Voltammetric and Spectroscopic Determination of Rare Earth Elements in Fresh and Surface Water Samples

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Water Sampling
2.2. Analysis of REEs in Water Samples
3. Results and Discussion
3.1. DPAdSV Analysis
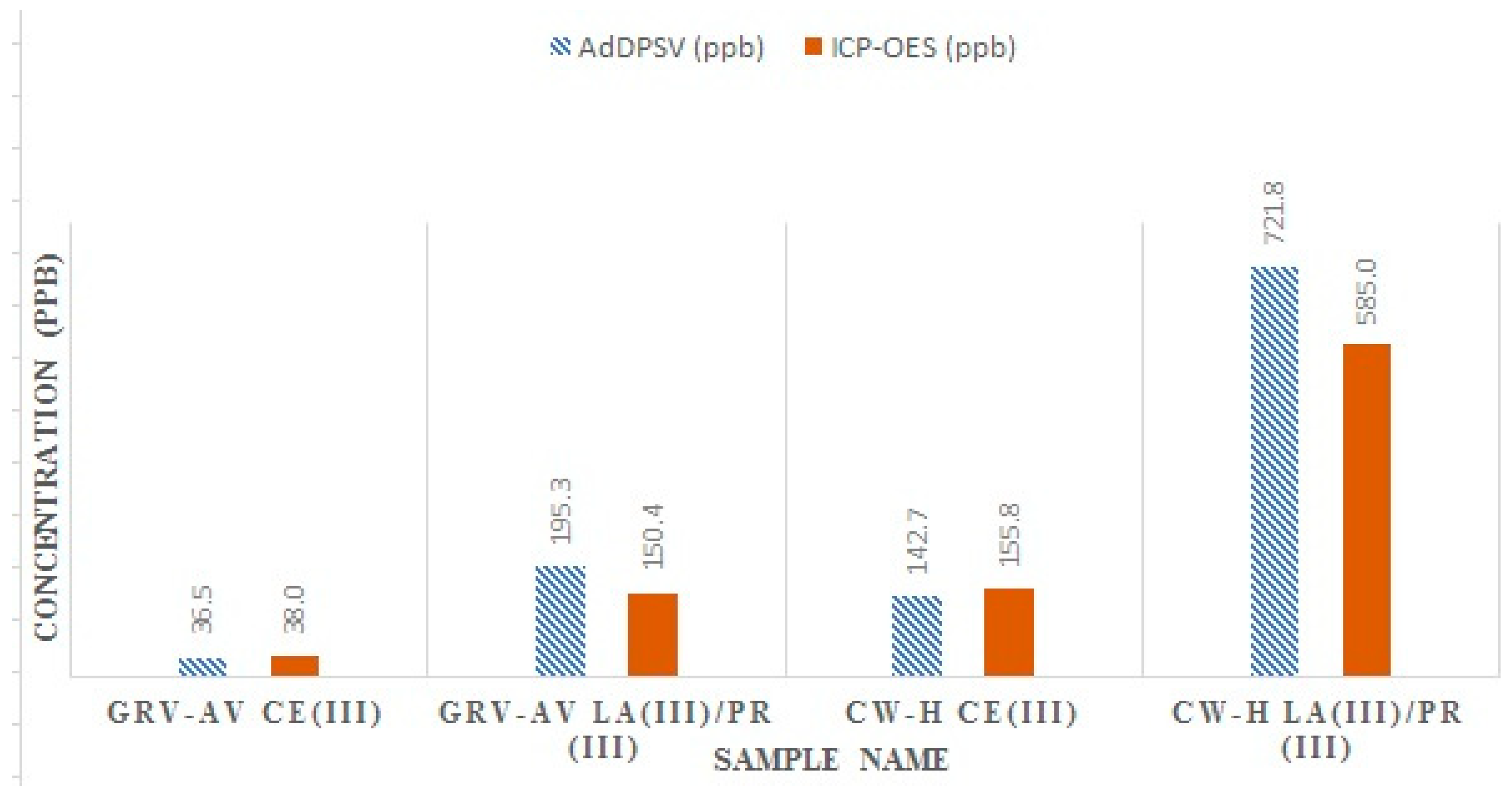
3.2. ICP-OES Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Iwashita, M.; Saito, A.; Arai, M.; Furusho, Y.; Shimamura, T. Determination of rare earth elements in water collected in suburban Tokyo. Geochem. J. 2011, 45, 187–197. [Google Scholar] [CrossRef]
- Norgate, T.E.; Jahanshahi, S.; Rankin, W.J. Assessing the environmental impact of metal production processes. J. Clean. Prod. 2007, 15, 838–848. [Google Scholar] [CrossRef]
- Castor, S.B.; Hedrick, J.B. Rare Earth Elements. In Industrial Minerals and Rocks—Commodities Markets and Uses, 7th ed.; Kogel, J.E., Trivedi, N.C., Barker, J.M., Krukowski, S.T., Eds.; Society for Mining, Metallurgy and Exploration, Inc. (SME): Englewood, CO, USA, 2006; pp. 769–792. [Google Scholar]
- Ali, S.H. Social and Environmental Impact of the Rare Earth Industries. Resources 2014, 3, 123–134. [Google Scholar] [CrossRef]
- Shyam, I.R.; Aery, I.C. Effect of cerium on growth, dry matter production, biochemical constituents and enzymatic activities of cowpea plants [Vigna unguiculata (L.) Walp]. J. Soil Sci. Plant Nutr. 2012, 12, 1–14. [Google Scholar] [CrossRef]
- Zhenggui, W.; Ming, Y.; Xun, Z.; Fashui, H.; Bing, L.; Ye, T.; Guiwen, Z.; Chunhua, Y. Rare earth elements in naturally grown fern-Dicranopteris linearis in relation to their variation in soils in South-Jiangxi region (Southern China). Environ. Pollut. 2001, 114, 345–355. [Google Scholar] [CrossRef]
- Paul, J.; Campbell, G. Investigating Rare Earth Element Mine, Development in EPA Region 8 and Potential Environmental Impacts; United States Environmental Protection Agency, (USEPA): Washington, DC, USA, 2011; pp. 1–8.
