Role of Internal Stress in the Early-Stage Nucleation of Amorphous Calcium Carbonate Gels




Abstract
Featured Application
Abstract
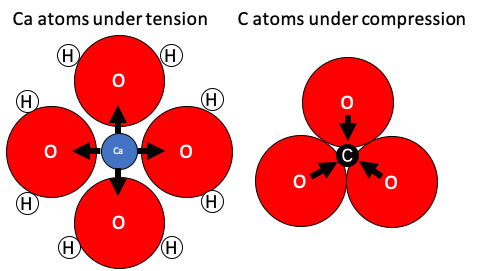
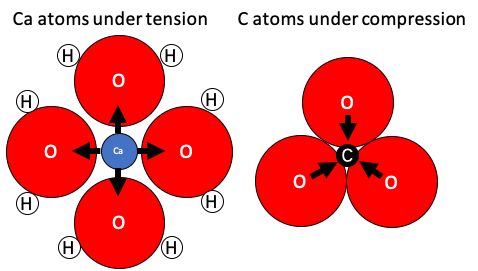{kind=link}
{kind=link}
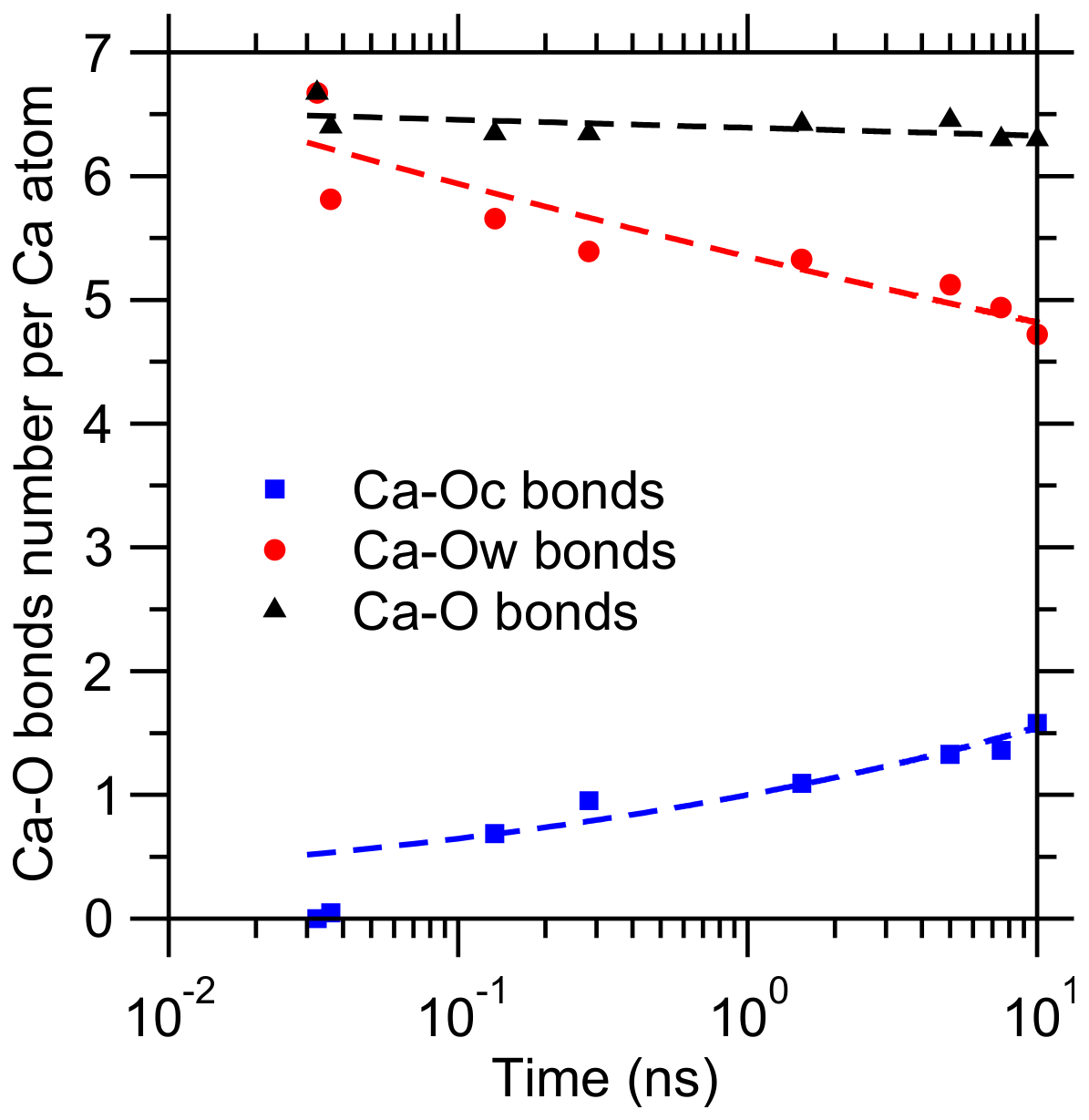{kind=link}
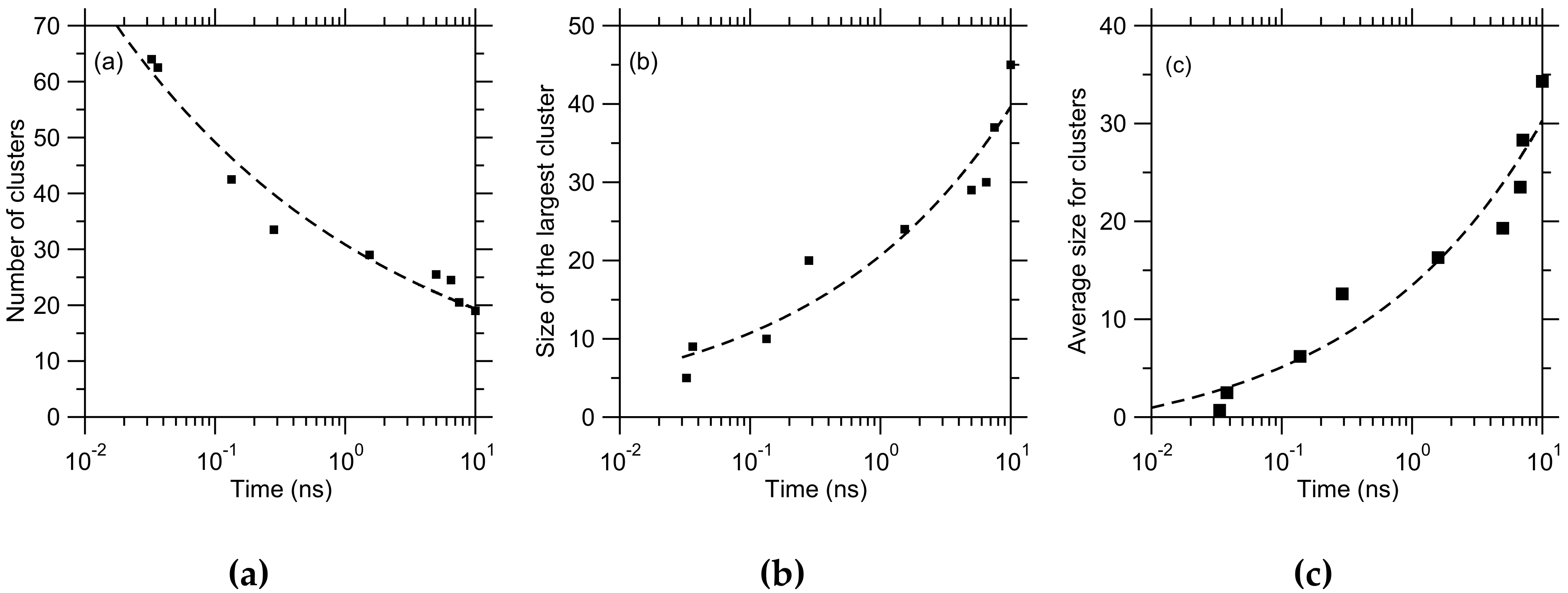{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
2.1. Simulation Methodology
2.2. Structural Analysis
2.3. Computation of the Local Atomic Stress
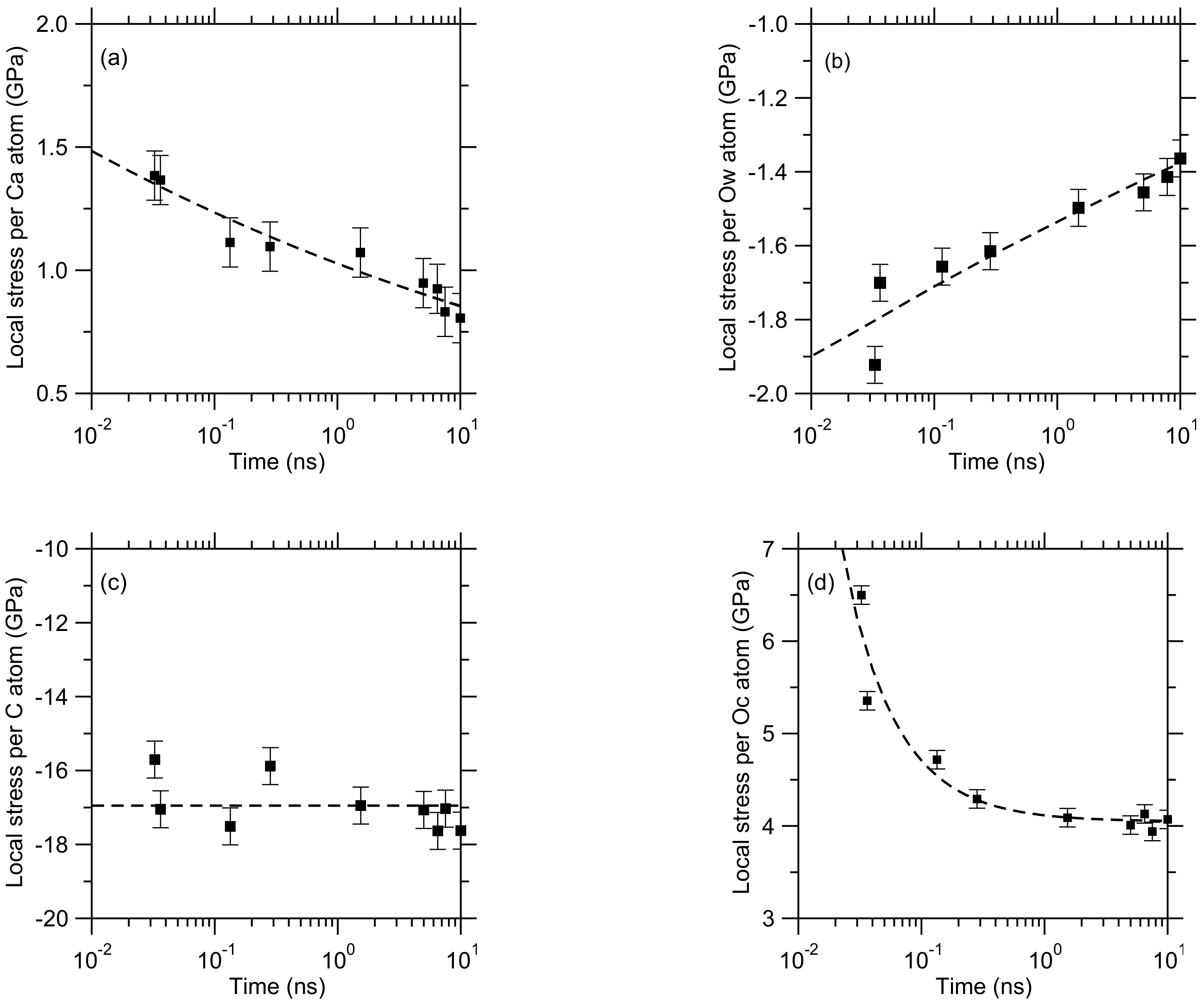
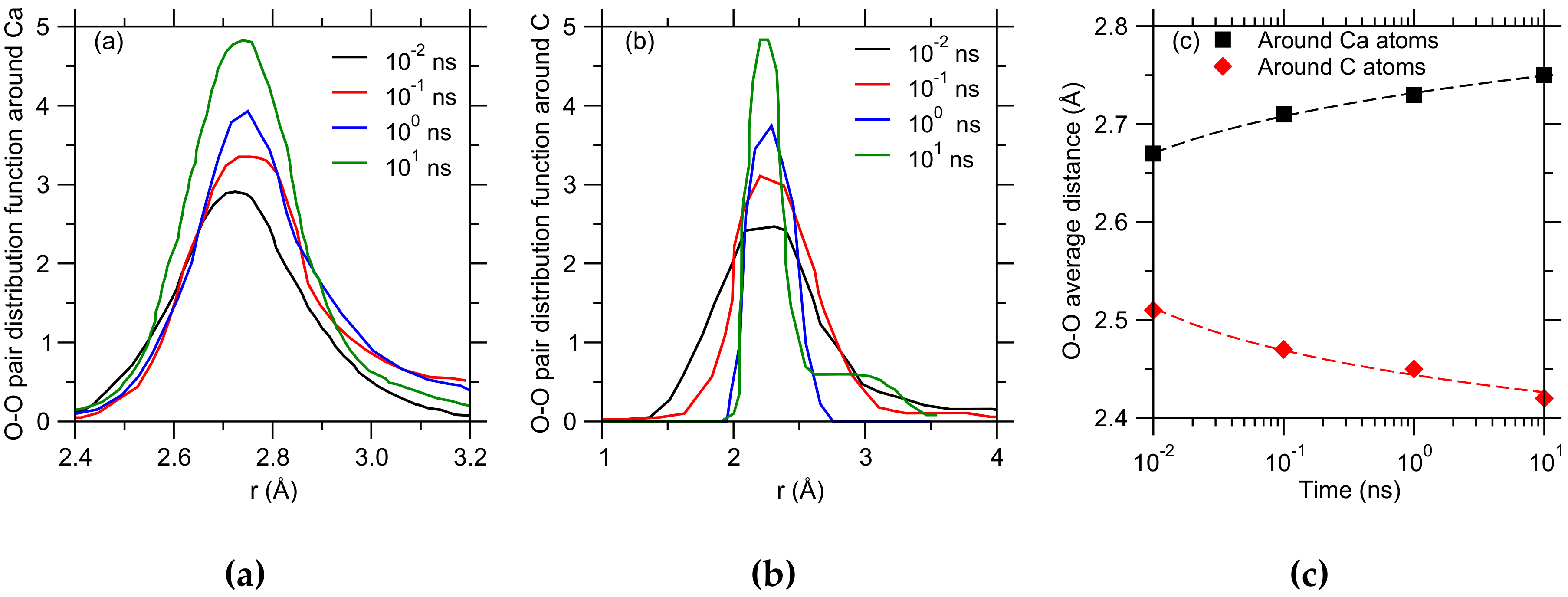
3. Results
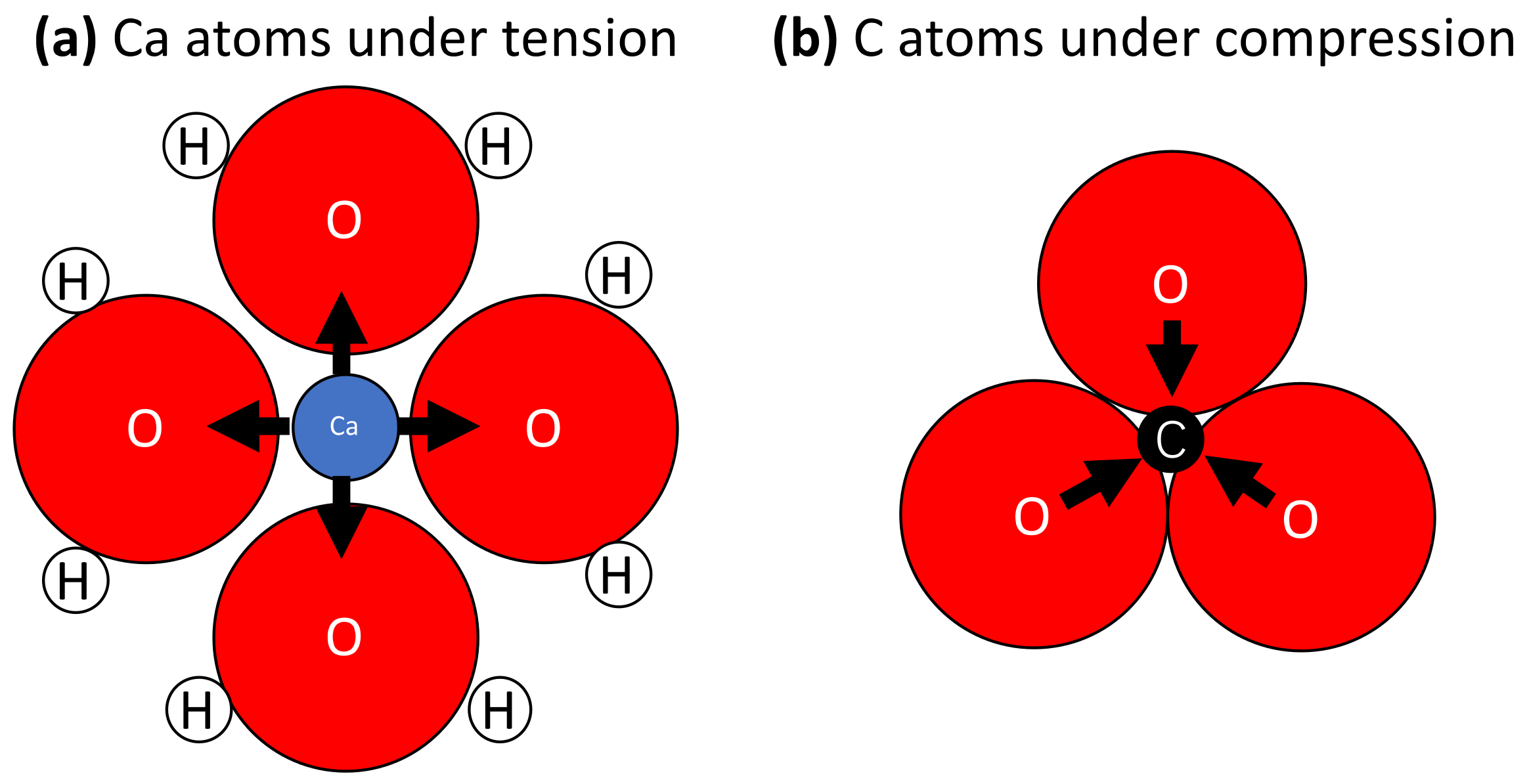
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Demichelis, R.; Raiteri, P.; Gale, J.D.; Quigley, D.; Gebauer, D. Stable prenucleation mineral clusters are liquid-like ionic polymers. Nat. Commun. 2011, 2, 590. [Google Scholar] [CrossRef] [PubMed]
- Mujah, D.; Shahin, M.A.; Cheng, L. State-of-the-Art Review of Biocementation by Microbially Induced Calcite Precipitation (MICP) for Soil Stabilization. Geomicrobiol. J. 2017, 34, 524–537. [Google Scholar] [CrossRef]
- Addadi, L.; Joester, D.; Nudelman, F.; Weiner, S. Mollusk Shell Formation: A Source of New Concepts for Understanding Biomineralization Processes. Chem. Eur. J. 2006, 12, 980–987. [Google Scholar] [CrossRef]
