Developmental Programming of PCOS Traits: Insights from the Sheep



Abstract
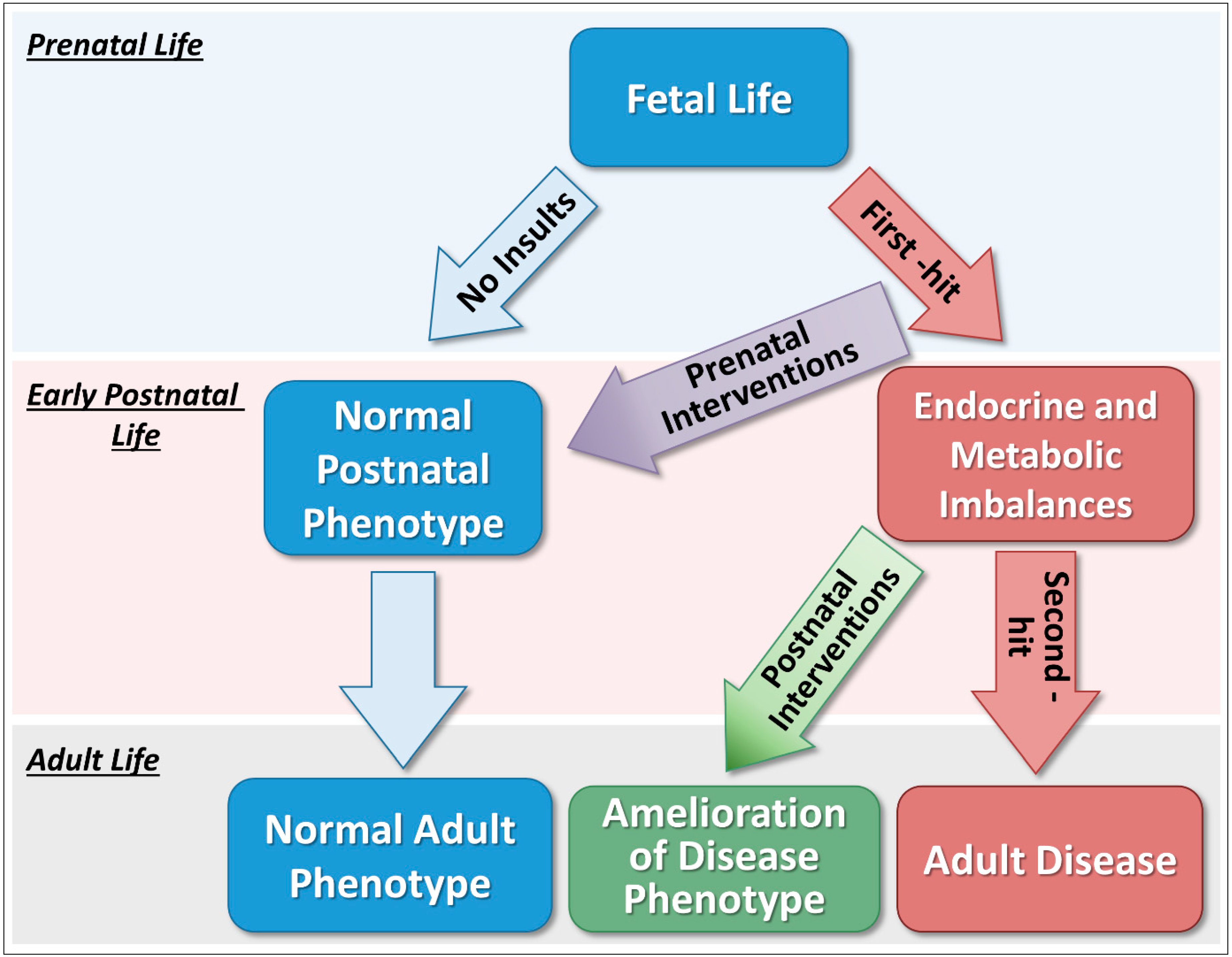
:1. Introduction
2. Sheep Model of Polycystic Ovary Syndrome Phenotype
3. Effects of Prenatal Interventions on Reproductive and Metabolic Phenotypes
4. Effects of Postnatal Interventions on Reproductive and Metabolic Phenotypes
5. Conclusions
Funding
Acknowledgements
Conflicts of Interest
References
- Boivin, J.; Bunting, L.; Collins, J.A.; Nygren, K.G. International estimates of infertility prevalence and treatment-seeking: Potential need and demand for infertility medical care. Hum. Reprod. 2007, 22, 1506–1512. [Google Scholar] [CrossRef] [PubMed]
- Azziz, R.; Woods, K.S.; Reyna, R.; Key, T.J.; Knochenhauer, E.S.; Yildiz, B.O. The prevalence and features of the polycystic ovary syndrome in an unselected population. J. Clin. Endocrinol. Metab. 2004, 89, 2745–2749. [Google Scholar] [CrossRef] [PubMed]
- Fauser, B.C.; Tarlatzis, B.C.; Rebar, R.W.; Legro, R.S.; Balen, A.H.; Lobo, R.; Carmina, E.; Chang, J.; Yildiz, B.O.; Laven, J.S. Consensus on women’s health aspects of polycystic ovary syndrome (PCOS): The Amsterdam ESHRE/ASRM-Sponsored 3rd PCOS Consensus Workshop Group. Fertil. Steril. 2012, 97, 28–38.e25. [Google Scholar] [CrossRef] [PubMed]
- Diamanti-Kandarakis, E.; Dunaif, A. Insulin resistance and the polycystic ovary syndrome revisited: An update on mechanisms and implications. Endocr. Rev. 2012, 33, 981–1030. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.R.; Goodarzi, M.O. Genetic determinants of polycystic ovary syndrome: Progress and future directions. Fertil. Steril. 2016, 106, 25–32. [Google Scholar] [CrossRef]
- Azziz, R. Introduction: Determinants of polycystic ovary syndrome. Fertil. Steril. 2016, 106, 4–5. [Google Scholar] [CrossRef]
- Xita, N.; Tsatsoulis, A. Fetal programming of polycystic ovary syndrome by androgen excess: Evidence from experimental, clinical, and genetic association studies. J. Clin. Endocrinol. Metab. 2006, 91, 1660–1666. [Google Scholar] [CrossRef]
- Abbott, D.H.; Dumesic, D.A.; Eisner, J.R.; Colman, R.J.; Kemnitz, J.W. Insights into the development of polycystic ovary syndrome (PCOS) from studies of prenatally androgenized female rhesus monkeys. Trends Endocrinol. Metab. 1998, 9, 62–67. [Google Scholar] [CrossRef]
- Barnes, R.B.; Rosenfield, R.L.; Ehrmann, D.A.; Cara, J.F.; Cuttler, L.; Levitsky, L.L.; Rosenthal, I.M. Ovarian hyperandrogynism as a result of congenital adrenal virilizing disorders: Evidence for perinatal masculinization of neuroendocrine function in women. J. Clin. Endocrinol. Metab. 1994, 79, 1328–1333. [Google Scholar]
- Sir-Petermann, T.; Codner, E.; Pérez, V.; Echiburú, B.; Maliqueo, M.; Ladrón de Guevara, A.; Preisler, J.; Crisosto, N.; Sánchez, F.; Cassorla, F. Metabolic and reproductive features before and during puberty in daughters of women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2009, 94, 1923–1930. [Google Scholar] [CrossRef]