- Pang, X.; Li, D.; Peng, A. Application of rare earth elements in the agriculture of China and its environment behaviour in soil. J. Soils Sediments 2001, 1, 124–129. [Google Scholar] [CrossRef]
- Emmanuel, E.C.; Ananthi, T.; Anandkumar, B.; Maruthamuthu, S. Accumulation of rare earth elements by Siderophore forming Arthrobacter lutecolus isolated from rare earth environment of Chavara India. J. Biosci. 2012, 37, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Turra, C.; Fernandes, E.A.N.; Bacchi, M.A. Evaluation on rare earth elements of Brazilian agricultural supplies. J. Environ. Chem. Ecotoxicol. 2011, 3, 86–92. [Google Scholar]
- Cheng, H.; Hao, F.; Ouyang, W.; Liu, S.; Chunye, L.I.N.; Wenjing, Y. Vertical distribution of rare earth elements in a wetland soil core from the Sanjiang Plain in China. J. Rare Earths 2012, 30, 731–738. [Google Scholar] [CrossRef]
- Chunye, L.; Mengchang, H.; Yanxia, L.; Linsheng, Y.; Ruimin, L.; Zhifeng, Y. Rare earth element content in the SPM of Daliao river system and its comparison with that in the sediments, loess and soils in China. J. Rare Earths 2008, 26, 414–420. [Google Scholar]
- Tao, L.; Shiming, D.; Wenchong, S.; Zhongyi, C.; Chaosheng, Z. A review of fractionations of rare earth elements in plants. J. Rare Earths 2008, 26, 7–15. [Google Scholar]
- Fernandez-Caliani, J.C.; Barba-Brioso, C.; De La Rosa, J.D. Mobility and separation of rare earth elements in acid mine soils and geochemical implications for river waters in the southwestern Iberian margin. Geoderma 2009, 149, 393–401. [Google Scholar] [CrossRef]
- Border, P. Postnote, POST-PN-392, Livestock Diseases, October 2011, London, UK. Available online: https://www.slideshare.net/isidorimasalu/livestock-diseases-post-pn392 (accessed on 5 July 2018).
