Evaluation of the Gas Content in Archived Shale Samples: A Carbon Isotope Study

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Shale Samples
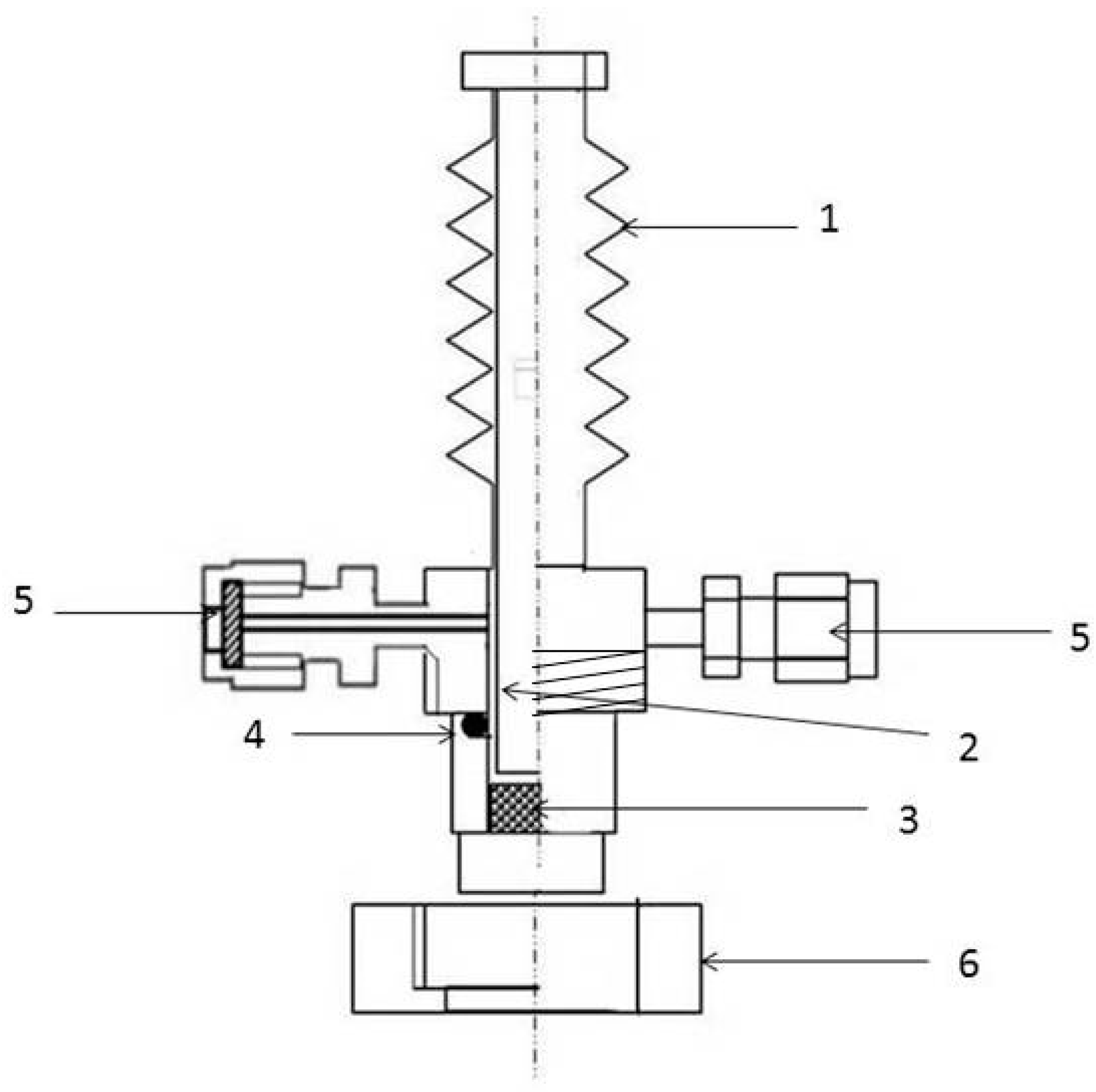
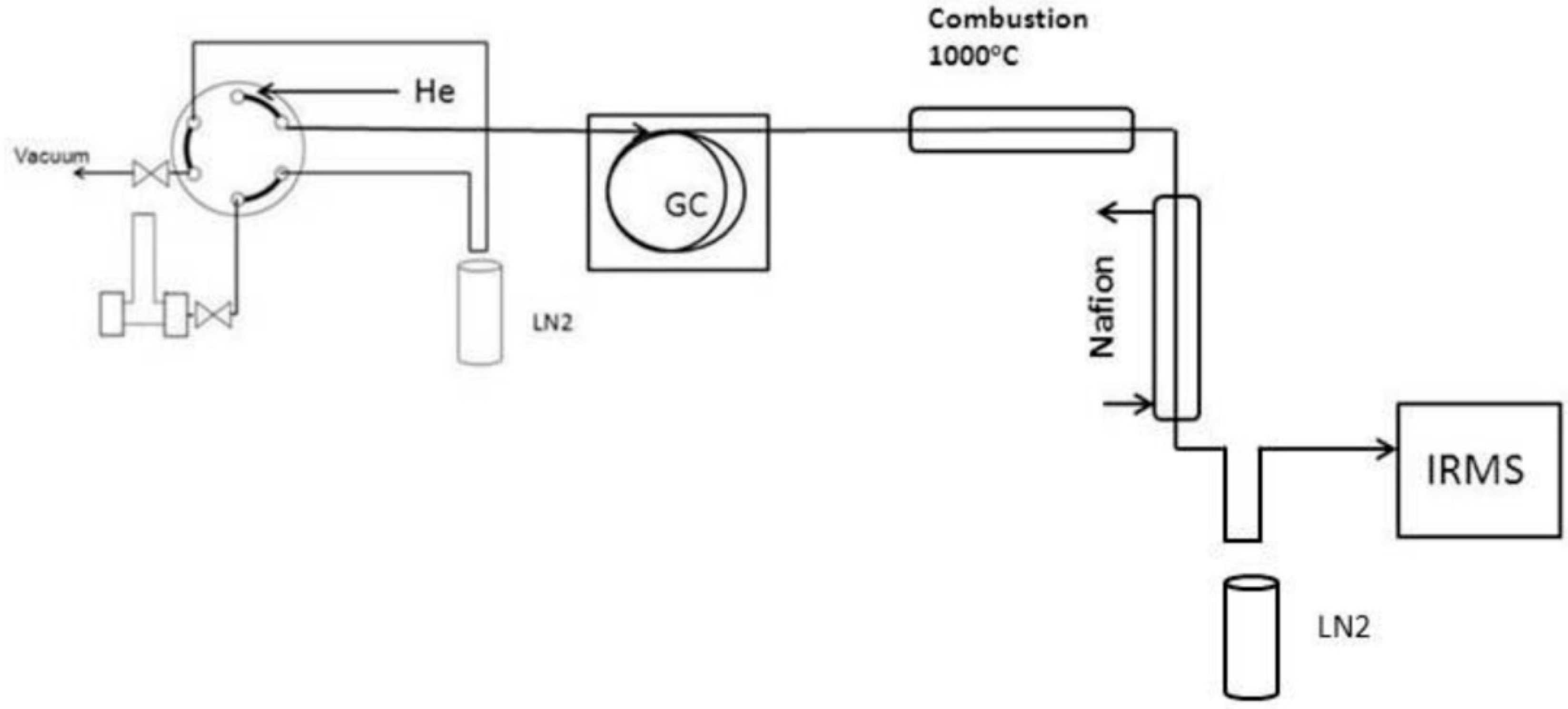
2.2. Analytical Methods
2.3. The Mechanism of Gas Release
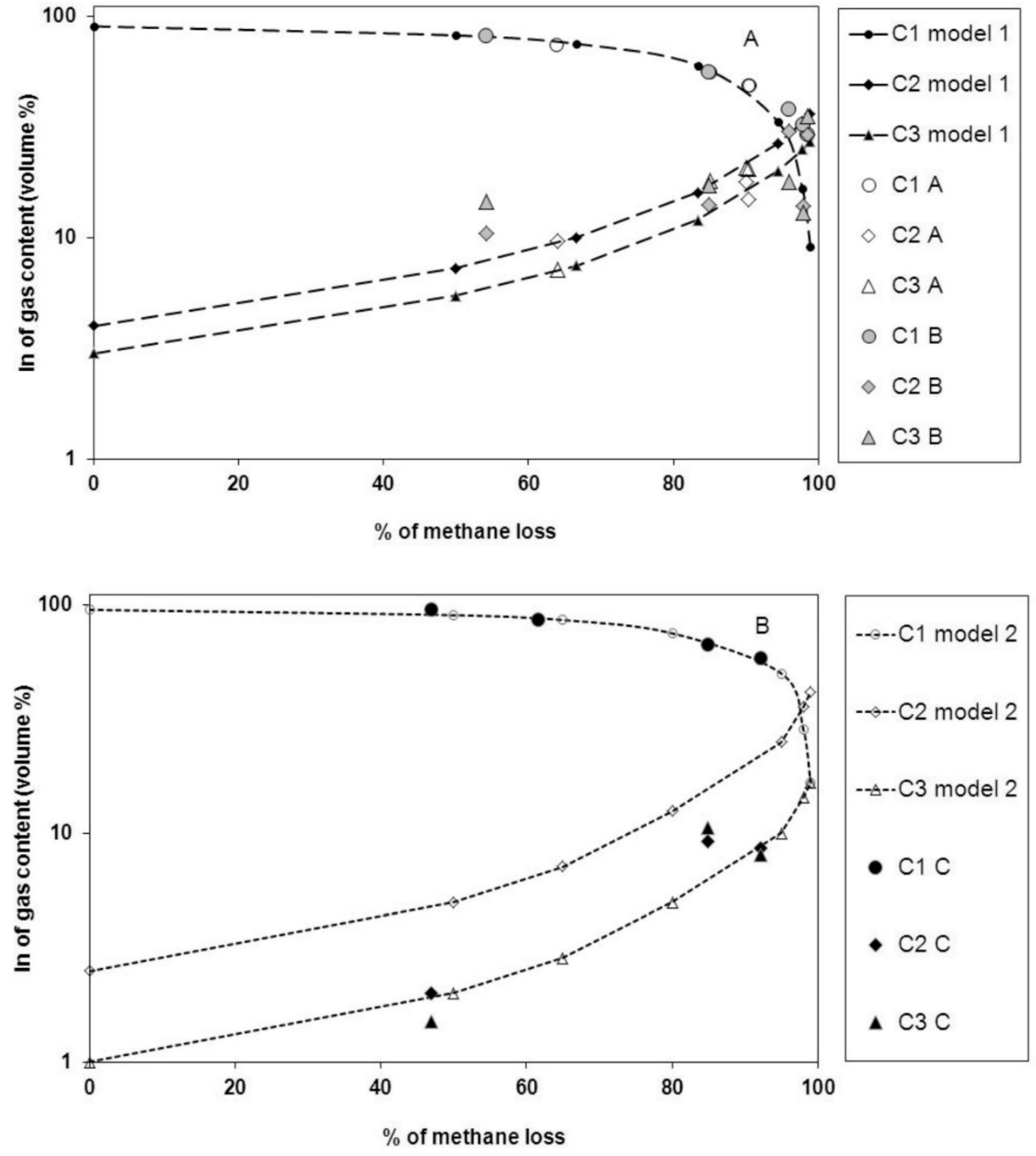
3. Results
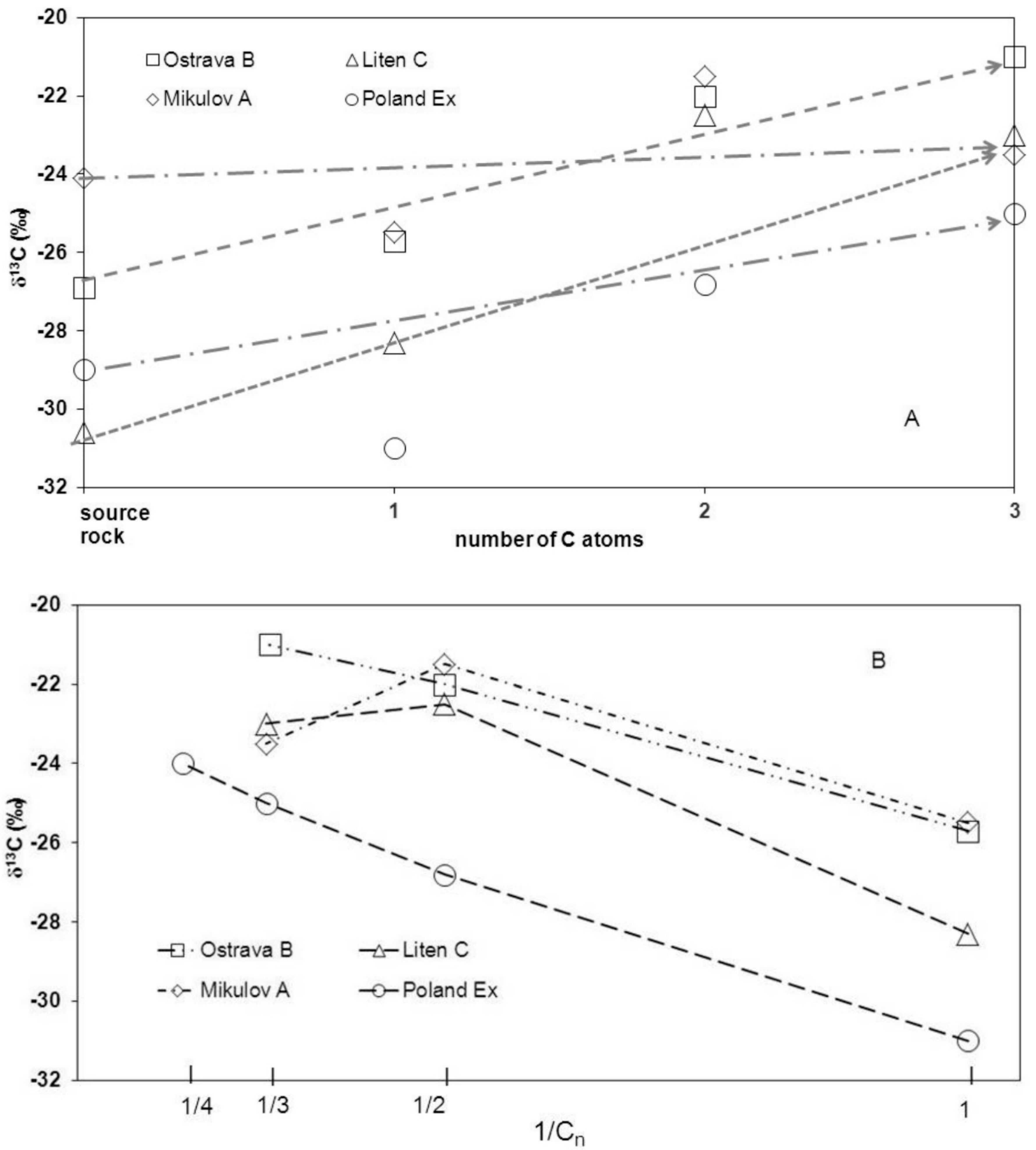
4. Discussion
4.1. Estimate of Gas at the Site
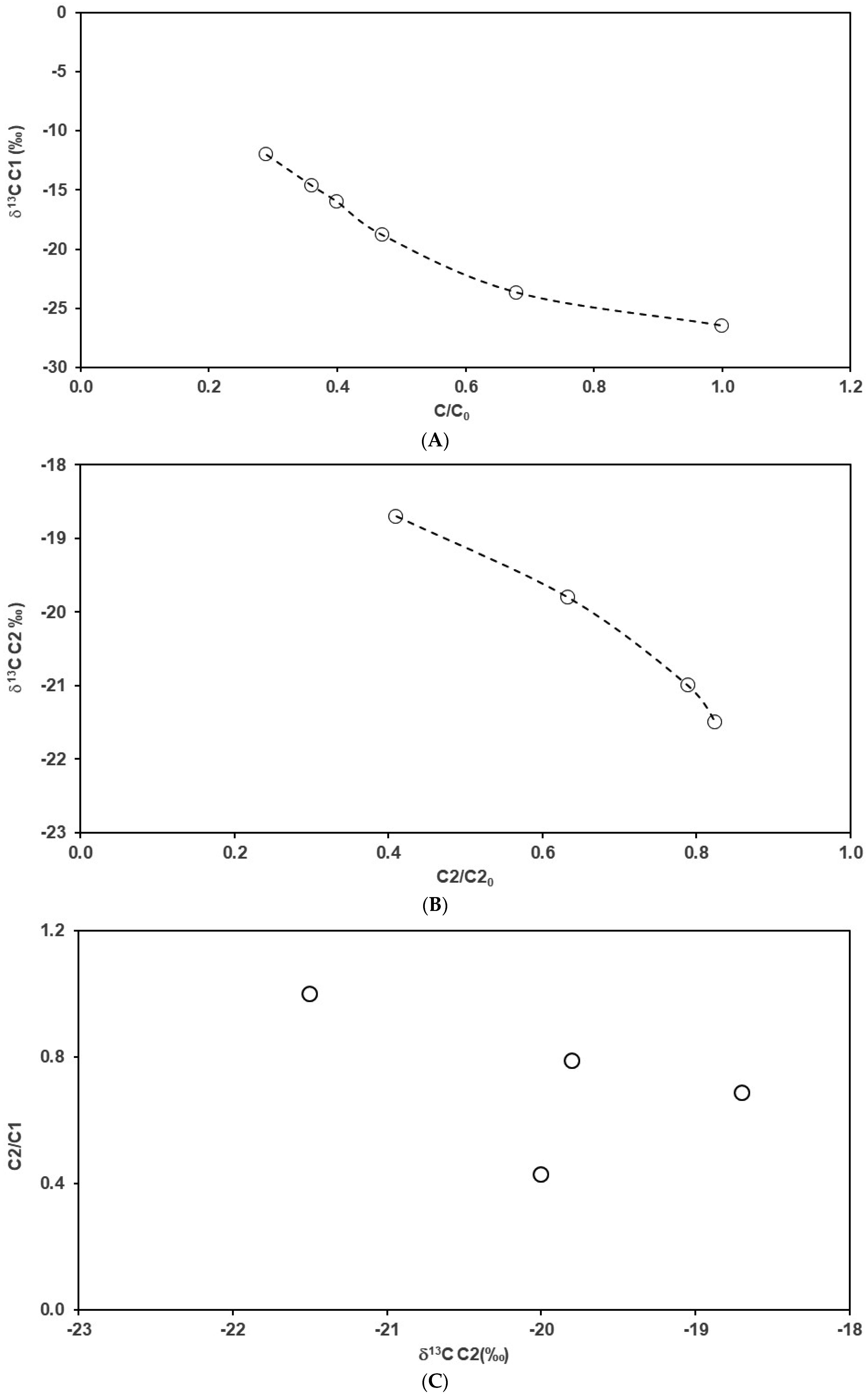
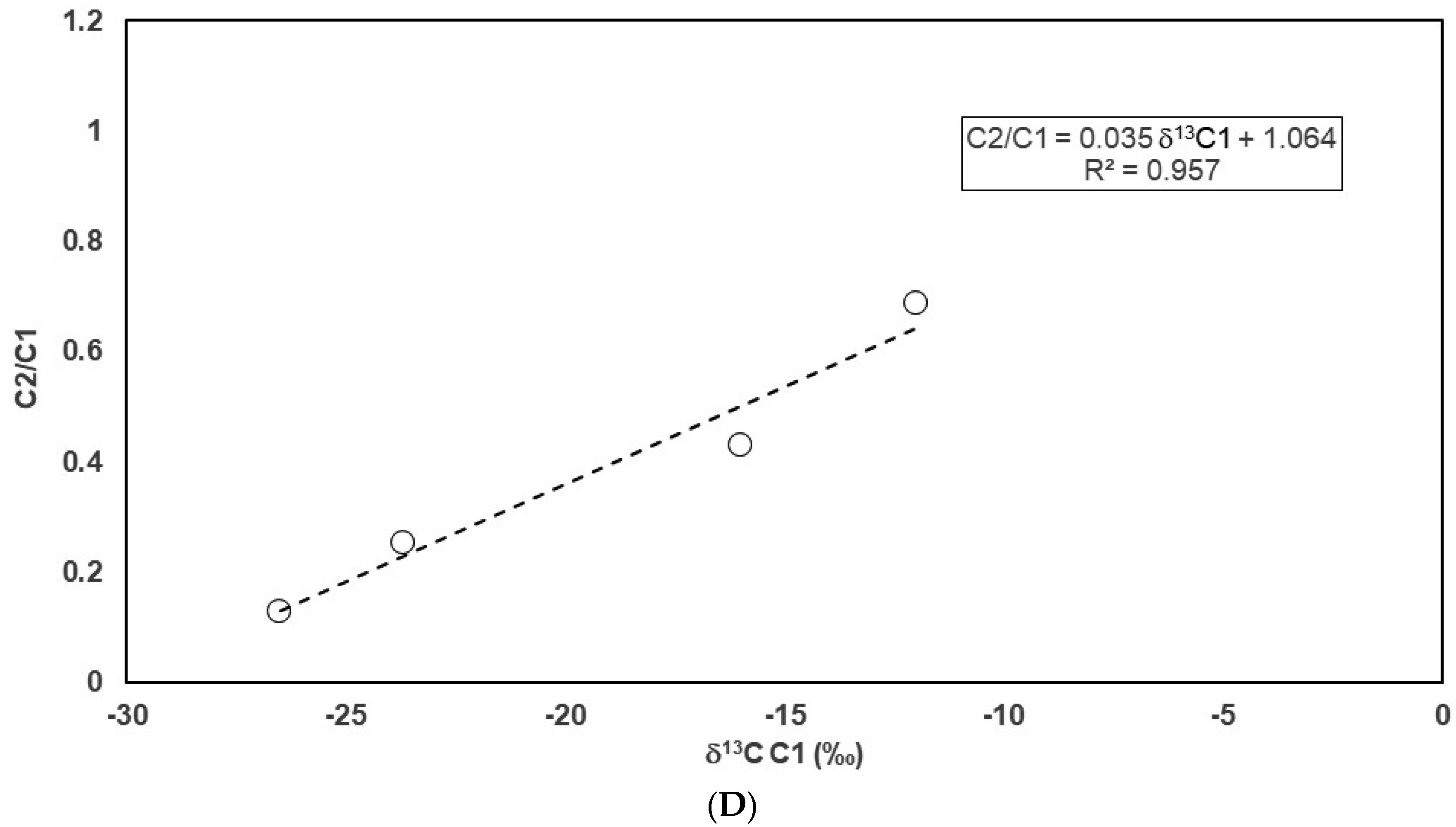
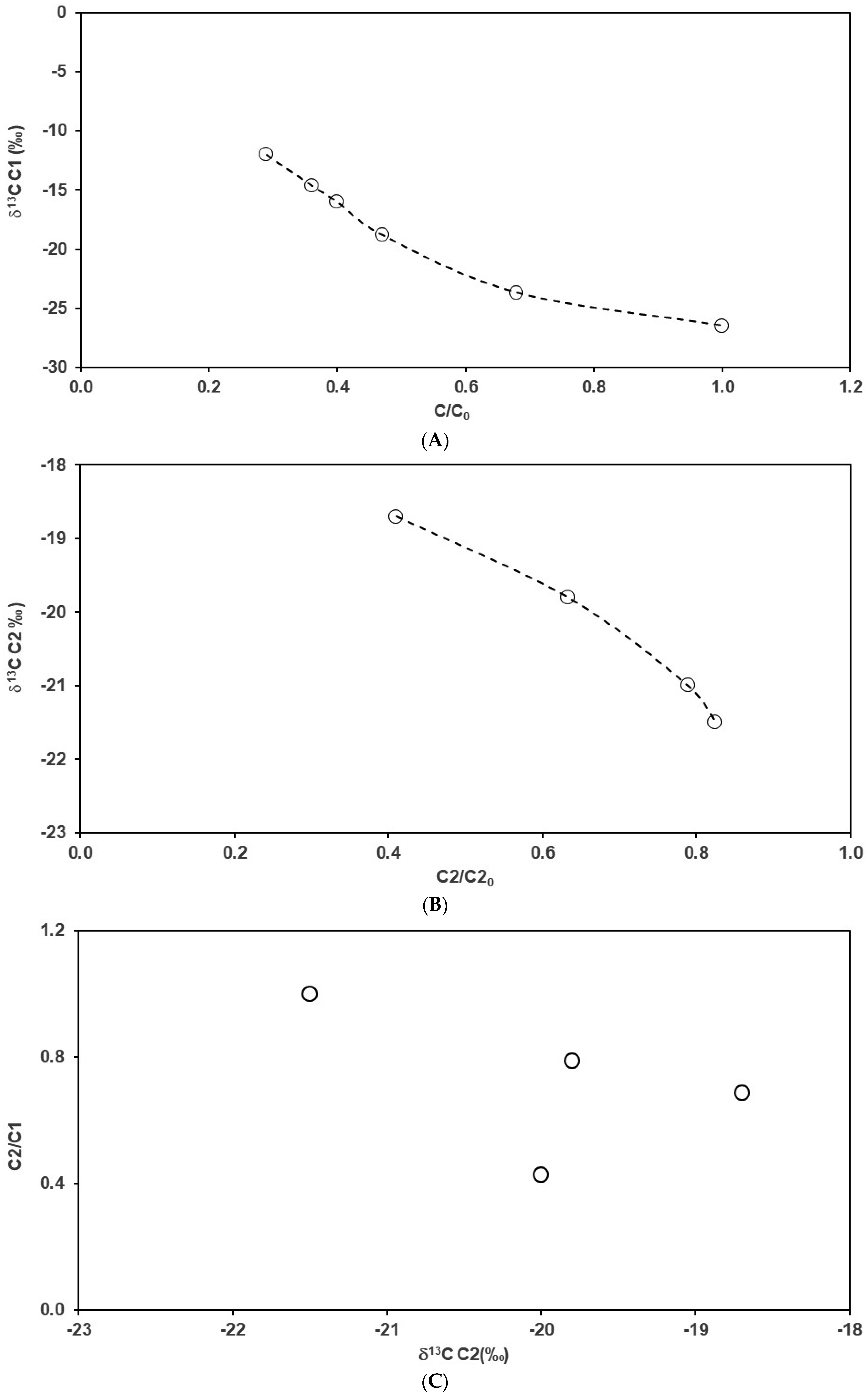
4.2. Carbon Isotope Fractionation during the Sample Degassing
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chaelrs, B.; John, L.; Reberto, S.-R.; Richard, E.L.; George, W. Producing Gas from Its Source. Oilfield Rev. 2006, 18, 36–49. [Google Scholar]
- John, B.C. Fractured shale-gas systems. AAPG Bull. 2002, 86, 1921–1938. [Google Scholar]
- Petroleum Formation and Occurrence; Tissot, B.P.; Welte, D.H. (Eds.) Springer: Heidelberg, Germany, 1984. [Google Scholar]
- Peters, K.E. Guidelines for Evaluating Petroleum Source Rock Using Programmed Pyrolysis. AAPG Bull. 1986, 70, 318–329. [Google Scholar]