- Committee on Developing a Research Agenda for Utilization of Gaseous Carbon Waste Streams; Board on Chemical Sciences and Technology; Division on Earth and Life Studies; National Academies of Sciences, Engineering, and Medicine. Gaseous Carbon Waste Streams Utilization: Status and Research Needs; National Academies Press: Washington, DC, USA, 2019; ISBN 978-0-309-48336-0. [Google Scholar]
- Vance, K.; Falzone, G.; Pignatelli, I.; Bauchy, M.; Balonis, M.; Sant, G. Direct Carbonation of Ca(OH)2 Using Liquid and Supercritical CO2: Implications for Carbon-Neutral Cementation. Ind. Eng. Chem. Res. 2015, 54, 8908–8918. [Google Scholar] [CrossRef]
- Wei, Z.; Wang, B.; Falzone, G.; La Plante, E.C.; Okoronkwo, M.U.; She, Z.; Oey, T.; Balonis, M.; Neithalath, N.; Pilon, L.; et al. Clinkering-free cementation by fly ash carbonation. J. CO2 Util. 2018, 23, 117–127. [Google Scholar] [CrossRef]
- Wang, Y.-W.; Kim, Y.-Y.; Stephens, C.J.; Meldrum, F.C.; Christenson, H.K. In Situ Study of the Precipitation and Crystallization of Amorphous Calcium Carbonate (ACC). Cryst. Growth Des. 2012, 12, 1212–1217. [Google Scholar] [CrossRef]
- An X-ray absorption spectroscopy study of the structure and transformation of amorphous calcium carbonate from plant cystoliths. Proc. R. Soc. Lond. B Biol. Sci. 1993, 252, 75–80. [CrossRef]