- Jinxia, L.; Mei, H.; Xiuqin, Y.; Jiliang, L. Effects of the accumulation of the rare earth elements on soil macrofauna community. J. Rare Earths 2010, 28, 657–964. [Google Scholar]
- Wood, S.A.; Shannon, W.M. Rare-earth elements in geothermal waters from Oregon, Nevada, and California. J. Solid State Chem. 2003, 171, 246–253. [Google Scholar] [CrossRef]
- Tranchida, G.; Oliveri, E.; Angelone, M.; Bellanca, A.; Gensi, P.; D’Elia, M.; Neri, R.; Placenti, F.; Sprovieri, M.; Mazzola, S. Distribution of rare earth elements in marine sediments from the strait of Sicily (Western Mediterranean Sea): Evidence of phosphogypsum waste contamination. Mar. Pollut. Bull. 2011, 62, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Tighe, M.; Lockwood, P.; Wilson, S.; Lisle, L. Comparison of digestion methods for ICP-OES analysis of wide range of analytes in heavy metal contaminated soil samples with specific reference to Arsenic and Antimony. Commun. Soil Sci. Plant Anal. 2004, 35, 1369–1385. [Google Scholar] [CrossRef]
- Makombe, M.; Van der Horst, C.; Silwana, B.; Iwuoha, E.; Somerset, V. Antimony film sensor for sensitive rare earth metal analysis in environmental samples. J. Environ. Sci. Health Part A 2016, 51, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Makombe, M.; Van der Horst, C.; Silwana, B.; Iwuoha, E.; Somerset, V. Optimisation of Parameters for Spectroscopic Analysis of Rare Earth Elements in Sediment Samples. In Rare Earth Element; Orjuela, J.E.A., Ed.; INTECH: Rijeka, Croatia, 2017; pp. 63–81. [Google Scholar]
- Van der Horst, C.; Silwana, B.; Iwuoha, E.; Somerset, V. Stripping voltammetric determination of palladium, platinum and rhodium in South African water resources. J. Environ. Sci. Health Part A 2012, 47, 2084–2093. [Google Scholar] [CrossRef] [PubMed]
- Silwana, B.; Van der Horst, C.; Iwuoha, E.; Somerset, V. Screen-printed electrodes modified with a bismuth film for stripping voltammetric analysis of platinum group metals in environmental samples. Electrochim. Acta 2014, 128, 119–127. [Google Scholar] [CrossRef]
- Somerset, V.; Silwana, B.; Van der Horst, C.; Iwuoha, E. Construction and Evaluation of a Carbon Paste Electrode Modified with Polyaniline-co-poly(dithiodianiline) for Enhanced Stripping Voltammetric Determination of Metals Ions. In Sensing Electroanalysis; Kalcher, K., Metelka, R., Švancara, I., Vytřas, K., Eds.; University Press Centre Pardubice: Pardubice, Czech Republic, 2014; pp. 143–154. [Google Scholar]
- Zawisza, B.; Pytlakowska, K.; Feist, B.; Polowniak, M.; Kita, A.; Sitko, R. Determination of rare earth elements by spectroscopic techniques: A review. J. Anal. At. Spectrom. 2011, 26, 2373–2390. [Google Scholar] [CrossRef]
- Brown, R.J.C.; Milton, M.J. Analytical techniques for trace element analysis: An overview. Trends Anal. Chem. 2005, 24, 266–274. [Google Scholar] [CrossRef]
- Somerset, V.; Van der Horst, C.; Silwana, B.; Iwuoha, E. Biomonitoring and evaluation of metal concentrations in sediment and crab samples from the North-West Province of South Africa. Water Air Soil Pollut. 2015, 226, 43. [Google Scholar] [CrossRef]
- Greaves, M.J.; Elderfield, H.; Klinkhammer, G.P. Determination of the rare earth elements in natural waters by isotope-dilution mass spectrometry. Anal. Chim. Acta 1989, 218, 265–280. [Google Scholar] [CrossRef]
- Whitely, J.D.; Murray, F. Anthropogenic platinum group element (Pt, Pd and Rh) concentrations in road dusts and roadside soils from Perth, Western Australia. Sci. Total Environ. 2003, 317, 121–135. [Google Scholar] [CrossRef]
- Elemental Analysis—Spectrometers. Available online: www.spectro.com (accessed on 18 May 2017).