- Schoell, M. Genetic Characterization of Natural Gases. AAPG Bull. 1983, 67, 2225–2238. [Google Scholar]
- Tang, Y.; Jenden, P.D.; Nigrini, A.; Teerman, S.C. Modeling Early Methane Generation in Coal. Energy Fuels 1996, 10, 659–671. [Google Scholar] [CrossRef]
- Tang, Y.; Perry, J.K.; Jenden, P.D.; Schoell, M. Mathematical modeling of stable carbon isotope ratios in natural gases. Geochim. Cosmochim Acta 2000, 64, 2673–2687. [Google Scholar] [CrossRef]
- Zhang, T.; Krooss, B.M. Experimental investigation of the carbon isotope fractionation of methane during gas migration by diffusion through sedimentary rocks at elevated temperature and pressure. Geochim. Cosmochim. Acta 2001, 65, 2723–2742. [Google Scholar] [CrossRef]
- Schloemer, S.; Krooss, B.M. Molecular transport of methane, ethane and nitrogen and the influence of diffusion on the chemical and isotopic composition of natural gas accumulations. Geofluids 2004, 4, 81–108. [Google Scholar] [CrossRef]
- Goddard, W.A., III; Tang, Y.; Wu, S.; Deev, A.; Ma, Q.; Li, G. Novel Gas Isotope Interpretation Tools to Optimize Gas Shale Production; Report Number 08122.15; Californian Institute of Technology: Pasadena, CA, USA, 2013. [Google Scholar]
- Xia, X.; Tang, Y. Isotope fractionation of methane during natural gas flow with coupled diffusion and adsorption/desorption. Geochim. Cosmochim. Acta 2012, 77, 489–503. [Google Scholar] [CrossRef]
- Strąpoć, D.; Schimmelmann, A.; Mastalerz, M. Carbon isotopic fractionation of CH4 and CO2 during canister desorption of coal. Org. Geochem. 2006, 37, 152–164. [Google Scholar] [CrossRef]
- Zhang, T.; Yang, R.; Milliken, K.L.; Ruppel, S.C.; Pottorf, R.J.; Sun, X. Chemical and isotopic composition of gases released by crush methods from organic rich mudrocks. Org. Geochem. 2014, 73, 16–28. [Google Scholar] [CrossRef]
- Geršlova, E.; Opletal, V.; Sýkorová, I.; Sedláková, I.; Geršl, M. A geochemical and petrographical characterization of organic matter in the Jurassic Mikulov Marls from the Czech Republic. Int. J. Coal Geol. 2015, 141, 42–50. [Google Scholar] [CrossRef]
- Geršlová, E.; Goldbach, M.; Geršl, M.; Skupien, P. Heat flow evolution, subsidence and erosion in Upper Silesian Coal Basin, Czech Republic. Int. J. Coal Geol. 2016, 154, 30–42. [Google Scholar] [CrossRef]