- Ogino, T.; Suzuki, T.; Sawada, K. The formation and transformation mechanism of calcium carbonate in water. Geochim. Cosmochim. Acta 1987, 51, 2757–2767. [Google Scholar] [CrossRef]
- Tribello, G.A.; Bruneval, F.; Liew, C.; Parrinello, M. A Molecular Dynamics Study of the Early Stages of Calcium Carbonate Growth. J. Phys. Chem. B 2009, 113, 11680–11687. [Google Scholar] [CrossRef]
- Michel, F.M.; MacDonald, J.; Feng, J.; Phillips, B.L.; Ehm, L.; Tarabrella, C.; Parise, J.B.; Reeder, R.J. Structural Characteristics of Synthetic Amorphous Calcium Carbonate. Chem. Mater. 2008, 20, 4720–4728. [Google Scholar] [CrossRef]
- Henzler, K.; Fetisov, E.O.; Galib, M.; Baer, M.D.; Legg, B.A.; Borca, C.; Xto, J.M.; Pin, S.; Fulton, J.L.; Schenter, G.K.; et al. Supersaturated calcium carbonate solutions are classical. Sci. Adv. 2018, 4, eaao6283. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, D.; Völkel, A.; Cölfen, H. Stable Prenucleation Calcium Carbonate Clusters. Science 2008, 322, 1819–1822. [Google Scholar] [CrossRef] [PubMed]