- Maliqueo, M.; Echiburú, B.; Crisosto, N.; Amigo, P.; Aranda, P.; Sánchez, F.; Sir-Petermann, T. Metabolic parameters in cord blood of newborns of women with polycystic ovary syndrome. Fertil. Steril. 2009, 92, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Sir-Petermann, T.; Maliqueo, M.; Codner, E.; Echiburú, B.; Crisosto, N.; Pérez, V.; Pérez-Bravo, F.; Cassorla, F. Early metabolic derangements in daughters of women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2007, 92, 4637–4642. [Google Scholar] [CrossRef] [PubMed]
- Sir-Petermann, T.; Maliqueo, M.; Angel, B.; Lara, H.; Perez-Bravo, F.; Recabarren, S.E. Maternal serum androgens in pregnant women with polycystic ovarian syndrome: Possible implications in prenatal androgenization. Hum. Reprod. 2002, 17, 2573–2579. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J. The origins of the developmental origins theory. J. Intern. Med. 2007, 261, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, G.P.; Stein, Z.A.; Susser, M.W. Obesity in young men after famine exposure in utero and early infancy. N. Engl. J. Med. 1976, 295, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Barker, D. The developmental origins of adult disease. J. Am. Coll. Nutr. 2004, 23, 588S–595S. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Hanson, M.A. Developmental origins of disease paradigm: A mechanistic and evolutionary perspective. Pediatric Res. 2004, 56, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Nijland, M.J.; Ford, S.P.; Nathanielsz, P.W. Prenatal origins of adult disease. Curr. Opin. Obstet. Gynecol. 2008, 20, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, V.; Cardoso, R.C.; Puttabyatappa, M. Developmental programming, a pathway to disease. Endocrinology 2016, 157, 1328–1340. [Google Scholar] [CrossRef]
- Tang, W.Y.; Ho, S.M. Epigenetic reprogramming and imprinting in origins of disease. Rev. Endocr. Metab. Disord. 2007, 8, 173–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puttabyatappa, M.; Cardoso, R.C.; Padmanabhan, V. Effect of maternal PCOS and PCOS-like phenotype on the offspring’s health. Mol. Cell Endocrinol. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, V.; Veiga-Lopez, A. Animal models of the polycystic ovary syndrome phenotype. Steroids 2013, 78, 734–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padmanabhan, V.; Veiga-Lopez, A. Sheep models of polycystic ovary syndrome phenotype. Mol. Cell. Endocrinol. 2013, 373, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, R.C.; Padmanabhan, V. Prenatal Steroids and Metabolic Dysfunction: Lessons from Sheep. Annu. Rev. Anim. Biosci. 2019, 7, 337–360. [Google Scholar] [CrossRef] [PubMed]
- Birch, R.A.; Padmanabhan, V.; Foster, D.L.; Unsworth, W.P.; Robinson, J.E. Prenatal programming of reproductive neuroendocrine function: Fetal androgen exposure produces progressive disruption of reproductive cycles in sheep. Endocrinology 2003, 144, 1426–1434. [Google Scholar] [CrossRef] [PubMed]
- Sarma, H.N.; Manikkam, M.; Herkimer, C.; Dell’Orco, J.; Welch, K.B.; Foster, D.L.; Padmanabhan, V. Fetal programming: Excess prenatal testosterone reduces postnatal luteinizing hormone, but not follicle-stimulating hormone responsiveness, to estradiol negative feedback in the female. Endocrinology 2005, 146, 4281–4291. [Google Scholar] [CrossRef] [PubMed]