- Suchy, V.; Sýkorová, I.; Stejskal, M.; Safanda, J.; Machovič, V.; Novotná, M. Dispersed organic matter from Silurian shales of the Barrandian Basin, Czech Republic: Optical properties, chemical composition and thermal maturity. Int. J. Coal Geol. 2002, 53, 1–25. [Google Scholar] [CrossRef]
- Picha, J.F.; Stranik, Z.; Krejci, O. Geology and hydrocarbon resources of the outer West Carpathians and their foreland, Czech Republic. In The Carpathians and Their Foreland: Geology and Hydrocarbon Resources; Golonka, J., Picha, J.F., Eds.; American Association of Petroleum Geologists (AAPG) Memoirs: Tulsa, OK, USA, 2006. [Google Scholar]
- Sedláková, I.; Geršlová, E.; Uhlík, P.; Opletal, V. Mineralogical characteristics of upper Jurassic Mikulov Marls, the Czech Republic, in relation to their thermal maturity. Geochemie der Erde 2017, 77, 159–167. [Google Scholar] [CrossRef]
- Transport Modeling for Environmental Engineers and Scientists; Clark, M.M. (Ed.) Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Chemodynamics: Environmental Movement of Chemicals in Air, Water, and Soil; Louis, J.T. (Ed.) Wiley: Hoboken, NJ, USA, 1979. [Google Scholar]
- Handbook of Environmental Isotope Geochemistry, vol. 1 a, Terrestrial Environment; Fritz, P.; Fontes, J.C. (Eds.) Elsevier: Amsterdam, The Netherlands, 1980. [Google Scholar]
- Niemann, M.; Whiticar, M.J. Stable Isotope Systematics of Coalbed Gas during Desorption and Production. Geoscience 2017, 7, 43. [Google Scholar] [CrossRef]
- Bacon, C.A.; Calver, C.R.; Boreham, C.J.; Leaman, D.E.; Morrison, K.C.; Revill, A.T.; Volkman, J.K. The petroleum potential of onshore Tasmania: A review. Geol Survey Bull. 2000, 71, 1–93. [Google Scholar]
- Prinzhofer, A.; Pernaton, E. Isotopically light methane in natural gas: Bacterial imprint or diffusive fractionation? Chem. Geol. 1997, 142, 193–200. [Google Scholar] [CrossRef]
- John, Z.; Kevin, F.; Stephen, B. Isotopic reversal (“rollover”) in shale gases produced from the Mississipian Barnett and Fayetteville formations. Mar. Petrol. Geol. 2012, 31, 43–52. [Google Scholar]