- Protano, G.; Riccobono, F. High contents of rare earth elements (REEs) in stream waters of a Cu-Pb-Zn mining area. Environ. Pollut. 2002, 117, 499–514. [Google Scholar] [CrossRef]
- Javanbakht, M.; Khoshafar, A.; Ganjali, M.R.; Norouzi, P.; Adib, M. Adsorptive stripping voltammetric determination of nanomolar concentration of cerium(III) at carbon paste modified by N′-[(2-Hydroxyphenyl) Methylidene]-2-Furohydrazide. Electroanalysis 2009, 21, 1605–1610. [Google Scholar] [CrossRef]
- Boumans, P.W.J.M. Measuring detection limits in inductively coupled plasma emission spectrometry using the SBR—RSDB approach: I. A tutorial discussion of the theory. Spectrochim. Acta B 1991, 46, 431–445. [Google Scholar] [CrossRef]
- Boumans, P.W.J.M.; Tielrooy, J.A.; Maessen, F.J.M.J. Mutual spectral interferences of rare earth elements in inductively coupled plasma atomic emission spectrometry: I. Rational line selection and correction procedure. Spectrochimica Acta B 1998, 43, 173–199. [Google Scholar] [CrossRef]
- Sturman, B.T.; Doidge, P.S. Semi–empirical approach to the calculation of instrumental detection limits in inductively coupled plasma atomic emission spectrometry. J. Anal. At. Spectrom. 1998, 13, 69–74. [Google Scholar] [CrossRef]
