The Carbon-Isotope Record of the Sub-Seafloor Biosphere


{kind=link}
{kind=link}
{kind=link}
{kind=link}
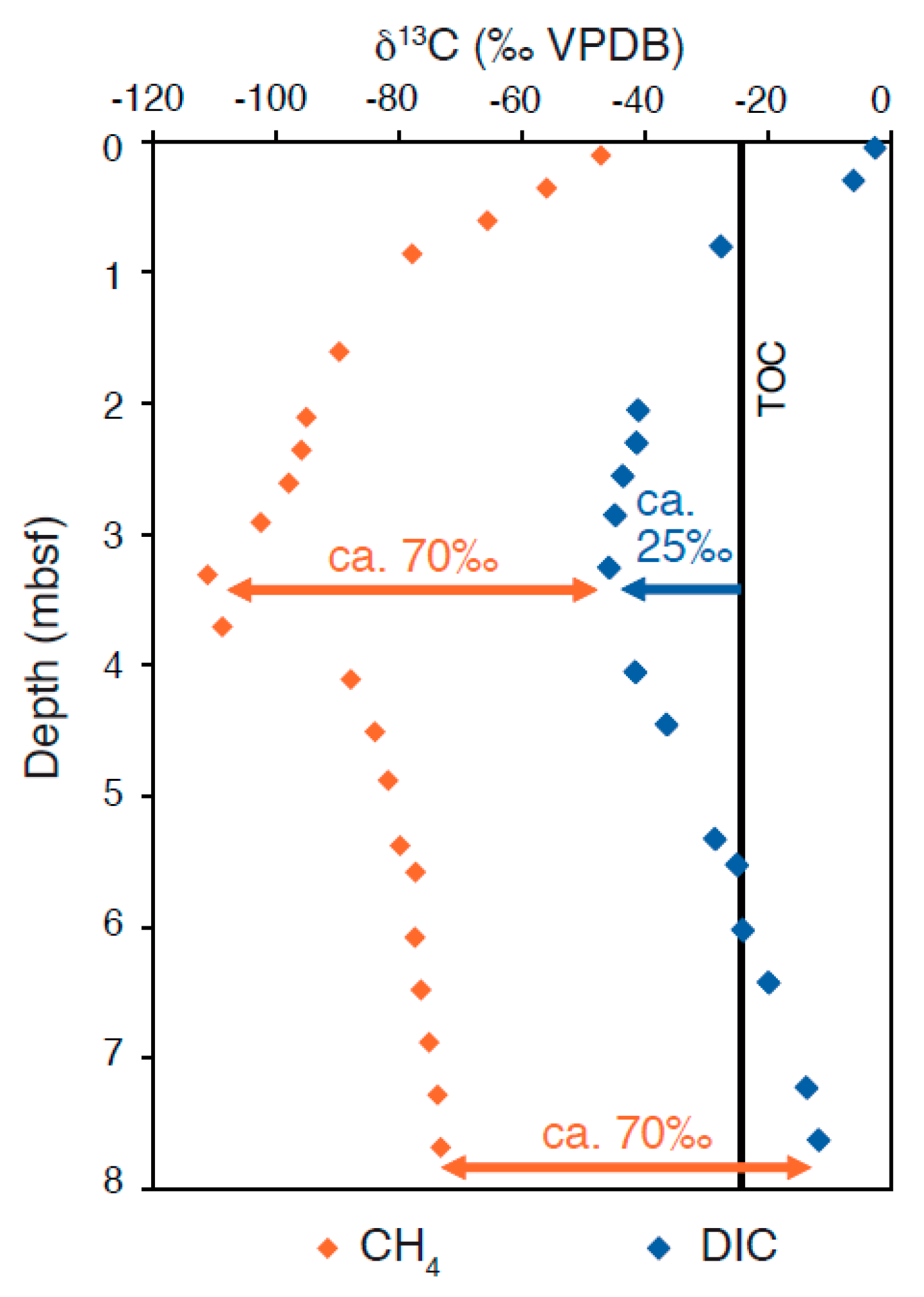{kind=link}
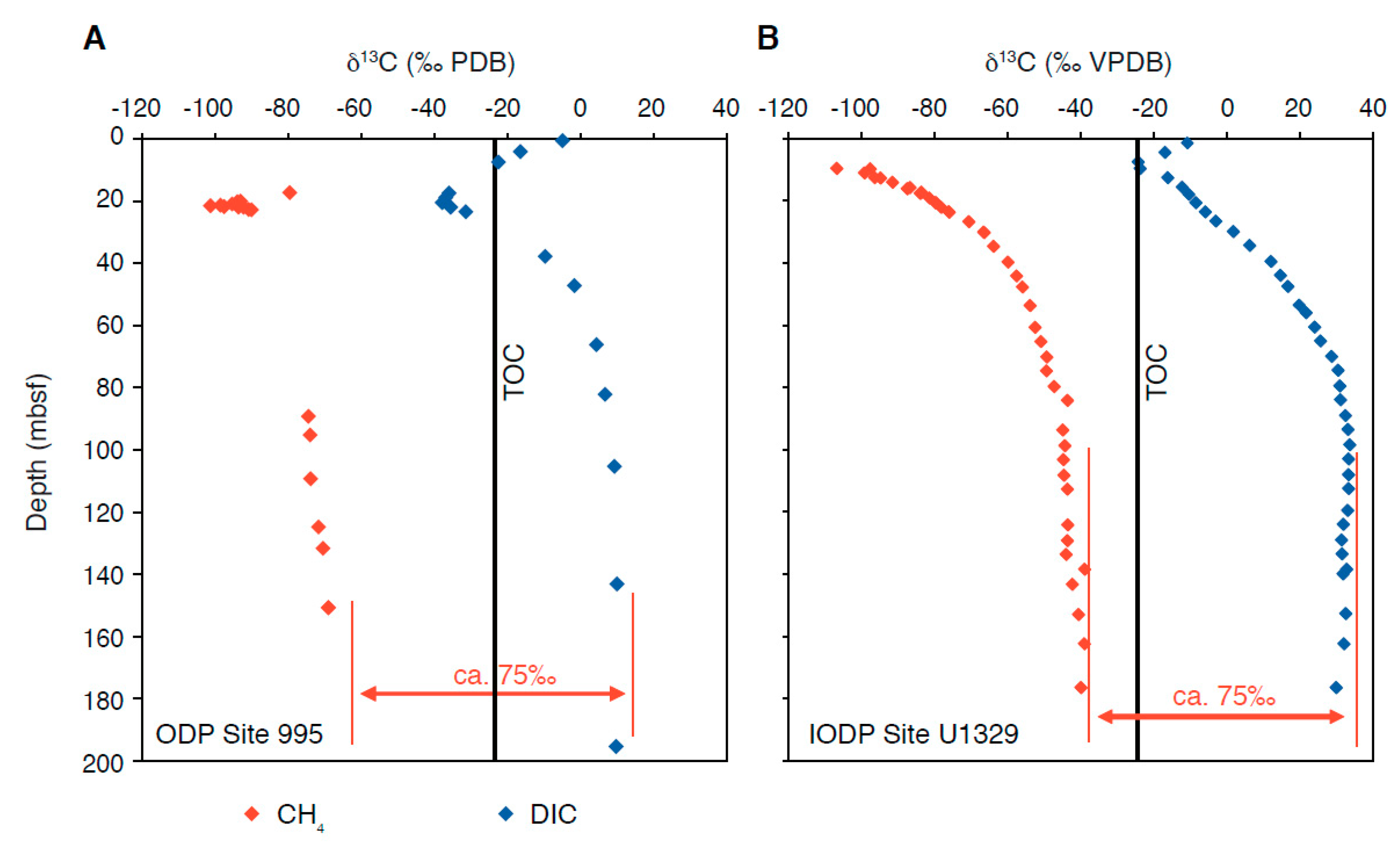{kind=link}
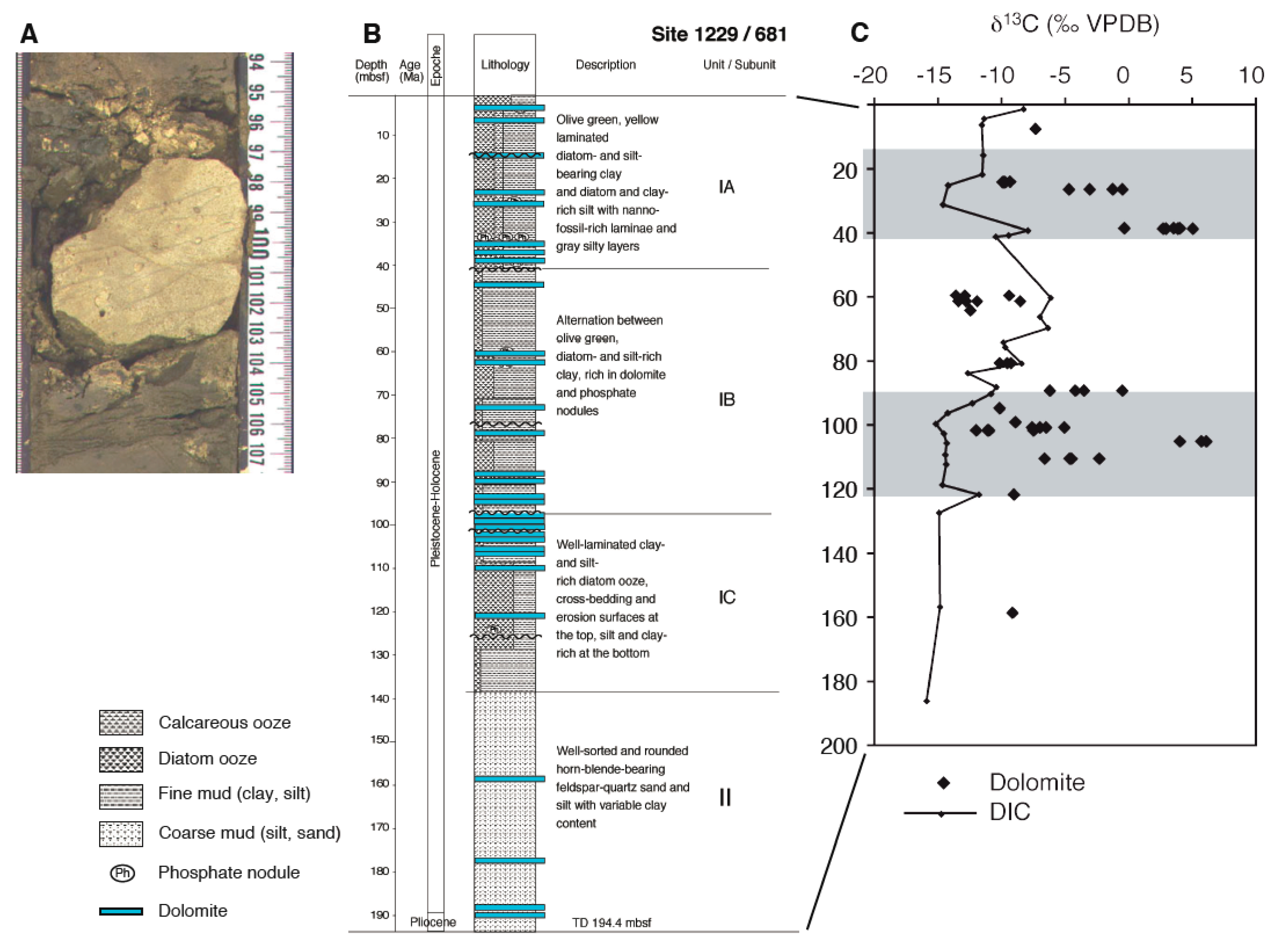{kind=link}
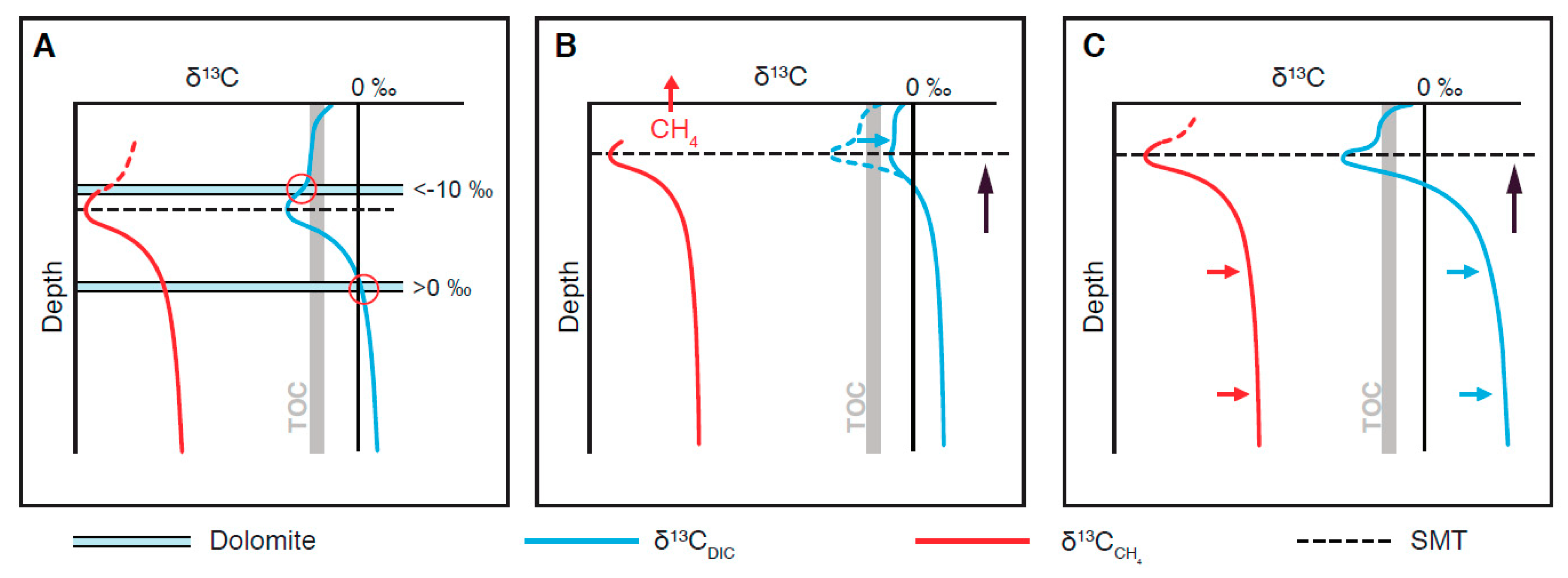{kind=link}
{kind=link}
Abstract
1. Introduction
2. Carbon-Isotope Fractionation by Different Microbial Pathways
2.1. Kinetic Fractionation
2.2. Equilibrium Fractionation
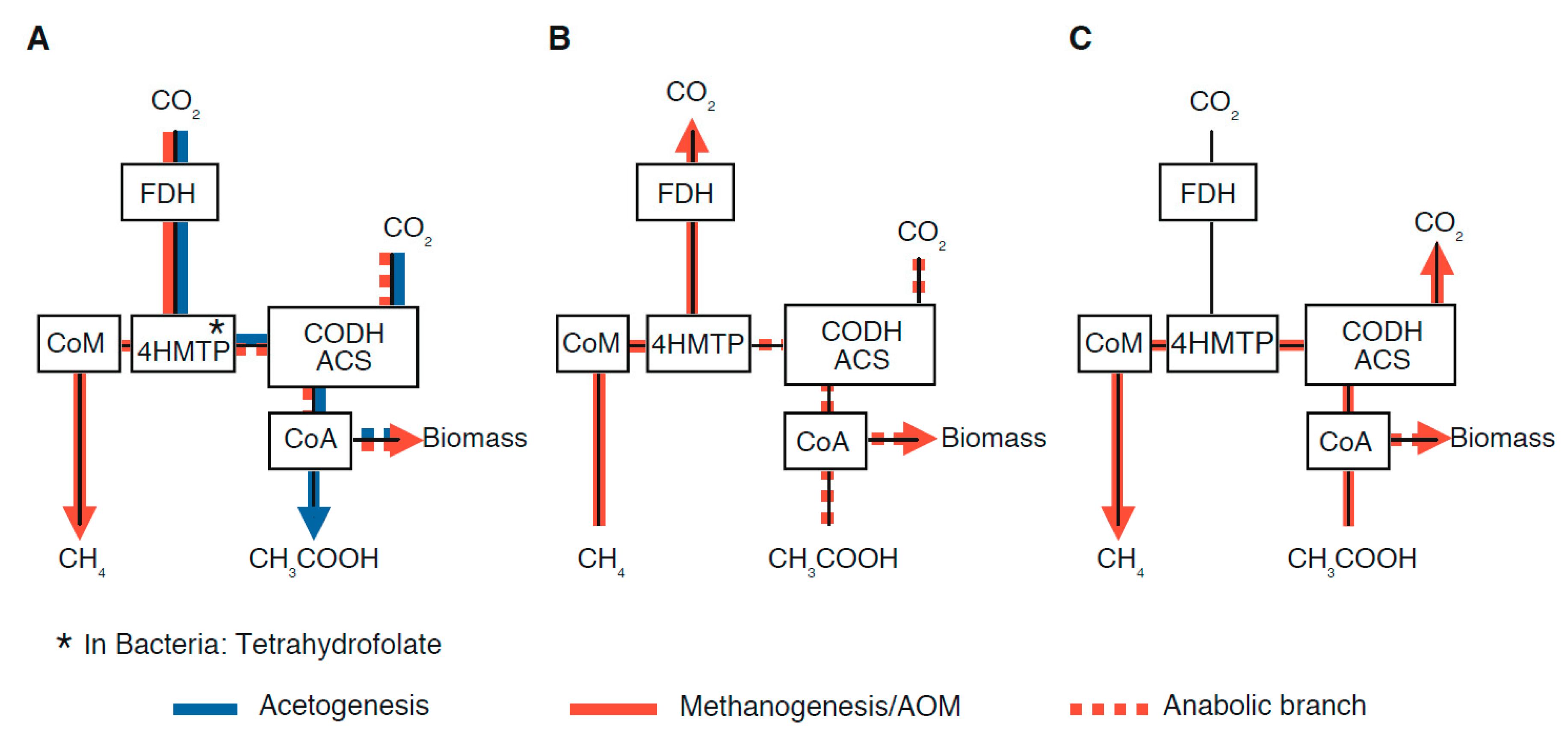
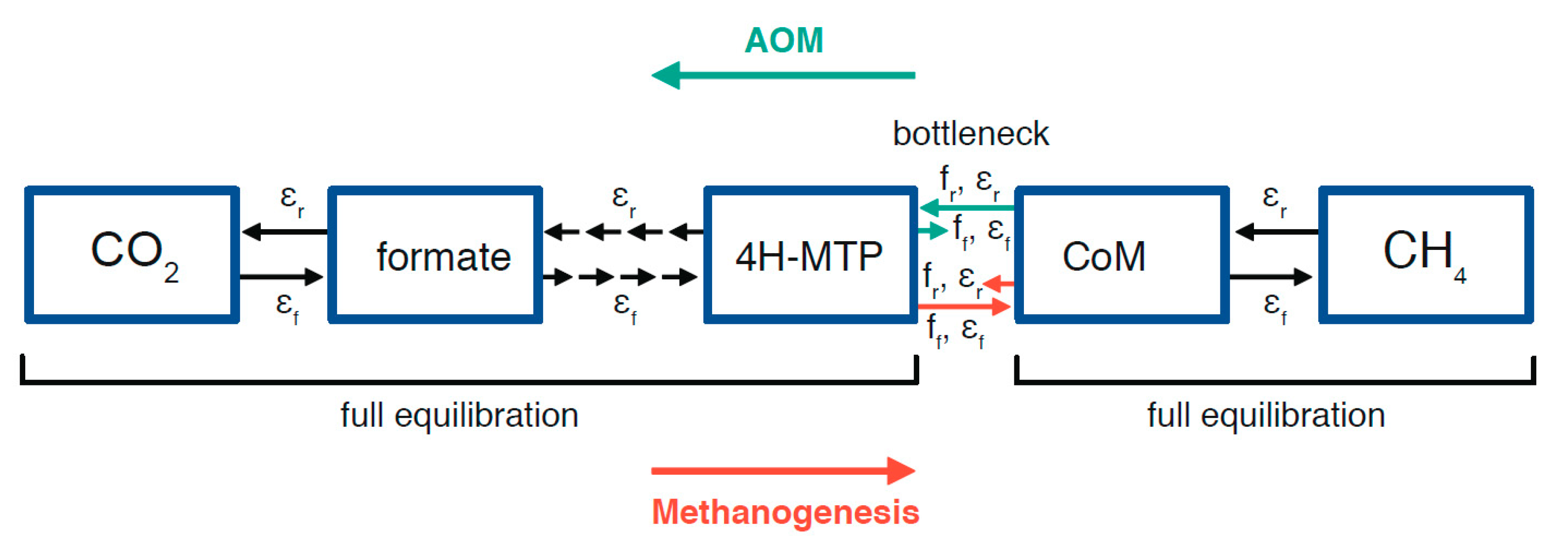
2.3. Potential Fractionation Effects within the Molecular Pathway
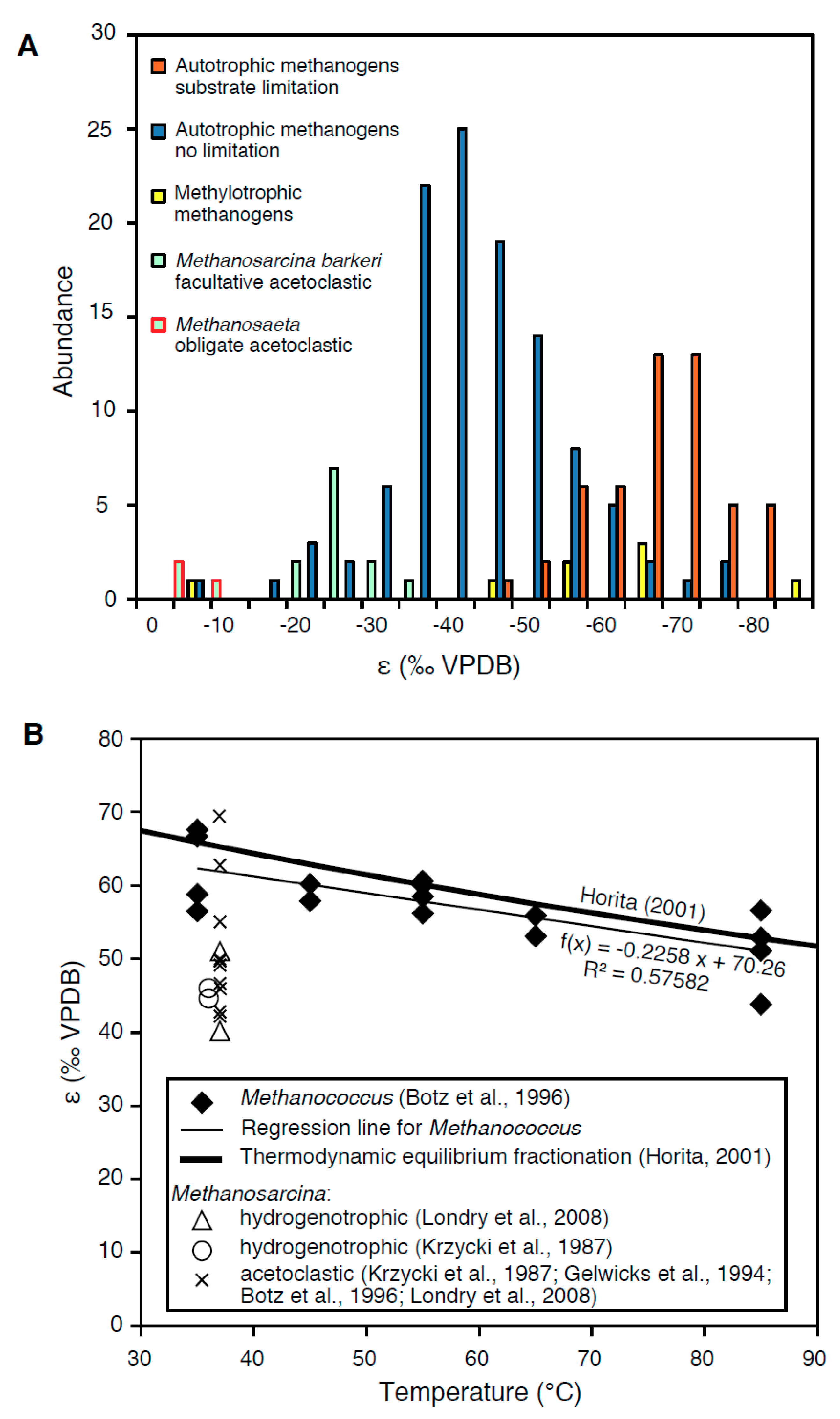
2.4. The Effects of Substrate and Carbon Limitation
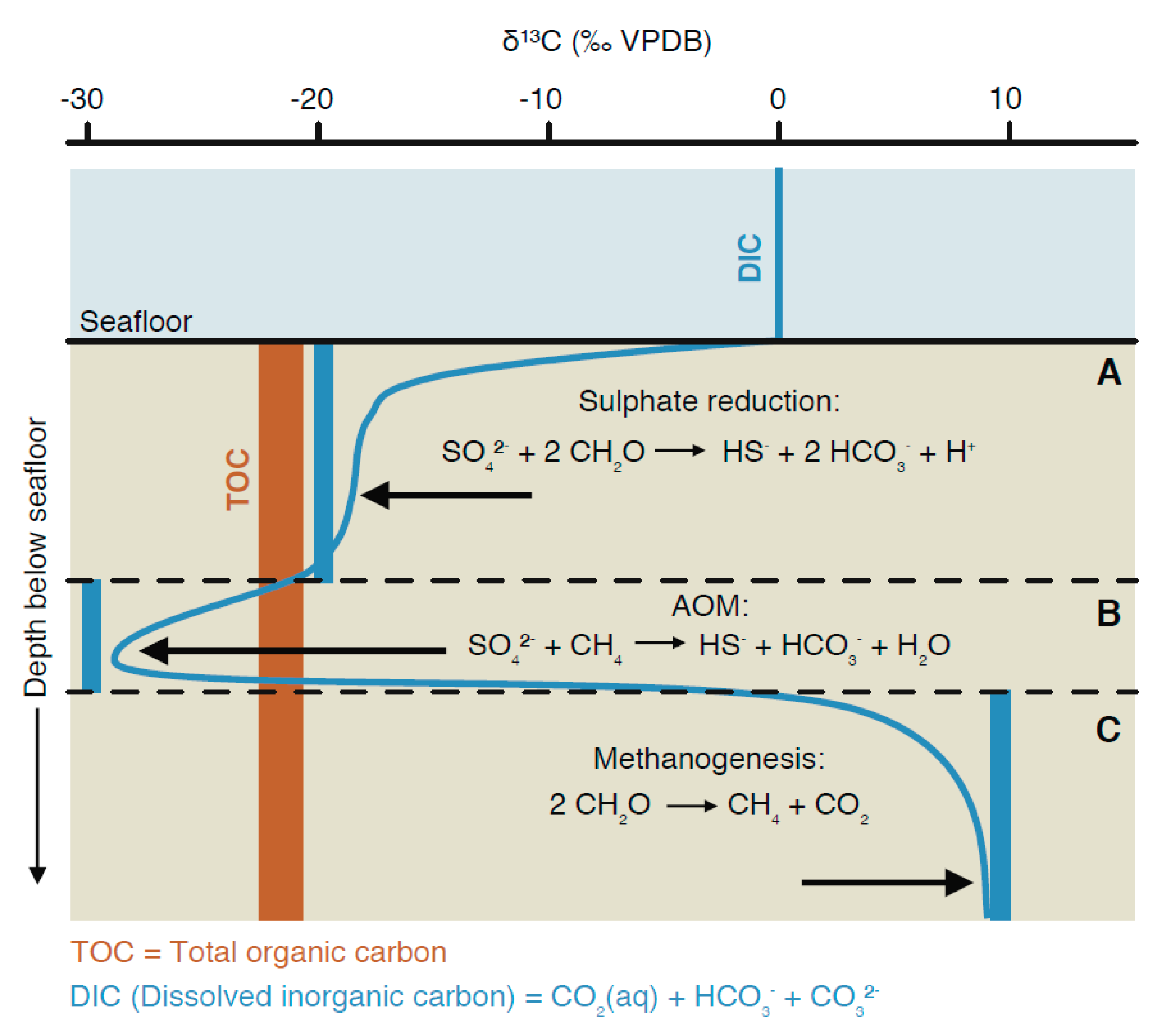
3. Carbon-Isotope Distribution in Porewater Systems
3.1. Isotope Effects in Diffusive Mixing Profiles
3.2. Towards a Simulation of Carbon-Isotope Profiles
3.3. Assessing the Factors Influencing Carbon-Isotope Profiles
3.4. Non-Steady-State Effects and Gas Transport
4. The Isotope Signature of Diagenetic Carbonates
4.1. Processes Inducing Carbonate Formation in the Deep Biosphere
4.2. Controls of δ13C Composition of Diagenetic Carbonates
4.3. Interpreting δ13C Archives Through Time
5. Implications for the Global Carbon Cycle
5.1. The Importance of Diagenetic Carbonates for Global Carbon Fluxes
5.2. Diagenetic vs. Atmospheric Signatures?
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Broeker, W.S. A boundary condition on the evolution of atmospheric oxygen. J. Geophys. Res. 1970, 75, 3553–3557. [Google Scholar] [CrossRef]
- Edwards, K.J.; Becker, K.; Colwell, F. The deep, dark energy biosphere: Intraterrestrial life on Earth. Annu. Rev. Earth Planet. Sci. 2012, 40, 551–568. [Google Scholar] [CrossRef]
- Froelich, P.N.; Klinkhammer, G.P.; Bender, M.L.; Luedtke, N.A.; Heath, G.R.; Cullen, D.; Dauphin, P.; Hammond, D.; Hartman, B.; Maynard, V. Early oxidation of organic matter in pelagic sediments of the eastern equatorial Atlantic: Suboxic diagenesis. Geochim. Cosmochim. Acta 1979, 43, 1075–1090. [Google Scholar] [CrossRef]
- D’Hondt, S.; Jørgensen, B.B.; Miller, J.; Batzke, A.; Blake, R.; Cragg, B.A.; Cypionka, H.; Dickens, G.R.; Ferdelman, T.; Hinrichs, K.U.; et al. Distributions of Microbial Activities in Deep Subseafloor Sediments. Science 2004, 306, 2216–2221. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.M.; Popp, B.N.; Takigiku, R.; Johnson, M.W. An isotopic study of biogeochemical relationships between carbonates and organic carbon in the Greenhorn Formation. Geochim. Cosmochim. Acta 1989, 53, 2961–2972. [Google Scholar] [CrossRef]
- Claypool, G.E.; Kaplan, I.R. The origin and distribution of methane in marine sediments. In Natural Gases in Marine Sediments; Kaplan, I.R., Ed.; Plenum Press: New York, NY, USA, 1974; pp. 99–140. [Google Scholar]
- Heuer, V.B.; Pohlman, J.W.; Torres, M.E.; Elvert, M.; Hinrichs, K.U. The stable carbon isotope biogeochemistry of acetate and other dissolved carbon species in deep sub-seafloor sediments at the northern Cascadia Margin. Geochim. Cosmochim. Acta 2009, 73, 3323–3336. [Google Scholar] [CrossRef]
- Schrag, D.P.; Higgins, J.A.; Macdonald, F.A.; Johnston, D.T. Authigenic carbonate and the history of the global carbon cycle. Science 2013, 339, 540–543. [Google Scholar] [CrossRef]
- Kelts, K.; McKenzie, J.A. A comparison of anoxic dolomite from deep-sea sediments: Quaternary Gulf of California and Messinian Tripoli Formation of Sicily. In Dolomites of the Monterey Formation and Other Organic-Rich Units; Garrison, R.E., Kastner, M., Zenger, D.H., Eds.; Pacific Section SEPM: Los Angeles, CA, USA, 1984; Volume 41, pp. 19–28. [Google Scholar]