- Sharma, T.P.; Herkimer, C.; West, C.; Ye, W.; Birch, R.; Robinson, J.E.; Foster, D.L.; Padmanabhan, V. Fetal programming: Prenatal androgen disrupts positive feedback actions of estradiol but does not affect timing of puberty in female sheep. Biol. Reprod. 2002, 66, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Steckler, T.L.; Herkimer, C.; Dumesic, D.A.; Padmanabhan, V. Developmental programming: Excess weight gain amplifies the effects of prenatal testosterone excess on reproductive cyclicity—Implication for polycystic ovary syndrome. Endocrinology 2009, 150, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Steckler, T.; Roberts, E.; Doop, D.; Lee, T.; Padmanabhan, V. Developmental programming in sheep: Administration of testosterone during 60–90 days of pregnancy reduces breeding success and pregnancy outcome. Theriogenology 2007, 67, 459–467. [Google Scholar] [CrossRef]
- Cardoso, R.C.; Burns, A.; Moeller, J.; Skinner, D.C.; Padmanabhan, V. Developmental programming: Insulin sensitizer prevents the GnRH-stimulated LH hypersecretion in a sheep model of PCOS. Endocrinology 2016, 157, 4641–4653. [Google Scholar] [CrossRef]
- Steckler, T.; Wang, J.; Bartol, F.F.; Roy, S.K.; Padmanabhan, V. Fetal programming: Prenatal testosterone treatment causes intrauterine growth retardation, reduces ovarian reserve and increases ovarian follicular recruitment. Endocrinology 2005, 146, 3185–3193. [Google Scholar] [CrossRef] [PubMed]
- West, C.; Foster, D.L.; Evans, N.P.; Robinson, J.; Padmanabhan, V. Intra-follicular activin availability is altered in prenatally-androgenized lambs. Mol. Cell. Endocrinol. 2001, 185, 51–59. [Google Scholar] [CrossRef]
- Smith, P.; Steckler, T.L.; Veiga-Lopez, A.; Padmanabhan, V. Developmental programming: Differential effects of prenatal testosterone and dihydrotestosterone on follicular recruitment, depletion of follicular reserve, and ovarian morphology in sheep. Biol. Reprod. 2009, 80, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Forsdike, R.A.; Hardy, K.; Bull, L.; Stark, J.; Webber, L.J.; Stubbs, S.; Robinson, J.E.; Franks, S. Disordered follicle development in ovaries of prenatally androgenized ewes. J. Endocrinol. 2007, 192, 421–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manikkam, M.; Steckler, T.L.; Welch, K.B.; Inskeep, E.K.; Padmanabhan, V. Fetal programming: Prenatal testosterone treatment leads to follicular persistence/luteal defects; partial restoration of ovarian function by cyclic progesterone treatment. Endocrinology 2006, 147, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Steckler, T.; Manikkam, M.; Inskeep, E.K.; Padmanabhan, V.J.E. Developmental programming: Follicular persistence in prenatal testosterone-treated sheep is not programmed by androgenic actions of testosterone. Endocrinology 2007, 148, 3532–3540. [Google Scholar] [CrossRef] [PubMed]
- Recabarren, S.E.; Padmanabhan, V.; Codner, E.; Lobos, A.; Durán, C.; Vidal, M.; Foster, D.L.; Sir-Petermann, T. Postnatal developmental consequences of altered insulin sensitivity in female sheep treated prenatally with testosterone. Am. J. Physiol. Regul. 2005, 289, E801–E806. [Google Scholar] [CrossRef] [PubMed]