- Khoo, S.B.; Zhu, J. Poly(catechol) film modified glassy carbon electrode for ultra-trace determination of cerium (III) by differential pulse anodic stripping voltammetry. Electroanalysis 1999, 11, 546–552. [Google Scholar] [CrossRef]
- Li, J.; Liu, S.; Yan, Z.; Mao, X.; Gao, P. Adsorptive Voltammetric Studies on the Cerium(III)–Alizarin Complexon Complex at a Carbon Paste Electrode. Microchim. Acta 2006, 154, 241–251. [Google Scholar] [CrossRef]
- Li, J.; Liu, S.; Mao, X.; Gao, P.; Yan, Z. Trace determination of rare earths by adsorption voltammetry at carbon paste electrode. J. Electroanal. Chem. 2004, 561, 137–142. [Google Scholar] [CrossRef]
- Lee, S.K.; Chung, T.D.; Kim, H. Indirect voltammetric determination of lanthanides in the presence of mordant Red 19. Electroanalysis 1997, 9, 527–532. [Google Scholar] [CrossRef]
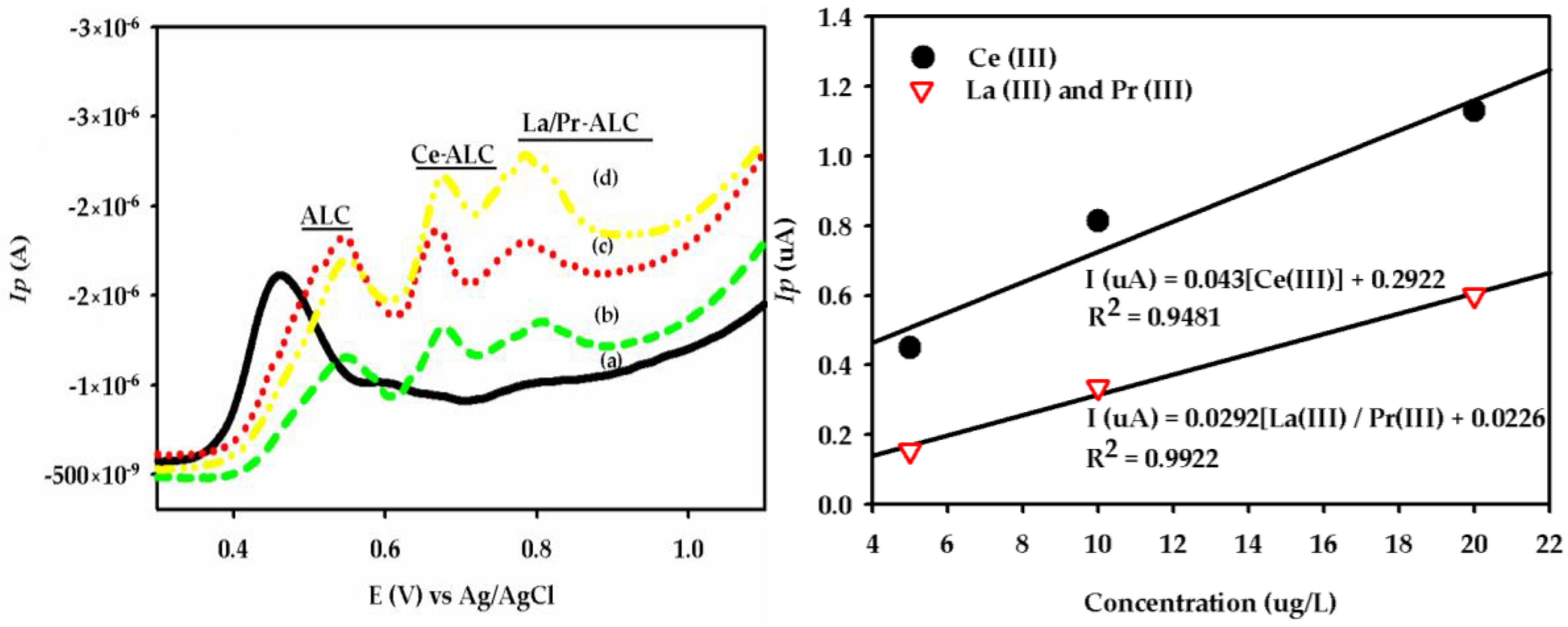

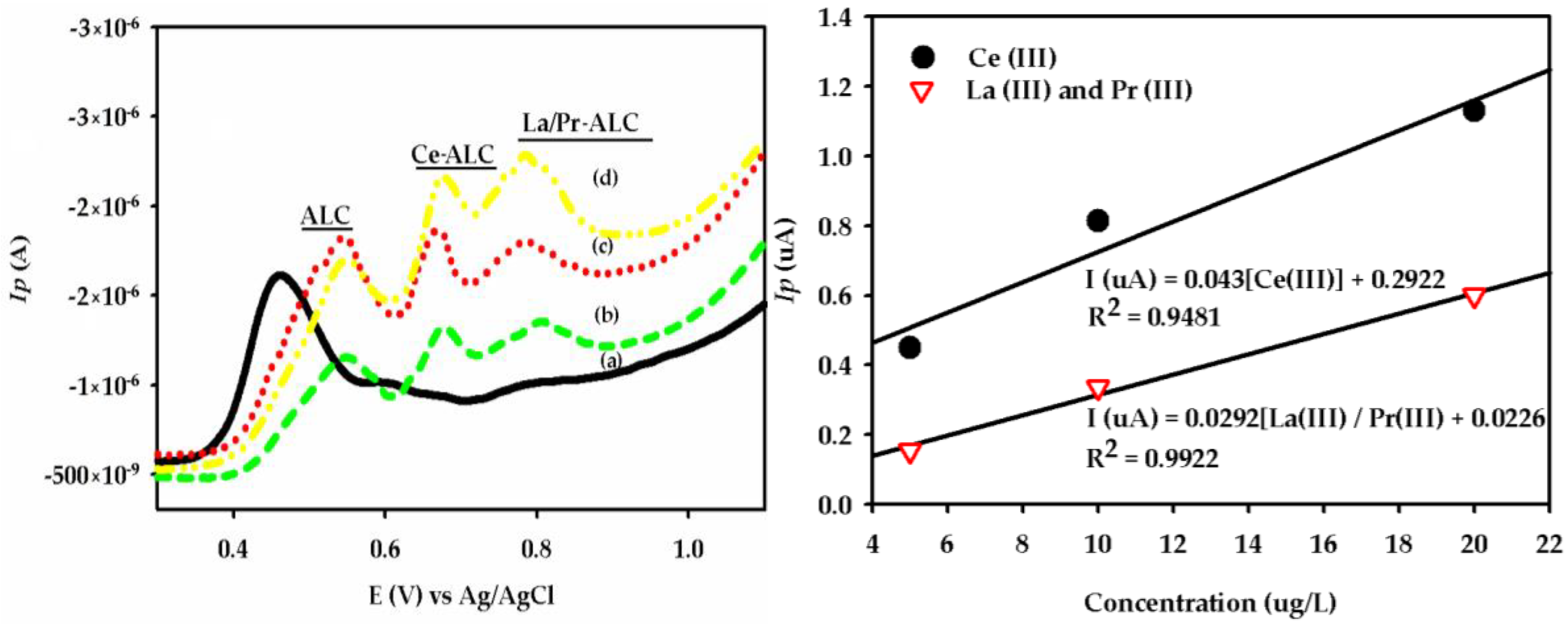{kind=link}
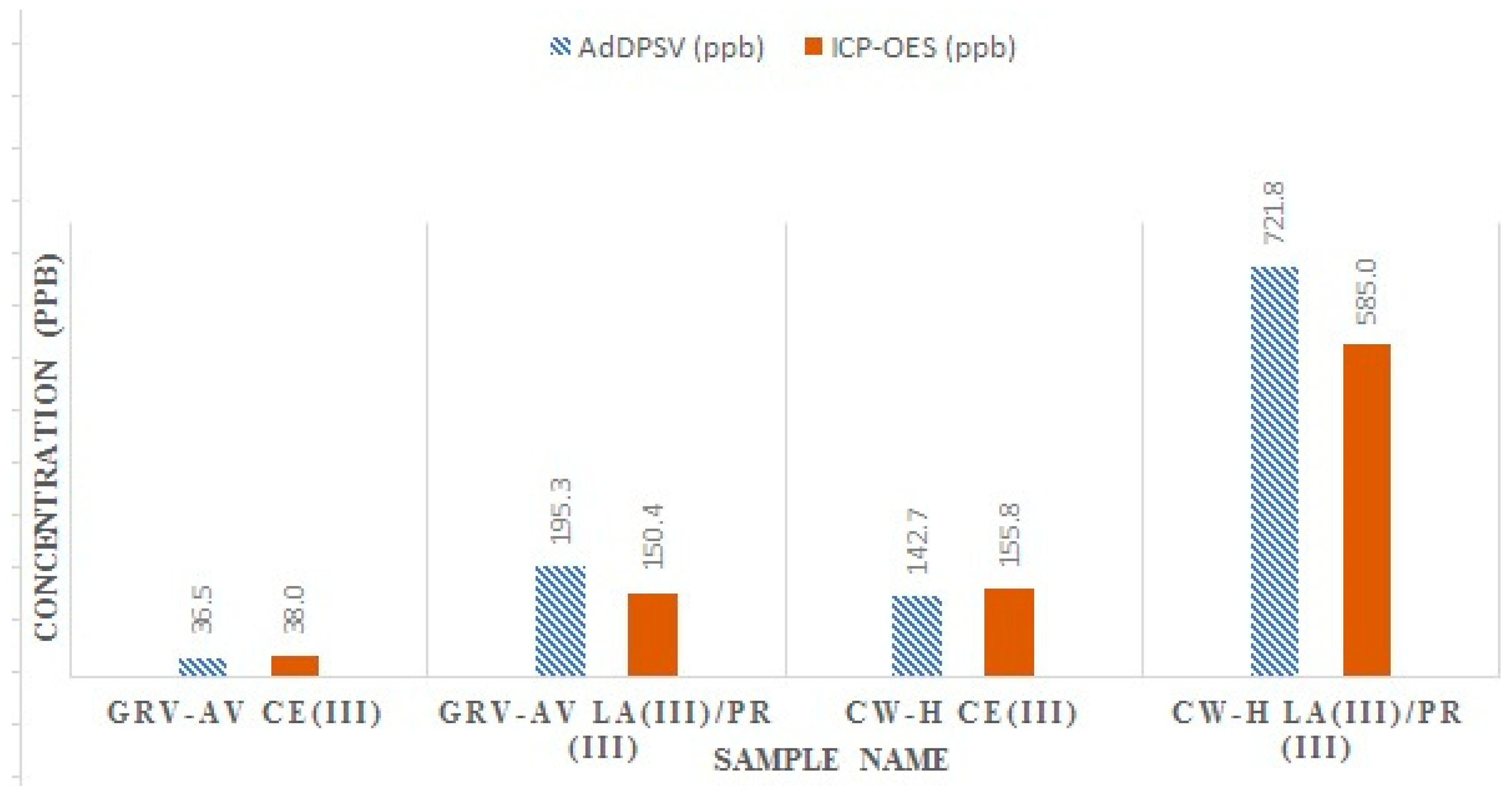{kind=link}
| Instrument Parameter | Condition |
|---|---|
| Plasma power (W) | 1400 |
| Pump speed (rpm) | 30 |
| Coolant flow (L/min) | 14.00 |
| Auxiliary flow(L/min) | 2.10 |
| Nebuliser flow (L/min) | 0.80 |
| Nebuliser | Crossflow |
| Torch Position | 0 |