- Malone, M.J.; Claypool, G.; Martin, J.B.; Dickens, G.R. Variable methane fluxes in shallow marine systems over geologic time: The composition and origin of pore waters and authigenic carbonates on the New Jersey shelf. Mar. Geol. 2002, 189, 175–196. [Google Scholar] [CrossRef]
- Meister, P.; Bernasconi, S.; McKenzie, J.A.; Vasconcelos, C.; Frank, M.; Gutjahr, M.; Schrag, D. Dolomite formation in the dynamic deep biosphere: Results from the Peru Margin (ODP Leg 201). Sedimentology 2007, 54, 1007–1032. [Google Scholar] [CrossRef]
- Meister, P. For the deep biosphere, the present is not always the key to the past: What we can learn from the geological record. Terra Nova 2015, 27, 400–408. [Google Scholar] [CrossRef]
- Meister, P.; Brunner, B.; Picard, A.; Böttcher, M.E.; Jørgensen, B.B. Sulphur and carbon isotopes as tracers of past sub-seafloor microbial activity. Nat. Sci. Rep. 2019, 9, 604. [Google Scholar] [CrossRef] [PubMed]
- Hoefs, J. Stable Isotope Geochemistry, 8th ed.; Springer: Berlin/Heidelberg, Germany, 2018; p. 437. [Google Scholar]
- Alperin, M.J.; Blair, N.E.; Albert, D.B.; Hoehler, T.M.; Martens, C.S. Factors that control the stable carbon isotopic composition of methane produced in an anoxic marine sediment. Glob. Biogeochem. Cycles 1992, 6, 271–291. [Google Scholar] [CrossRef]
- Hayes, J. Factors controlling 13C contents of sedimentary organic compounds: Principles and evidence. Mar. Geol. 1993, 113, 111–125. [Google Scholar] [CrossRef]
- Hayes, J.M. An Introduction to Isotopic Calculations; Woods Hole Oceanographic Institution: Woods Hole, MA, USA, 2004. [Google Scholar]
- Londry, K.L.; Des Marais, D.J. Stable carbon isotope fractionation by sulfate-reducing bacteria. Appl. Environ. Microbiol. 2003, 69, 2942–2949. [Google Scholar] [CrossRef] [PubMed]
- Whiticar, M.J.; Faber, E. Methane oxidation in sediment and water column environments—Isotope evidence. Org. Geochem. 1986, 10, 759–768. [Google Scholar] [CrossRef]
- Alperin, M.J.; Reeburgh, W.S.; Whiticar, M.J. Carbon and hydrogen isotope fractionation resulting from anaerobic methane oxidation. Glob. Biogeochem. Cycles 1988, 2, 279–288. [Google Scholar] [CrossRef]
- Holler, T.; Wegener, G.; Knittel, K.; Boetius, A.; Brunner, B.; et al. Substantial 13C/12C and D/H fractionation during anaerobic oxidation of methane by marine consortia enriched in vitro. Environ. Microbiol. Rep. 2009, 1, 370–376. [Google Scholar] [CrossRef]
- Games, L.M.; Hayes, J.M.; Gunsalus, R.P. Methane-producing bacteria: Natural fractionations of the stable carbon isotopes. Geochim. Cosmochim. Acta 1978, 42, 1295–1297. [Google Scholar] [CrossRef]
- Fuchs, G.; Thauer, R.; Ziegler, H.; Stichler, W. Carbon isotope fractionation by Methanobacterium thermoautotrophicum. Arch. Microbiol. 1979, 120, 135–139. [Google Scholar] [CrossRef]
- Belyaev, S.S.; Wolkin, R.; Kenealy, W.R.; DeNiro, M.J.; Epstein, S.; Zeikus, J.G. Methanogenic bacteria from the Bondyuzhskoe oil field: General characterization and analysis of stable-carbon isotopic fractionation. Appl. Environ. Microbiol. 1983, 45, 691–697. [Google Scholar]
- Balabane, M.; Galimov, E.; Hermann, M.; Létolle, R. Hydrogen and carbon isotope fractionation during experimental production of bacterial methane. Org. Geochem. 1987, 11, 115–119. [Google Scholar] [CrossRef]
- Krzycki, J.A.; Kenealy, W.R.; DeNiro, M.J.; Zeikus, J.G. Stable carbon isotope fractionation by Methanosarcina barkeri during methanogenesis from acetate, methanol or carbon dioxide-hydrogen. Appl. Environ. Microbiol. 1987, 53, 2597–2599. [Google Scholar] [PubMed]
- Gelwicks, J.T.; Risatti, J.B.; Hayes, J.M. Carbon isotope effects associated with aceticlastic methanogenesis. Appl. Environ. Microbiol. 1994, 60, 467–472. [Google Scholar] [PubMed]