- Wallace, A.F.; Hedges, L.O.; Fernandez-Martinez, A.; Raiteri, P.; Gale, J.D.; Waychunas, G.A.; Whitelam, S.; Banfield, J.F.; Yoreo, J.J.D. Microscopic Evidence for Liquid-Liquid Separation in Supersaturated CaCO3 Solutions. Science 2013, 341, 885–889. [Google Scholar] [CrossRef]
- Du, T.; Li, H.; Sant, G.; Bauchy, M. New insights into the sol–gel condensation of silica by reactive molecular dynamics simulations. J. Chem. Phys. 2018, 148, 234504. [Google Scholar] [CrossRef]
- Wang, M.; Smedskjaer, M.M.; Mauro, J.C.; Bauchy, M. Modifier clustering and avoidance principle in borosilicate glasses: A molecular dynamics study. J. Chem. Phys. 2019, 150, 044502. [Google Scholar] [CrossRef] [PubMed]
- Du, T.; Li, H.; Zhou, Q.; Wang, Z.; Sant, G.; Ryan, J.V.; Bauchy, M. Atomistic origin of the passivation effect in hydrated silicate glasses. NPJ Mater. Degrad. 2019, 3, s41529–s019. [Google Scholar] [CrossRef]
- Raiteri, P.; Demichelis, R.; Gale, J.D. Thermodynamically Consistent Force Field for Molecular Dynamics Simulations of Alkaline-Earth Carbonates and Their Aqueous Speciation. J. Phys. Chem. C 2015, 119, 24447–24458. [Google Scholar] [CrossRef]
- Raiteri, P.; Gale, J.D. Water Is the Key to Nonclassical Nucleation of Amorphous Calcium Carbonate. J. Am. Chem. Soc. 2010, 132, 17623–17634. [Google Scholar] [CrossRef]