| Stripping Step | Condition REE [Ce(III), La(III) and Pr(III)] |
|---|---|
| pH | 5.8 |
| Reduction potential (V) | −0.1 |
| Deposition time (s) | 360 |
| Supporting electrolyte | 0.08 M NaOAc |
| Measurement technique | DPAdSV |
| Potential window | −0.2 to +1.2 V |
| Complexing agent | Alizarin Complexione (ALC) |
| Site | Metal Ions | |
|---|---|---|
| Ce(III) (µg/L) | La(III) (µg/L) + Pr(III) (µg/L) | |
| W-M | ND | ND |
| CW-H | 142.7 ± 5.7 | 721.8 ± 16.7 |
| GRV-AV | 36.5 ± 0.6 | 195.3 ± 1.5 |
| Site ID | Metal Ions | ||
|---|---|---|---|
| Ce(III) (µg/L) | La(III) (µg/L) | Pr(III) (µg/L) | |
| W-M | ND | ND | ND |
| CW-H | 155.8 ± 1.5 | 298.0 ± 1.6 | 287.0 ± 5.5 |
| GRV-AV | 38.0 ± 1.2 | 68.0 ± 1.9 | 82.4 ± 5.2 |
| Method | Metal Ion | R2 | RSD (%) | LOQ (µg/L) | LOD (µg/L) | References |
|---|---|---|---|---|---|---|
| GCE/PC | Ce(III) | 3.4 | 0.03 | [36] | ||
| CPE/NHMF | Ce(III) | 0.9982 | 5.6 | 0.11 | [32] | |
| CPE/ALC | Ce(III) | 3.5 | 0.27 | [37] | ||
| GCE/SbF | Ce(III) | 0.9938 | 4.0 | 0.19 | 0.06 | [20] |
| La(III) | 0.9928 | 5.0 | 1.42 | 0.42 | ||
| Pr(III) | 0.9888 | 5.0 | 2.43 | 0.71 | ||
| GCE/SbF | Ce(III) | 0.9481 | 5.2 | 2.12 | 0.10 | This work |
| La(III) + Pr(III) | 0.9922 | 4.4 | 10.42 | 2.10 | ||