- Botz, R.; Pokojski, H.D.; Schmitt, M.; Thomm, M. Carbon isotope fractionation during bacterial methanogenesis by CO2 reduction. Org. Geochem. 1996, 25, 255–262. [Google Scholar] [CrossRef]
- Summons, R.E.; Franzmann, P.D.; Nichols, P.D. Carbon isotopic fractionation associated with methylotrophic methanogenesis. Org. Geochem. 1998, 28, 465–475. [Google Scholar] [CrossRef]
- Valentine, D.L.; Chidthaisong, A.; Rice, A.; Reeburgh, W.S.; Tyler, S.C. Carbon and hydrogen isotope fractionation by moderately thermophilic methanogens. Geochim. Cosmochim. Acta 2004, 68, 1571–1590. [Google Scholar] [CrossRef]
- Londry, K.L.; Dawson, K.G.; Grover, H.D.; Summons, R.E.; Bradley, A.S. Stable carbon isotope fractionation between substrates and products of Methanosarcina barkeri. Org. Geochem. 2008, 39, 608–621. [Google Scholar] [CrossRef]
- Okumura, T.; Kawagucci, S.; Saito, Y.; Matsui, Y.; Takai, K.; Imachi, H. Hydrogen and carbon isotope systematics in hydrogenotrophic methanogenesis under H2-limited and H2-enriched conditions: Implications for the origin of methane and its isotopic diagnosis. Prog. Earth Planet. Sci. 2016, 3, 1–19. [Google Scholar] [CrossRef]
- Miller, H.M.; Chaudhry, N.; Conrad, M.E.; Bill, M.; Kopf, S.H.; Templeton, A.S. Large carbon isotope variability during methanogenesis under alkaline conditions. Geochim. Cosmochim. Acta 2018, 237, 18–31. [Google Scholar] [CrossRef]
- Horita, J. Carbon isotope exchange in the system CO2-CH4 at elevated temperatures. Geochim. Cosmochim. Acta 2001, 65, 1907–1919. [Google Scholar] [CrossRef]
- Sugimoto, A.; Wada, E. Carbon isotopic composition of bacterial methane in a soil incubation experiment: Contributions of acetate and CO2/H2. Geochim. Cosmochim. Acta 1993, 57, 4015–4027. [Google Scholar] [CrossRef]
- Whiticar, M.J.; Faber, E.; Schoell, M. Biogenic methane formation in marine and freshwater environments: CO2 reduction vs. acetate fermentation—Isotope evidence. Geochim. Cosmochim. Acta 1986, 50, 693–709. [Google Scholar] [CrossRef]
- Jørgensen, B.B.; D’Hondt, S. A starving majority deep beneath the seafloor. Science 2006, 314, 932–934. [Google Scholar] [CrossRef] [PubMed]
- Holler, T.; Wegener, G.; Niemann, H.; Deusner, C.; Ferdelman, T.G.; Boetius, A.; Brunner, B.; Widdel, F. Carbon and sulfur back flux during anaerobic microbial oxidation of methane and coupled sulfate reduction. Proc. Natl. Acad. Sci. USA 2011, 108, 1484–1490. [Google Scholar] [CrossRef]
- Yoshinaga, M.Y.; Holler, T.; Goldhammer, T.; Wegener, G.; Pohlman, J.W.; Brunner, B.; Kuypers, M.M.M.; Hinrichs, K.U.; Elvert, M. Carbon isotope equilibration during sulphate-limited anaerobic oxidation of methane. Nat. Geosci. 2014, 7, 190–194. [Google Scholar] [CrossRef]
- Urey, H.C.; Greiff, L.J. Isotopic exchange equilbria. J. Am. Chem. Soc. 1935, 57, 321–327. [Google Scholar] [CrossRef]
- Giggenbach, W.F. Carbon-13 exchange between CO2 and CH4 under geothermal conditions. Geochim. Cosmochim. Acta 1982, 46, 159–165. [Google Scholar] [CrossRef]
- Richet, P.; Bottinga, Y.; Javay, M. A review of hydrogen, carbon, nitrogen, oxygen, sulphur, and chlorine stable isotope fractionation among gaseous molecules. Ann. Rev. Earth Planet. Sci. 1977, 5, 65–110. [Google Scholar] [CrossRef]
- Bottinga, Y. Calculated fractionation factors for carbon and hydrogen isotope exchange in the system calcite-carbon dioxide-graphite-methane-hydrogen-water vapour. Geochim. Cosmochim. Acta 1969, 33, 49–64. [Google Scholar] [CrossRef]
- Penning, H.; Plugge, C.M.; Galand, P.E.; Conrad, R. Variation of carbon isotope fractionation in hydrogenotrophic methanogenic microbial cultures and environmental samples at different energy status. Glob. Chang. Biol. 2005, 11, 2103–2113. [Google Scholar] [CrossRef]