- Veiga-Lopez, A.; Moeller, J.; Patel, D.; Ye, W.; Pease, A.; Kinns, J.; Padmanabhan, V. Developmental programming: Impact of prenatal testosterone excess on insulin sensitivity, adiposity, and free fatty acid profile in postpubertal female sheep. Endocrinology 2013, 154, 1731–1742. [Google Scholar] [CrossRef]
- Puttabyatappa, M.; Lu, C.; Martin, J.D.; Chazenbalk, G.; Dumesic, D.; Padmanabhan, V. Developmental Programming: Impact of Prenatal Testosterone Excess on Steroidal Machinery and Cell Differentiation Markers in Visceral Adipocytes of Female Sheep. Reprod. Sci. 2018, 25, 1010–1023. [Google Scholar] [CrossRef]
- King, A.J.; Olivier, N.B.; MohanKumar, P.S.; Lee, J.S.; Padmanabhan, V.; Fink, G.D. Hypertension caused by prenatal testosterone excess in female sheep. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1837–E1841. [Google Scholar] [CrossRef]
- Cardoso, R.C.; Veiga-Lopez, A.; Moeller, J.; Beckett, E.; Pease, A.; Keller, E.; Madrigal, V.; Chazenbalk, G.; Dumesic, D.; Padmanabhan, V. Developmental Programming: Impact of Gestational Steroid and Metabolic Milieus on Adiposity and Insulin Sensitivity in Prenatal Testosterone-Treated Female Sheep. Endocrinology 2016, 157. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, V.; Veiga-Lopez, A.; Abbott, D.; Recabarren, S.; Herkimer, C. Developmental programming: Impact of prenatal testosterone excess and postnatal weight gain on insulin sensitivity index and transfer of traits to offspring of overweight females. Endocrinology 2010, 151, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Weyer, C.; Foley, J.; Bogardus, C.; Tataranni, P.; Pratley, R. Enlarged subcutaneous abdominal adipocyte size, but not obesity itself, predicts type II diabetes independent of insulin resistance. Diabetologia 2000, 43, 1498–1506. [Google Scholar] [CrossRef] [PubMed]
- Salans, L.B.; Knittle, J.L.; Hirsch, J. The role of adipose cell size and adipose tissue insulin sensitivity in the carbohydrate intolerance of human obesity. J. Clin. Investig. 1968, 47, 153. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, T.; Deng, A.; Yee, G.; Lamendola, C.; Reaven, G.; Tsao, P.; Cushman, S.; Sherman, A. Inflammation in subcutaneous adipose tissue: Relationship to adipose cell size. Diabetologia 2010, 53, 369–377. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, T.; Sherman, A.; Tsao, P.; Gonzalez, O.; Yee, G.; Lamendola, C.; Reaven, G.; Cushman, S. Enhanced proportion of small adipose cells in insulin-resistant vs insulin-sensitive obese individuals implicates impaired adipogenesis. Diabetologia 2007, 50, 1707–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, G. Let’s shift lipid burden—From large to small adipocytes. Eur. J. Pharmacol. 2011, 656, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Virtue, S.; Vidal-Puig, A. Adipose tissue expandability, lipotoxicity and the metabolic syndrome—An allostatic perspective. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2010, 1801, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-H.; Pratley, R.E. The evolving role of inflammation in obesity and the metabolic syndrome. Curr. Diabetes Rep. 2005, 5, 70–75. [Google Scholar] [CrossRef]