- Adkins, L.; Cormack, A. Large-scale simulations of sodium silicate glasses. J. Non-Cryst. Solids 2011, 357, 2538–2541. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short–Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, W.; Shiga, M.; Mikami, M. Rapid estimation of elastic constants by molecular dynamics simulation under constant stress. Phys. Rev. B 2004, 69. [Google Scholar] [CrossRef]
- Tuckerman, M.E.; Alejandre, J.; López-Rendón, R.; Jochim, A.L.; Martyna, G.J. A Liouville-operator derived measure-preserving integrator for molecular dynamics simulations in the isothermal–isobaric ensemble. J. Phys. Math. Gen. 2006, 39, 5629–5651. [Google Scholar] [CrossRef]
- Gale, J.; Raiteri, P.; Van Duin, A.C.T. A reactive force field for aqueous-calcium carbonate systems. Phys. Chem. Chem. Phys. 2011, 13, 16666–16679. [Google Scholar] [CrossRef]
- Wang, N.; Feng, Y.; Guo, X.; Van Duin, A.C.T. Insights into the Role of H2O in the Carbonation of CaO Nanoparticle with CO2. J. Phys. Chem. C 2018, 122, 21401–21410. [Google Scholar] [CrossRef]
- Costa, M.F. Molecular dynamics of molten Li2CO3–K2CO3. J. Mol. Liq. 2008, 138, 61–68. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO—the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2010, 18, 015012. [Google Scholar] [CrossRef]