- Moran, J.J.; House, C.H.; Freeman, K.H.; Ferry, J.G. Trace methane oxidation studied in several Euryarchaeota under diverse conditions. Archaea 2005, 1, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Takai, K.; Nakamura, K.; Toki, T.; Tsunogai, U.; Miyazaki, M.; Miyazaki, J.; Hirayama, H.; Nakagawa, S.; Nunoura, T.; Horikoshi, K. Cell proliferation at 122 °C and isotopically heavy CH4 production by a hyperthermophilic methanogen under high-pressure cultivation. Proc. Natl. Acad. Sci. USA 2008, 105, 10949–10954. [Google Scholar] [CrossRef] [PubMed]
- Adam, P.S.; Borrel, G.; Gribaldo, S. Evolutionary history of carbon monoxide dehydrogenase/acetyl-CoA synthase, one of the oldest enzymatic complexes. Proc. Natl. Acad. Sci. USA 2018, 115, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.; Russel, M.J. On the origin of biochemistry at an alkaline hydrothermal vent. Phil. Trans. R. Soc. B 2007, 362, 1887–1925. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.C.; Sousa, F.L.; Mrnjavac, N.; Neukirchen, S.; Roettger, M.; Nelson-Sathi, S.; Martin, W.F. The physiology and habitat of the last universal common ancestor. Nat. Microbiol. 2016, 1, 16116. [Google Scholar] [CrossRef]
- Berghuis, B.A.; Yu, F.B.; Schulz, F.; Blainey, P.C.; Woyke, T.; Quake, S.R. Hydrogenotrophic methanogenesis in archaeal phylum Verstraetearchaeota reveals the shared ancestry of all methanogens. Proc. Natl. Acad. Sci. USA 2019, 116, 5037–5044. [Google Scholar] [CrossRef]
- Kellermann, M.Y.; Wegener, G.; Elvert, M.; Yoshinaga, M.Y.; Lin, Y.S.; Thomas, H.; Prieto Mollar, X.; Knittel, K.; Hinrichs, K.U. Autotrophy as a predominant mode of carbon fixation in anaerobic methane-oxidizing microbial communities. Proc. Natl. Acad. Sci. USA 2012, 109, 19321–19326. [Google Scholar] [CrossRef]
- Alperin, M.J.; Hoehler, T.M. Anaerobic methane oxidation by Archaea/sulfate-reducing Bacteria aggregates: 2. isotopic constraints. Am. J. Sci. 2009, 309, 958–984. [Google Scholar] [CrossRef]
- Skennerton, C.T.; Chourey, K.; Iyer, R.; Hettich, R.L.; Tyson, G.W.; Orphan, V.J. Methane-fueled syntrophy through extracellular electron transfer: Uncovering the genomic traits conserved within diverse bacterial partners of anaerobic methanotrophic Archaea. mBio 2017, 8, e00530-17. [Google Scholar] [CrossRef]
- Thauer, R.K.; Kaster, A.K.; Seedorf, H.; Buckel, W.; Hedderich, R. Methanogenic archaea: Ecologically relevant differences in energy conservation. Nat. Rev. 2008, 6, 579–591. [Google Scholar] [CrossRef]
- Meister, P.; Liu, B.; Khalili, A.; Böttcher, M.E.; Jørgensen, B.B. Factors controlling the carbon isotope composition of dissolved inorganic carbon and methane in marine porewater: An evaluation by reactive-transport modelling. J. Mar. Syst. 2019, 200, 103227. [Google Scholar] [CrossRef]
- Blair, N.E.; Carter, W.D., Jr. The carbon isotope biogeochemistry of acetate from a methanogenic marine sediment. Geochim. Cosmochim. Acta 1992, 56, 1247–1258. [Google Scholar] [CrossRef]
- Hinrichs, K.U.; Summons, R.E.; Orphan, V.; Sylva, S.P.; Hayes, J.M. Molecular and isotopic analyses of anaerobic methane-oxidizing communities in marine sediments. Org. Geochem. 2000, 31, 1685–1701. [Google Scholar] [CrossRef]
- Pancost, R.D.; Sinninghe Damsté, J.S.; de Lint, S.; van der Maarel, M.J.; Gottschal, J.C. The Medinaut Shipboard Scientific Party Biomarker evidence for widespread anaerobic methane oxidation in Mediterranean sediments by a consortium of methanogenic Archaea and bacteria. Appl. Environ. Microbiol. 2000, 66, 1126–1132. [Google Scholar] [CrossRef]
- Contreras, S.; Meister, P.; Liu, B.; Prieto-Mollar, X.; Hinrichs, K.U.; Khalili, A.; Ferdelman, T.G.; Kuypers, M.; Jørgensen, B.B. Strong glacial-interglacial variation of sub-seafloor microbial activity on the Peruvian shelf. Proc. Natl. Acad. Sci. USA 2013, 110, 18098–18103. [Google Scholar] [CrossRef]