- Sørensen, T.I.; Virtue, S.; Vidal-Puig, A. Obesity as a clinical and public health problem: Is there a need for a new definition based on lipotoxicity effects? Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2010, 1801, 400–404. [Google Scholar] [CrossRef]
- Puttabyatappa, M.; Andriessen, V.; Mesquitta, M.; Zeng, L.; Pennathur, S.; Padmanabhan, V. Developmental Programming: Impact of Gestational Steroid and Metabolic Milieus on Mediators of Insulin Sensitivity in Prenatal Testosterone–Treated Female Sheep. Endocrinology 2017, 158, 2783–2798. [Google Scholar] [CrossRef]
- Dumesic, D.A.; Phan, J.D.; Leung, K.L.; Grogan, T.R.; Ding, X.; Li, X.; Hoyos, L.R.; Abbott, D.H.; Chazenbalk, G.D. Adipose Insulin Resistance in Normal-Weight Women With Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2019, 104, 2171–2183. [Google Scholar] [CrossRef]
- Hakim, C.; Padmanabhan, V.; Vyas, A.K. Gestational hyperandrogenism in developmental programming. Endocrinology 2016, 158, 199–212. [Google Scholar] [CrossRef]
- Vyas, A.K.; Hoang, V.; Padmanabhan, V.; Gilbreath, E.; Mietelka, K.A. Prenatal programming: Adverse cardiac programming by gestational testosterone excess. Sci. Rep. 2016, 6, 28335. [Google Scholar] [CrossRef]
- Veiga-Lopez, A.; Steckler, T.L.; Abbott, D.H.; Welch, K.B.; MohanKumar, P.S.; Phillips, D.J.; Refsal, K.; Padmanabhan, V. Developmental programming: Impact of excess prenatal testosterone on intrauterine fetal endocrine milieu and growth in sheep. Biol. Reprod. 2011, 84, 87–96. [Google Scholar] [CrossRef]
- Abi Salloum, B.; Veiga-Lopez, A.; Abbott, D.H.; Burant, C.F.; Padmanabhan, V. Developmental programming: Exposure to testosterone excess disrupts steroidal and metabolic environment in pregnant sheep. Endocrinology 2015, 156. [Google Scholar] [CrossRef]
- Padmanabhan, V.; Veiga-Lopez, A.; Herkimer, C.; Abi Salloum, B.; Moeller, J.; Beckett, E.; Sreedharan, R.J.E. Developmental programming: Prenatal and postnatal androgen antagonist and insulin sensitizer interventions prevent advancement of puberty and improve LH surge dynamics in prenatal testosterone-treated sheep. Endocrinology 2015, 156, 2678–2692. [Google Scholar] [CrossRef]
- Wood, R.I.; Foster, D.L. Sexual differentiation of reproductive neuroendocrine function in sheep. Rev. Reprod. 1998, 3, 130–140. [Google Scholar] [CrossRef]
- Veiga-Lopez, A.; Astapova, O.I.; Aizenberg, E.F.; Lee, J.S.; Padmanabhan, V. Developmental programming: Contribution of prenatal androgen and estrogen to estradiol feedback systems and periovulatory hormonal dynamics in sheep. Biol. Reprod. 2009, 80, 718–725. [Google Scholar] [CrossRef]
- Puttabyatappa, M.; Cardoso, R.C.; Herkimer, C.; Veiga-Lopez, A.; Padmanabhan, V.J.R. Developmental programming: Postnatal estradiol modulation of prenatally organized reproductive neuroendocrine function in sheep. Reproduction 2016, 152, 139–150. [Google Scholar] [CrossRef]
- Jackson, L.M.; Timmer, K.M.; Foster, D.L. Sexual differentiation of the external genitalia and the timing of puberty in the presence of an antiandrogen in sheep. Endocrinology 2008, 149, 4200–4208. [Google Scholar] [CrossRef]