- Vitek, V.; Egami, T. Atomic Level Stresses in Solids and Liquids. Phys. Status Solidi B 1987, 144, 145–156. [Google Scholar] [CrossRef]
- Egami, T. Atomic level stresses. Prog. Mater. Sci. 2011, 56, 637–653. [Google Scholar] [CrossRef]
- Thompson, A.P.; Plimpton, S.J.; Mattson, W. General formulation of pressure and stress tensor for arbitrary many-body interaction potentials under periodic boundary conditions. J. Chem. Phys. 2009, 131, 154107. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, M.; Anoop Krishnan, N.M.; Smedskjaer, M.M.; Deenamma Vargheese, K.; Mauro, J.C.; Balonis, M.; Bauchy, M. Hardness of silicate glasses: Atomic-scale origin of the mixed modifier effect. J. Non-Cryst. Solids 2018, 489, 16–21. [Google Scholar] [CrossRef]
- Li, X.; Song, W.; Smedskjaer, M.M.; Mauro, J.C.; Bauchy, M. Quantifying the internal stress in over-constrained glasses by molecular dynamics simulations. J. Non-Cryst. Solids X 2019, 1, 100013. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, M.; Smedskjaer, M.M.; Mauro, J.C.; Sant, G.; Bauchy, M. Thermometer Effect: Origin of the Mixed Alkali Effect in Glass Relaxation. Phys. Rev. Lett. 2017, 119, 095501. [Google Scholar] [CrossRef]
- Yu, Y.; Mauro, J.C.; Bauchy, M. Stretched exponential relaxation of glasses: Origin of the mixed-alkali effect. Am. Ceram. Soc. Bull. 2017, 96, 34–36. [Google Scholar]