- Carr, S.A.; Schubotz, F.; Dunbar, R.B.; Mills, C.T.; Dias, R.; Summons, R.E.; Mandernack, K.W. Acetoclastic Methanosaeta are dominant methanogens in organic-rich Antarctic marine sediments. Int. Soc. Microb. Ecol. J. 2018, 12, 330–342. [Google Scholar] [CrossRef]
- McCollom, T.M.; Bach, W. Thermodynamic constraints on hydrogen generation during serpentinization of ultramafic rocks. Geochim. Cosmochim. Acta 2009, 73, 856–875. [Google Scholar] [CrossRef]
- Meister, P.; Wiedling, J.; Lott, C.; Bach, W.; Kuhfuß, H.; Wegener, G.; Böttcher, M.E.; Deusner, C.; Lichtschlag, A.; Bernasconi, S.M.; et al. Anaerobic methane oxidation inducing carbonate precipitation at abiogenic methane seeps in the Tuscan archipelago (Italy). PLoS ONE 2018, 13, e0207305. [Google Scholar] [CrossRef]
- Ijiri, A.; Harada, N.; Hirota, A.; Tsunogai, U.; Ogawa, N.O.; Itaki, T.; Khim, B.K.; Uchida, M. Biogeochemical processes involving acetate in sub-seafloor sediments from the Bering Sea shelf break. Org. Geochem. 2012, 48, 47–55. [Google Scholar] [CrossRef]
- Zeebe, R.E. Modeling CO2 chemistry, δ13C, and oxidation of organic carbon and methane in sediment porewater: Implications for paleo-proxies in benthic foraminifera. Geochim. Cosmochim. Acta 2007, 71, 3238–3256. [Google Scholar] [CrossRef]
- Drake, H.; Åström, M.E.; Heim, C.; Broman, C.; Åström, J.; Whitehouse, M.; Ivarsson, M.; Siljeström, S.; Sjövall, P. Extreme 13 C depletion of carbonates formed during oxidation of biogenic methane in fractured granite. Nat. Commun. 2015, 6, 7020. [Google Scholar] [CrossRef] [PubMed]
- Drake, H.; Heim, C.; Roberts, N.M.W.; Zack, T.; Tillberg, M.; Broman, C.; Ivarsson, M.; Whitehouse, M.J.; Åström, M.E. Isotopic evidence for microbial production and consumption of methane in the upper continental crust throughout the Phanerozoic eon. Earth Planet. Sci. Lett. 2017, 470, 108–118. [Google Scholar] [CrossRef]
- Nissenbaum, A.; Presley, B.J.; Kaplan, I.R. Early diagenesis in a reducing fjord, Saanich Inlet, British Columbia–I. Chemical and isotopic changes in major components of interstitial water. Geochim. Cosmochim. Acta 1972, 36, 1007–1027. [Google Scholar] [CrossRef]
- Paull, C.K.; Lorenson, T.D.; Borowski, W.S.; Ussler, W., III; Olsen, K.; Rodriguez, N.M. Isotopic composition of CH4, CO2 species, and sedimentary organic matter within samples from the Blake Ridge: Gas source implications. Proc. ODP Sci. Results 2000, 164, 67–78. [Google Scholar]
- Chatterjee, S.; Dickens, G.R.; Bhatnagar, G.; Chapman, W.G.; Dugan, B.; Snyder, G.T.; Hirasaki, G.J. Pore water sulfate, alkalinity, and carbon isotope profiles in shallow sediment above marine gas hydrate systems: A numerical modeling perspective. J. Geophys. Res. 2011, 116, B9. [Google Scholar] [CrossRef]
- Pohlman, J.W.; Ruppel, C.; Hutchinson, D.R.; Coffin, R.B. Assessing sulfate reduction and methane cycling in a high salinity pore water system in the northern Gulf of Mexico. Mar. Pet. Geol. 2008, 25, 942–951. [Google Scholar] [CrossRef]
- Wu, Z.; Liu, B.; Escher, P.; Kowalski, N.; Böttcher, M.E. Carbon diagenesis in different sedimentary environments of the subtropical Beibu Gulf, South China Sea. J. Mar. Syst. 2018, 186, 68–84. [Google Scholar] [CrossRef]
- Chuang, P.C.; Yang, T.F.; Wallmann, K.; Matsumoto, R.; Hu, C.Y.; Chen, H.W.; Lin, S.; Sun, C.H.; Li, H.C.; Wang, Y.; et al. Carbon isotope exchange during anaerobic oxidation of methane (AOM) in sediments of the northeastern South China Sea. Geochim. Cosmochim. Acta 2019, 246, 138–155. [Google Scholar] [CrossRef]