- Pak, T.R.; Chung, W.C.; Hinds, L.R.; Handa, R.J. Estrogen receptor-β mediates dihydrotestosterone-induced stimulation of the arginine vasopressin promoter in neuronal cells. Endocrinology 2007, 148, 3371–3382. [Google Scholar] [CrossRef]
- Legro, R.S.; Zaino, R.J.; Demers, L.M.; Kunselman, A.R.; Gnatuk, C.L.; Williams, N.I.; Dodson, W.C. The effects of metformin and rosiglitazone, alone and in combination, on the ovary and endometrium in polycystic ovary syndrome. Am. J. Obstet. Gynecol. 2007, 196, 402.e1–402.e11. [Google Scholar] [CrossRef]
- Roy, K.; Baruah, J.; Sharma, A.; Sharma, J.; Kumar, S.; Kachava, G.; Karmakar, D. A prospective randomized trial comparing the clinical and endocrinological outcome with rosiglitazone versus laparoscopic ovarian drilling in patients with polycystic ovarian disease resistant to ovulation induction with clomiphene citrate. Arch. Gynecol. Obstet. 2010, 281, 939–944. [Google Scholar] [CrossRef]
- Veiga-Lopez, A.; Lee, J.S.; Padmanabhan, V. Developmental programming: Insulin sensitizer treatment improves reproductive function in prenatal testosterone-treated female sheep. Endocrinology 2010, 151, 4007–4017. [Google Scholar] [CrossRef]
- Samuelsson, A.-M.; Matthews, P.A.; Argenton, M.; Christie, M.R.; McConnell, J.M.; Jansen, E.H.; Piersma, A.H.; Ozanne, S.E.; Twinn, D.F.; Remacle, C.J.H. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: A novel murine model of developmental programming. Hypertension 2008, 51, 383–392. [Google Scholar] [CrossRef]
- Fernandez-Twinn, D.; Wayman, A.; Ekizoglou, S.; Martin, M.; Hales, C.; Ozanne, S.E. Maternal protein restriction leads to hyperinsulinemia and reduced insulin-signaling protein expression in 21-mo-old female rat offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R368–R373. [Google Scholar] [CrossRef]
- Kinder, J.; Bergfeld, E.; Wehrman, M.; Peters, K.; Kojima, F.N. Endocrine basis for puberty in heifers and ewes. J. Reprod. Fertil. Suppl. 1995, 49, 393–407. [Google Scholar] [CrossRef]
- Amstalden, M.; Alves, B.R.; Liu, S.; Cardoso, R.C.; Williams, G.L. Neuroendocrine pathways mediating nutritional acceleration of puberty: Insights from ruminant models. Front. Endocrinol. 2011, 2, 109. [Google Scholar] [CrossRef]
- Abi Salloum, B.; Herkimer, C.; Lee, J.S.; Veiga-Lopez, A.; Padmanabhan, V. Developmental programming: Prenatal and postnatal contribution of androgens and insulin in the reprogramming of estradiol positive feedback disruptions in prenatal testosterone-treated sheep. Endocrinology 2012, 153, 2813–2822. [Google Scholar] [CrossRef]
- Sheppard, K.M.; Padmanabhan, V.; Coolen, L.M.; Lehman, M.N. Prenatal programming by testosterone of hypothalamic metabolic control neurones in the ewe. J. Neuroendocrinol. 2011, 23, 401–411. [Google Scholar] [CrossRef]
- De Leo, V.; Lanzetta, D.; D’Antona, D.; La Marca, A.; Morgante, G. Hormonal effects of flutamide in young women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 1998, 83, 99–102. [Google Scholar] [CrossRef]
- Eagleson, C.A.; Gingrich, M.B.; Pastor, C.L.; Arora, T.K.; Burt, C.M.; Evans, W.S.; Marshall, J.C. Polycystic ovarian syndrome: Evidence that flutamide restores sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J. Clin. Endocrinol. Metab. 2000, 85, 4047–4052. [Google Scholar]