- Yu, Y.; Wang, M.; Zhang, D.; Wang, B.; Sant, G.; Bauchy, M. Stretched Exponential Relaxation of Glasses at Low Temperature. Phys. Rev. Lett. 2015, 115, 165901. [Google Scholar] [CrossRef] [PubMed]
- Ioannidou, K.; Del, G.E.; Ulm, F.-J.; Pellenq Roland, J.-M. Inhomogeneity in Cement Hydrates: Linking Local Packing to Local Pressure. J. Nanomech Micromech 2017, 7, 04017003. [Google Scholar] [CrossRef]
- Mähler, J.; Persson, I. A Study of the Hydration of the Alkali Metal Ions in Aqueous Solution. Inorg. Chem. 2012, 51, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Bismayer, U.; Epple, M.; Fabritius, H.; Hasse, B.; Shi, J.; Ziegler, A. Structural characterisation of X-ray amorphous calcium carbonate (ACC) in sternal deposits of the crustacea Porcellio scaber. Dalton Trans. 2003, 551–555. [Google Scholar] [CrossRef]
- Levi-Kalisman, Y.; Raz, S.; Weiner, S.; Addadi, L.; Sagi, I. Structural Differences Between Biogenic Amorphous Calcium Carbonate Phases Using X-ray Absorption Spectroscopy. Adv. Funct. Mater. 2002, 12, 43–48. [Google Scholar] [CrossRef]