- Meister, P.; Liu, B.; Ferdelman, T.G.; Jørgensen, B.B.; Khalili, A. Control of sulphate and methane distributions in marine sediments by organic matter reactivity. Geochim. Cosmochim. Acta 2013, 104, 183–193. [Google Scholar] [CrossRef]
- Dale, A.W.; Aguilera, D.R.; Regnier, P.; Fossing, H.; Knab, N.J.; Jørgensen, B.B. Seasonal dynamics of the depth and rate of anaerobic oxidation of methane in Aarhus Bay (Denmark) sediments. J. Mar. Res. 2008, 66, 127–155. [Google Scholar] [CrossRef]
- Mogollón, J.M.; L’Heureux, I.; Dale, A.W.; Regnier, P. Methane gas-phase dynamics in marine sediments: A model study. Am. J. Sci. 2009, 309, 189–220. [Google Scholar] [CrossRef]
- Smrzka, D.; Kraemer, S.M.; Zwicker, J.; Birgel, D.; Fischer, D.; Kasten, S.; Goedert, J.L.; Peckmann, J. Constraining silica diagenesis in methane-seep deposits. Palaeogeogr. Palaeoclim. Palaeoecol. 2015, 420, 13–26. [Google Scholar] [CrossRef]
- Boudreau, B.P. The physics of bubbles in surficial, soft, cohesive sediments. Mar. Pet. Geol. 2012, 38, 1–18. [Google Scholar] [CrossRef]
- Dale, A.W.; Van Cappellen, P.; Aguilera, D.R.; Regnier, P. Methane efflux from marine sediments in passive and active margins: Estimations from bioenergetic reaction-transport simulations. Earth Planet. Sci. Lett. 2008, 265, 329–344. [Google Scholar] [CrossRef]
- Zeebe, R.E. On the molecular diffusion coefficients of dissolved CO2, HCO3−, and CO32−, and their dependence on isotopic mass. Geochim. Cosmochim. Acta 2011, 75, 2483–2498. [Google Scholar] [CrossRef]
- Borowski, W.S.; Paull, C.K.; Ussler, W., III. Global and local variations of interstitial sulfate gradients in deep-water, continental margin sediments: Sensitivity to underlying methane and gas hydrates. Mar. Geol. 1999, 159, 131–154. [Google Scholar] [CrossRef]
- Kennett, J.P.; Cannariato, K.G.; Hendy, I.L.; Behl, R.J. Carbon isotopic evidence for methane hydrate instability during Quaternary Interstadials. Science 2000, 288, 128–133. [Google Scholar] [CrossRef]
- Whiticar, M.J. Carbon and hydrogen isotope systematics of bacterial formation and oxidation of methane. Chem. Geol. 1999, 161, 291–314. [Google Scholar] [CrossRef]
- Baker, P.A.; Burns, S.J. Occurrence and formation of dolomite in organic-rich continental margin sediments. Am. Assoc. Pet. Geol. Bull. 1985, 69, 1917–1930. [Google Scholar]
- Meister, P.; Gutjahr, M.; Frank, M.; Bernasconi, S.; McKenzie, J.A.; Vasconcelos, C. Dolomite formation within the methanogenic zone induced by tectonically-driven fluids in the Peru accretionary prism. Geology 2011, 39, 563–566. [Google Scholar] [CrossRef]
- Meister, P. Two opposing effects of sulfate reduction on calcite and dolomite precipitation in marine, hypersaline and alkaline environments. Geology 2013, 41, 499–502. [Google Scholar] [CrossRef]
- Moore, T.S.; Murray, R.W.; Kurtz, A.C.; Schrag, D.P. Anaerobic methane oxidation and the formation of dolomite. Earth Planet. Sci. Lett. 2004, 229, 141–154. [Google Scholar] [CrossRef]
- Ussler, W., III; Paull, C.K. Rates of anaerobic oxidation of methane and authigenic carbonate mineralization in methane-rich deep-sea sediments inferred from models and geochemical profiles. Earth Planet. Sci. Lett. 2008, 266, 271–287. [Google Scholar] [CrossRef]
- Wallmann, K.; Aloisi, G.; Haeckel, M.; Tishchenko, P.; Pavlova, G.; Greinert, J.; Kutterolf, S.; Eisenhauer, A. Silicate weathering in anoxic marine sediments. Geochim. Cosmochim. Acta 2008, 72, 3067–3090. [Google Scholar] [CrossRef]
- Wehrmann, L.M.; Ockert, C.; Mix, A.; Gussone, N.; Teichert, B.M.A.; Meister, P. Multiple onset of methanogenic zones, diagenetic dolomite formation, and silicate alteration under varying organic carbon deposition in Bering Sea sediments (Bowers Ridge, IODP Exp. 323 Site U1341). Deep Sea Res. II 2016, 125, 117–132. [Google Scholar] [CrossRef]