| ICP-OES | Ce | 1.0000 | 2.9 | 7.61 | 2.45 | This work |
| La | 1.0000 | 5.3 | 4.63 | 3.12 | ||
| Pr | 0.9998 | 4.7 | 16.23 | 3.90 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makombe, M.; Van der Horst, C.; Silwana, B.; Iwuoha, E.; Somerset, V. Voltammetric and Spectroscopic Determination of Rare Earth Elements in Fresh and Surface Water Samples. Environments 2018, 5, 112. https://doi.org/10.3390/environments5100112
Makombe M, Van der Horst C, Silwana B, Iwuoha E, Somerset V. Voltammetric and Spectroscopic Determination of Rare Earth Elements in Fresh and Surface Water Samples. Environments. 2018; 5(10):112. https://doi.org/10.3390/environments5100112
Chicago/Turabian StyleMakombe, Martin, Charlton Van der Horst, Bongiwe Silwana, Emmanuel Iwuoha, and Vernon Somerset. 2018. "Voltammetric and Spectroscopic Determination of Rare Earth Elements in Fresh and Surface Water Samples" Environments 5, no. 10: 112. https://doi.org/10.3390/environments5100112
APA StyleMakombe, M., Van der Horst, C., Silwana, B., Iwuoha, E., & Somerset, V. (2018). Voltammetric and Spectroscopic Determination of Rare Earth Elements in Fresh and Surface Water Samples. Environments, 5(10), 112. https://doi.org/10.3390/environments5100112