- Chung, H.; Gormly, J.; Squires, R. Origin of gaseous hydrocarbons in subsurface environments: Theoretical considerations of carbon isotope distribution. Chem. Geol. 1988, 71, 97–104. [Google Scholar] [CrossRef]
- Berner, U.; Faber, E. Empirical carbon isotope/maturity relationships for gases from algal kerogens and terrigenous organic matter, based on dry, open-system pyrolysis. Org. Geochem. 1996, 24, 947–955. [Google Scholar] [CrossRef]
- Meng, Q.; Wang, X.; Wang, X.; Zhang, L.; Jiang, C.; Li, X.; Shi, B. Variation in the carbon isotopic composition of alkanes during shale gas desorption process and its geological significance. J. Nat. Gas Geosci. 2016, 1, 139–146. [Google Scholar] [CrossRef]
- Strapozc, D.; Michael, G.E.; Roper, J.; Maguire, M. Insight into shale gas production & storage from gas chemistry—what is it telling us? In Proceedings of the AAPG Hedberg Conference, Austin, TX, USA, 5–10 December 2010. [Google Scholar]
- Hao, F.; Zou, H. Cause of shale gas geochemical anomalies and mechanisms for gas enrichment and depletion in high-maturity shales. Mar. Petrol. Geol. 2013, 44, 1–12. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