- Turner, J.V. Kinetic fractionation of carbon-13 during calcium carbonate precipitation. Geochim. Cosmochim. Acta 1982, 46, 1183–1191. [Google Scholar] [CrossRef]
- Deines, P.; Langmuir, D.; Harmon, R.S. Stable Carbon isotope ratios and the existence of a gas phase in the evolution of carbonate ground waters. Geochim. Cosmochim. Acta 1974, 38, 1147–1164. [Google Scholar] [CrossRef]
- Ohmoto, H.; Rye, R.O. Isotopes of sulphur and carbon. In Geochemistry of Hydrothermal Deposits; Barnes, H.L., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 1979; pp. 509–567. [Google Scholar]
- Golyshev, S.I.; Padalko, N.L.; Pechenkin, S.A. Fractionation of stable oxygen and carbon isotopes in carbonate systems. Geochem. Int. 1981, 18, 85–99. [Google Scholar]
- Mook, W.G.; Bommerson, J.C.; Staverman, W.H. Carbon isotope fractionation between dissolved bicarbonate and gaseous carbon dioxide. Earth Planet. Sci. Lett. 1974, 22, 169–176. [Google Scholar] [CrossRef]
- Michaelis, J.; Usdowski, E.; Menschel, G. Partitioning of 13C and 12C on the degassing of CO2 and the precipitation of calcite—Rayleigh-type fractionation and a kinetic model. Am. J. Sci. 1985, 285, 318–327. [Google Scholar] [CrossRef]
- Meister, P.; Bernasconi, S.M.; Aiello, I.W.; Vasconcelos, C.; McKenzie, J.A. Depth and controls of Ca-rhodochrosite precipitation in bioturbated sediments of the Eastern Equatorial Pacific, ODP Leg 201, Site 1226 and DSDP Leg 68, Site 503. Sedimentology 2009, 56, 1552–1568. [Google Scholar] [CrossRef]
- Kasina, M.; Bock, S.; Wuerdemann, H.; Pudlo, D.; Picard, A.; Lichtschlag, A.; März, C.E.; Wagenknecht, L.; Wehrmann, L.M.; Vogt, C.; et al. Mineralogical and geochemical analysis of Fe-phases in drill-cores from the Triassic Stuttgart Formation at Ketzin CO2 storage site before CO2 arrival. Environ. Earth Sci. 2017, 161, 161. [Google Scholar] [CrossRef]
- Bernasconi, S.M. Geochemical and microbial controls on dolomite formation in anoxic environments: A case study from the Middle Triassic (Ticino, Switzerland). In Contributions to Sedimentology; E. Schweizerbart Science Publishers: Stuttgart, Germany, 1994; Volume 19, pp. 1–109. [Google Scholar]
- Meister, P.; McKenzie, J.A.; Bernasconi, S.M.; Brack, P. Dolomite formation in the shallow seas of the Alpine Triassic. Sedimentology 2013, 60, 270–291. [Google Scholar] [CrossRef]
- Heimhofer, U.; Meister, P.; Bernasconi, S.M.; Ariztegui, D.; Martill, D.M.; Rios-Netto, A.M.; Schwark, L. Isotope and elemental geochemistry of black shale-hosted fossiliferous concretions (Early Cretaceous Santana Formation, Brazil). Sedimentology 2017, 64, 150–167. [Google Scholar] [CrossRef]
- Thang, N.M.; Brüchert, V.; Formolo, M.; Wegener, G.; Ginters, L.; Jørgensen, B.B.; Ferdelman, T.G. The impact of sediment and carbon fluxes on the biogeochemistry of methane and sulfur in littoral Baltic Sea sediments (Himmerfjärden, Sweden). Estuar. Coasts 2013, 36, 98–115. [Google Scholar] [CrossRef]
- Jørgensen, N.O. Methane-derived carbonate cementation of marine sediments from the Kattegat, Denmark: Geochemical and geological evidence. Mar. Geol. 1992, 103, 1–13. [Google Scholar] [CrossRef]
- Pisciotto, K.A.; Mahoney, J.J. Isotopic survey of diagenetic carbonates, Deep Sea Drilling Project Leg 63. In Initial Reports of the Deep Sea Drilling Project; Yeats, R.S., Haq, B.U., Eds.; U.S. Govt. Printing Office: Washington, DC, USA, 1981; Volume 63, pp. 595–609. [Google Scholar]
- Kelts, K.; McKenzie, J.A. Diagenetic dolomite formation in Quaternary anoxic diatomaceous muds of Deep Sea Drilling Project Leg 64, Gulf of California. In Init. Repts. DSDP; Curray, J.R., Moore, D.G., Eds.; U.S. Govt. Printing Office: Washington, DC, USA, 1982; Volume 64, pp. 553–569. [Google Scholar]
