifki, R. Oncolytic potency and reduced virus tumor-specificity in oncolytic virotherapy. A mathematical modelling approach. PLoS ONE 2017, 12, e0184347. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Units | Description | Value |
|---|---|---|---|
| day | decay rate of short-term immature DCs | 1.562 | |
| day | decay rate of long-term immature DCs | 0.318 | |
| f | dimensionless | proportion of short-term injected/released DCs | 0.325 |
| DCs/day | gradient of release rate | 9.725 | |
| DCs/day | constant release rate | 1.463 |
| Experiment | |||||
|---|---|---|---|---|---|
| PBS & Gel | Ad/I/G | DC | DC+Ad/I/G | DC+Ad/I/G+Gel | |
| Relevant equations | Equation (1) | Equation (1) | Equation (1) | Equation (1) | Equation (1) |
| Equation (2) | Equation (2) | Equation (2) | |||
| Equation (3) | Equation (3) | Equation (3) | |||
| Equation (5) | Equation (5) | Equation (5) | |||
| Equation (4) | Equation (4) | Equation (4) | |||
| Equation (6) | Equation (6) | ||||
| Equation (7) | Equation (7) | Equation (7) | Equation (7) | ||
| Equation (8) | Equation (8) | Equation (8) | Equation (8) | ||
| Equation (9) | Equation (9) | Equation (9) | Equation (9) | ||
| Variables | U | U,I,V | U | U,I,V | U,I,V |
| A,H | AI,AM, H | AI,AM, H | AI,AM, H | ||
| K | K | K | K | ||
| Parameters fit | r,L,U0 | β,U0 | sAU,U0 | rAI,SAI,U0 | aV,bV,U0 |
| k | k | sAU | |||
| Parameters fixed to Table 1 and Table 3 | - | r,L | r,L | r,L,β | r,L,β |
| dS,dL,dAI | dS,dL,dAI,κ | dS,dL,aD,bD | |||
| dAI,rAI,sAI,k | |||||
| Parameters fixed from [36] | dV,α,sH,dH | sH,dH | dV,α,sH,dH | dV,α,sH,dH | |
| dI,sA,dA | dA | dI,sAU,dA | dI,sAU,dA | ||
| sKH,sKA,dK | sKH,sKA,dK | sKH,sKA,dK | sKH,sKA,dK | ||
| Param | Units | Description | PBS& Gel | Ad/I/G | DC | DC + Ad/I/G | DC + Ad/I/G + gel | |
|---|---|---|---|---|---|---|---|---|
| Fit | day | Immature DCs decay rate | 1.562 | 1.562 | 1.562 | |||
| L | cells | carrying capacity | 13572 | 13572 | 13572 | 13572 | 135720 | |
| r | day | growth rate | 0.1045 | 0.1045 | 0.1045 | 0.1045 | 0.1045 | |
| cells | initial tumour size | 20.35 | 85.90 | 85.90 | 85.90 | 47.45 | ||
| day | infection rate | - | 0.7295 | - | 0.7295 | 0.7295 | ||
| day | killing rate | - | 0.8225 | 0.218147 | 0.3626 | 0.3626 | ||
| day | APC activation rate by U | - | - | 0.001047 | 0.0022 | 0.05278 | ||
| day | recruitment rate of | - | - | - | 0.0192 | 0.0192 | ||
| day | APC activation rate by I | - | - | - | 0.0011 | 0.0011 | ||
| linear release slope (virus) | - | - | - | - | 0.33791 | |||
| initial linear release (virus) | - | - | - | - | 2.35971 | |||
| [36] | virus | viral burst size | - | 3500 | - | 3500 | 3500 | |
| day | burst rate | - | 1 | - | 1 | 1 | ||
| day | viral decay rate | - | 2.3 | - | 2.3 | 2.3 | ||
| day | decay of mature APCs | - | 0.23 | 0.23 | 0.23 | 0.23 | ||
| day | decay of immature APCs | - | 1.562 | 1.562 | 1.562 | 1.562 | ||
| day | decay of helper T cells | - | 0.23 | 0.23 | 0.23 | 0.23 | ||
| day | decay of killer T cells | - | 0.35 | 0.35 | 0.35 | 0.35 | ||
| day | APC activation rate | - | 1.2 | - | 7.1 | 7.1 | ||
| day | APC activatet killer T cell | - | 7.1 | 7.1 | ||||
| day | helper T cell activation | - | 0.78 | 0.78 | 0.78 | 0.78 | ||
| day | helper T cell activate K | - | 1.6 | 1.6 | 1.6 | 1.6 |
| Residual | Coefficient of | Pearson’s Correlation | |
|---|---|---|---|
| Norm | Determination () | Coefficient | |
| PBS and gel | 33,688 | 0.8998 | 0.9487 |
| Ad/IL12/GMCSF | 10,905 | 0.6465 | 0.8041 |
| DC | 11,931 | 0.8980 | 0.9441 |
| DC+Ad/IL12/GMCSF | 7659.4 | 0.7785 | 0.8830 |
| DC+Ad/IL12/GMCSF+gel | 5249.7 | 0.5926 | 0.7736 |
| Constant | Linear (Increasing) | Sigmoidal | |||||
|---|---|---|---|---|---|---|---|
| Ad/IL12/GMCSF | 0.1261 | 0.1252 | 14.9893 | 0.05 | 9.2789 | −6.0499 | |
| DCs | 13.7732 | 13.1629 | 2.8866 | 13.6311 | 9.1951 | −8.3333 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jenner, A.L.; Frascoli, F.; Yun, C.-O.; Kim, P.S. Optimising Hydrogel Release Profiles for Viro-Immunotherapy Using Oncolytic Adenovirus Expressing IL-12 and GM-CSF with Immature Dendritic Cells. Appl. Sci. 2020, 10, 2872. https://doi.org/10.3390/app10082872
Jenner AL, Frascoli F, Yun C-O, Kim PS. Optimising Hydrogel Release Profiles for Viro-Immunotherapy Using Oncolytic Adenovirus Expressing IL-12 and GM-CSF with Immature Dendritic Cells. Applied Sciences. 2020; 10(8):2872. https://doi.org/10.3390/app10082872
Chicago/Turabian StyleJenner, Adrianne L., Federico Frascoli, Chae-Ok Yun, and Peter S. Kim. 2020. "Optimising Hydrogel Release Profiles for Viro-Immunotherapy Using Oncolytic Adenovirus Expressing IL-12 and GM-CSF with Immature Dendritic Cells" Applied Sciences 10, no. 8: 2872. https://doi.org/10.3390/app10082872
APA StyleJenner, A. L., Frascoli, F., Yun, C.-O., & Kim, P. S. (2020). Optimising Hydrogel Release Profiles for Viro-Immunotherapy Using Oncolytic Adenovirus Expressing IL-12 and GM-CSF with Immature Dendritic Cells. Applied Sciences, 10(8), 2872. https://doi.org/10.3390/app10082872