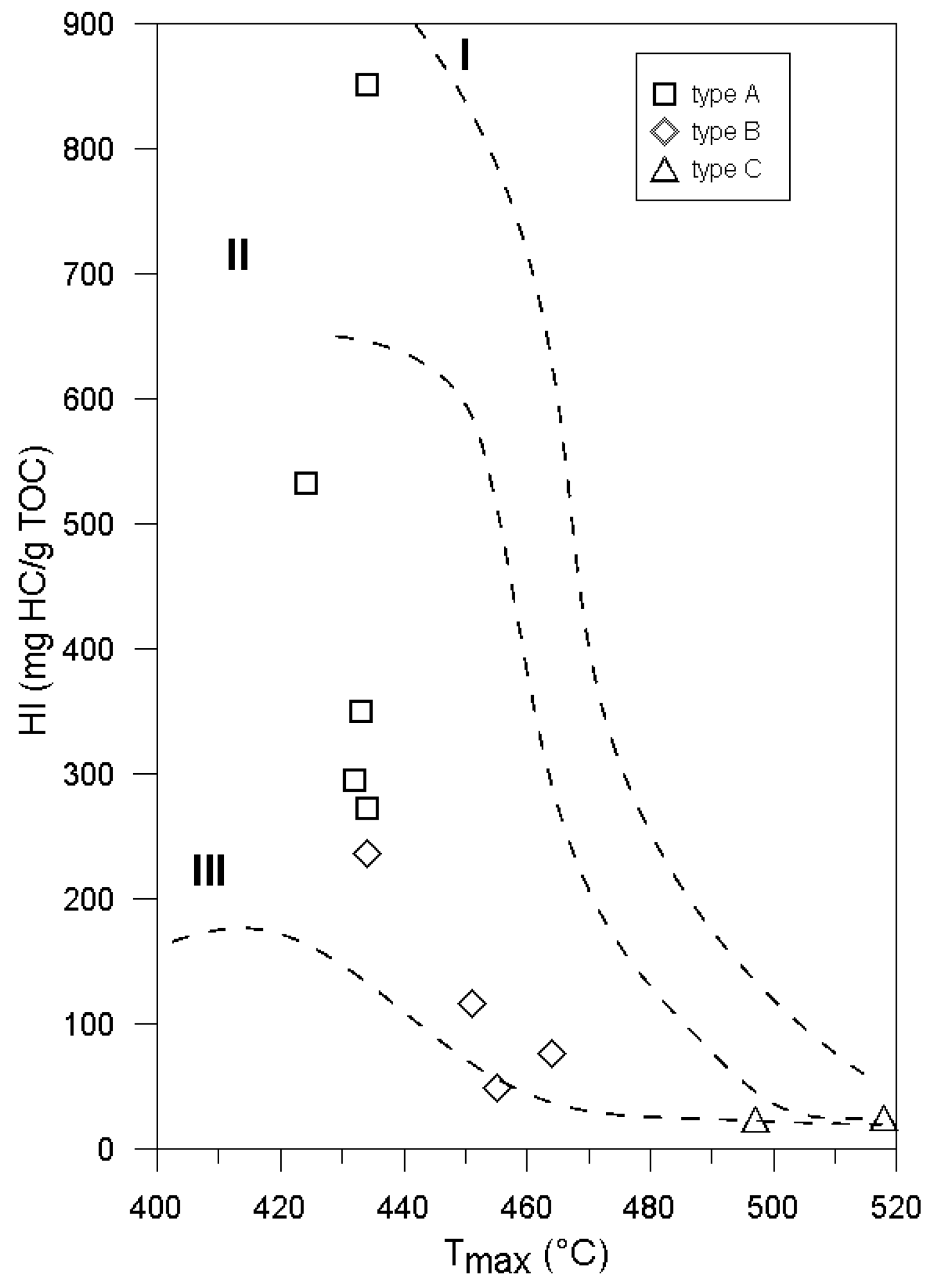
| Sample | Corg (%) | δ13C (‰) | TOC Calculated (%) | Tmax (°C) | S2 (mg HC/g Rock) | S1 (mg HC/g Rock) | HI (mg HC/g TOC) | OI (mg CO2/g TOC) | SR Type | PI = S1/(S1 + S2) | Rr (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 2.56 | −26.7 | 1.46 | 432 | 4.31 | 0.11 | 295 | 47 | Very good | 0.02 | 0.69 |
| A | 2.44 | −27.2 | 1.39 | 434 | 3.78 | 0.09 | 272 | 22 | Very good | 0.02 | 0.72 |
| A | 2.47 | −26.8 | 1.23 | 433 | 4.30 | 0.07 | 350 | 33 | Very good | 0.02 | 0.68 |
| A | 3.57 | −26.7 | 0.96 | 434 | 8.17 | 0.23 | 851 | 90 | Very good | 0.03 | 0.65 |
| A | 0.74 | −24.3 | 2.4 | 424 | 12.79 | 0.23 | 533 | 29 | Fair | 0.02 | 0.66 |
| B | 1.08 | −23.1 | 1.09 | 455 | 0.52 | 0.04 | 48 | 26 | Good | 0.07 | 1.20 |
| B | 4.57 | −24.2 | 1.48 | 434 | 3.50 | 0.12 | 236 | 13 | Very good | 0.03 | 0.64 |
| B | 1.45 | −24 | 1.44 | 451 | 1.60 | 0.08 | 117 | Good | 0.05 | 1.00 | |
| B | 0.98 | −24.5 | 1.67 | 464 | 1.20 | 0.08 | 77 | Good | 0.06 | 1.05 | |
| C | 3.18 | −31.5 | 3.89 | 501 | 1.00 | 0.05 | 27 | 1 | Very good | 0.05 | 1.79 |