- Murata, K.J.; Friedman, I.; Madsen, B.H. Isotopic Composition of Diagenetic Carbonates in Marine Miocene Formations of California and Oregon; USGS Prof. Paper; U.S. Govt. Printing Office: Washington, DC, USA, 1969; p. 24.
- Rodriguez, N.M.; Paull, C.K.; Borowski, W.S. Zonation of authigenic carbonates within gas hydrate-bearing sedimentary sections on the blake ridge: Offshore southeastern North America. In Proc. ODP, Sci. Results; Paull, C.K., Matsumoto, R., Wallace, P.J., Dillon, W.P., Eds.; Ocean Drilling Program: College Station, TX, USA, 2000; Volume 164, pp. 301–312. [Google Scholar]
- Compton, J.S. Sediment composition and precipitation of dolomite and pyrite in the Neogene Monterey and Sisquoc Formations, Santa Maria Basin area, California. In Sedimentology and Geochemistry of Dolostones; Shukla, V., Baker, P.A., Eds.; SEPM Spec. Pub.: Tulsa, OK, USA, 1988; Volume 43, pp. 53–64. [Google Scholar]
- Meister, P.; Bernasconi, S.; McKenzie, J.A.; Vasconcelos, C. Sea-level changes control diagenetic dolomite formation in hemipelagic sediments of the Peru Margin. Mar. Geol. 2008, 252, 166–173. [Google Scholar] [CrossRef]
- Alt, J.C.; Teagle, D.A.H. The uptake of carbon during alteration of ocean crust. Geochim. Cosmochim. Acta 1999, 63, 1527–1535. [Google Scholar] [CrossRef]
- Dickens, G.R. A blast of gas in the latest Paleocene: Simulating first-order effects of massive dissociation of oceanic methane hydrate. Geology 1997, 25, 259–262. [Google Scholar] [CrossRef]
- Knoll, A.H.; Hayes, J.M.; Kaufmann, A.J.; Swett, K.; Lambert, I.B. Secular variation in carbon isotope ratios from Upper Proterozoic successions of Svalbard and East Greenland. Nature 1986, 321, 832–838. [Google Scholar] [CrossRef]
- Payne, J.L.; Lehrmann, D.J.; Wei, J.; Orchard, M.J.; Schrag, D.P.; Knoll, A.H. Large perturbations of the carbon cycle during recovery from the end-Permian extinction. Science 2004, 305, 506–509. [Google Scholar] [CrossRef] [PubMed]
- Wefer, G.; Heinze, P.M.; Berger, W.H. Clues to ancient methane release. Nature 1994, 369, 282. [Google Scholar] [CrossRef]
- Louis-Schmid, B.; Rais, P.; Logvinovich, D.; Bernasconi, S.M.; Weissert, H. Impact of methane seeps on the local carbon-isotope record: A case study from a Late Jurassic hemipelagic section. Terra Nova 2007, 19, 259–265. [Google Scholar] [CrossRef]
- Schidlowski, M.; Matzigkeit, U.; Krumbein, W.E. Superheavy organic carbon from hypersaline microbial mats: Assimilatory pathway and geochemical implications. Naturwissenschaften 1984, 71, 303–308. [Google Scholar] [CrossRef]
- Hayes, J.M.; Waldbauer, J., Jr. The carbon cycle and associated redox processes through time. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 931–950. [Google Scholar] [CrossRef]
- Birgel, D.; Meister, P.; Lundberg, R.; Horat, T.; Bontognali, T.; Bahniuk, A.; Rezende, C.; Vasconcelos, C.; McKenzie, J.A. Methanogenesis produces strong 13C-enrichment in stromatolites of Lagoa Salgada, Brazil: A modern analogue for Palaeo-/Neoproterozoic stromatolites? Geobiology 2015, 13, 245–266. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meister, P.; Reyes, C. The Carbon-Isotope Record of the Sub-Seafloor Biosphere. Geosciences 2019, 9, 507. https://doi.org/10.3390/geosciences9120507
Meister P, Reyes C. The Carbon-Isotope Record of the Sub-Seafloor Biosphere. Geosciences. 2019; 9(12):507. https://doi.org/10.3390/geosciences9120507
Chicago/Turabian StyleMeister, Patrick, and Carolina Reyes. 2019. "The Carbon-Isotope Record of the Sub-Seafloor Biosphere" Geosciences 9, no. 12: 507. https://doi.org/10.3390/geosciences9120507
APA StyleMeister, P., & Reyes, C. (2019). The Carbon-Isotope Record of the Sub-Seafloor Biosphere. Geosciences, 9(12), 507. https://doi.org/10.3390/geosciences9120507