- Downing, J.; Scaramuzzi, R.J.T. The effect of the infusion of insulin during the luteal phase of the estrous cycle on the ovulation rate and on plasma concentrations of LH, FSH and glucose in ewes. Theriogenology 1997, 47, 747–759. [Google Scholar] [CrossRef]
- Ibáñez, L.; de Zegher, F. Low-dose combination of flutamide, metformin and an oral contraceptive for non-obese, young women with polycystic ovary syndrome. Hum. Reprod. 2003, 18, 57–60. [Google Scholar] [CrossRef]

{kind=link}
| Traits | Prenatal Testosterone | Prenatal DHT | Interventions—Pathology Manifestations | |||
|---|---|---|---|---|---|---|
| Prenatal Testosterone + Prenatal Androgen Antagonist | Prenatal Testosterone + Prenatal Insulin Sensitizer | Prenatal Testosterone + Postnatal Androgen Antagonist | Prenatal Testosterone + Postnatal Insulin Sensitizer | |||
| Reproductive Traits | ||||||
| Advanced puberty | Yes [57] | Yes Ψ [58] | No [57] | No [57] | No [57] | No [57] |
| Functional Hyperandrogenism | Yes [23] | Yes [23] | Not tested | Not tested | Not tested | Not tested |
| PCO morphology | Yes [31] | No [31] | Not tested | Not tested | Not tested | Not tested |
| Disrupted preovulatory LH surge | Yes [57] | No [59] | Partially [57] | Yes [57] | Yes [57] | Yes [57] |
| Disrupted estradiol positive feedback | Yes [60] | No [58,59] | Yes [60] | Yes [60] | Partially [60] | Partially [60] |
| Disrupted estradiol negative feedback | Yes [26] | Yes [59] | No Σ [61] | Not tested | Not tested | Not tested |
| GnRH-stimulated LH hypersecretion | Yes [30] | Yes [30] | Yes [30] | Partially [30] | Yes [30] | No [30] |
| Increased follicular recruitment | Yes [33] | Yes [33] | Not tested | Not tested | Not tested | Not tested |
| Follicular persistence | Yes [35] | No [36] | Not tested | Not tested | Not tested | Not tested |
| Metabolic Traits | ||||||
| Insulin resistance | Yes [38,41] | Yes [41] | Yes [41] | No [41] | Yes # | No # |
| Altered visceral adiposity | Yes [38] | Not tested | Not tested | Not tested | Not tested | Not tested |
| Altered adipocyte size | Yes [38,41] | Not tested | Partially [41] | Partially [41] | Not tested | Not tested |
| Adipocyte differentiation | Reduced [39] | Not tested | Partially [39] | Partially [39] | Not tested | Not tested |
| Hypertension | Yes [40] | Not tested | Not tested | Not tested | Not tested | Not tested |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardoso, R.C.; Padmanabhan, V. Developmental Programming of PCOS Traits: Insights from the Sheep. Med. Sci. 2019, 7, 79. https://doi.org/10.3390/medsci7070079
Cardoso RC, Padmanabhan V. Developmental Programming of PCOS Traits: Insights from the Sheep. Medical Sciences. 2019; 7(7):79. https://doi.org/10.3390/medsci7070079
Chicago/Turabian StyleCardoso, Rodolfo C., and Vasantha Padmanabhan. 2019. "Developmental Programming of PCOS Traits: Insights from the Sheep" Medical Sciences 7, no. 7: 79. https://doi.org/10.3390/medsci7070079
APA StyleCardoso, R. C., & Padmanabhan, V. (2019). Developmental Programming of PCOS Traits: Insights from the Sheep. Medical Sciences, 7(7), 79. https://doi.org/10.3390/medsci7070079