- Avaro, J.; Moon, E.M.; Rose, J.; Rose, A.L. Calcium coordination environment in precursor species to calcium carbonate mineral formation. Geochim. Cosmochim. Acta 2019, 259, 344–357. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Zhao, C.; Zhou, W.; Zhou, Q.; Zhang, Y.; Liu, H.; Sant, G.; Liu, X.; Guo, L.; Bauchy, M. Precipitation of Calcium–Alumino–Silicate–Hydrate Gels: The Role of the Internal Stress. J. Chem. Phys. 2020, 124, 105806. [Google Scholar]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Q.; Du, T.; Guo, L.; Sant, G.; Bauchy, M. Role of Internal Stress in the Early-Stage Nucleation of Amorphous Calcium Carbonate Gels. Appl. Sci. 2020, 10, 4359. https://doi.org/10.3390/app10124359
Zhou Q, Du T, Guo L, Sant G, Bauchy M. Role of Internal Stress in the Early-Stage Nucleation of Amorphous Calcium Carbonate Gels. Applied Sciences. 2020; 10(12):4359. https://doi.org/10.3390/app10124359
Chicago/Turabian StyleZhou, Qi, Tao Du, Lijie Guo, Gaurav Sant, and Mathieu Bauchy. 2020. "Role of Internal Stress in the Early-Stage Nucleation of Amorphous Calcium Carbonate Gels" Applied Sciences 10, no. 12: 4359. https://doi.org/10.3390/app10124359
APA StyleZhou, Q., Du, T., Guo, L., Sant, G., & Bauchy, M. (2020). Role of Internal Stress in the Early-Stage Nucleation of Amorphous Calcium Carbonate Gels. Applied Sciences, 10(12), 4359. https://doi.org/10.3390/app10124359