| C | 3.18 | −30.4 | 3.89 | 518 | 0.90 | 0.06 | 25 | 1 | Very good | 0.06 | |
| C | 5.69 | −29.8 | 6.32 | 504 | 1.35 | 0.07 | 22 | 1 | Very good | 0.05 | |
| C | 3.26 | −30 | 5.07 | 497 | 1.10 | 0.09 | 23 | 1 | Very good | 0.08 | |
| Ex | 6.43 | −29 | 2.59 | 455 | 1.65 | 0.44 | 24 | 0 | Very good | 0.21 |
| Sample | Depth (m) | Gas Volume (m3∙t−1) | C1 (%) | δ13C1 (‰) | C2 (%) | δ13C2 (‰) | C3 (%) | δ13C3 (‰) | C4 (%) | δ13C4 (‰) |
|---|---|---|---|---|---|---|---|---|---|---|
| A | 3342 | 0.00050 | 48.5 | −19.6 | 14.8 | −20.6 | 24.4 | 10.9 | ||
| A | 3491 | 0.00065 | 74.1 | −22.4 | 9.6 | −21.4 | 7.2 | −21 | 6.3 | |
| A | 2997 | 0.00045 | 48.4 | −18.8 | 17.8 | −17.9 | 20.7 | 10.3 | ||
| A | 3411 | 0.00055 | 55.5 | −18.4 | 12.6 | −16.9 | 18.3 | 10.1 | ||
| A | 2546 | 0.00090 | 81.3 | −26.5 | 10.4 | −24.6 | 4.6 | 1.8 | ||
| B | 4575 | 0.00040 | 55.5 | −23.7 | 14.1 | −22.3 | 17.3 | −23 | 10 | |
| B | 4034 | 0.00017 | 38.0 | −18.8 | 20.0 | −19.8 | 24.4 | 14 | ||
| B | 1374 | 0.00015 | 29.2 | −14.6 | 29.2 | −21.5 | 35.3 | 6.3 | ||
| B | 4760 | 0.00030 | 32.3 | −16.0 | 13.9 | −20 | 13 | 22.8 | ||
| C | 0.0014 | 58.2 | −22.0 | 8.6 | −19.4 | 10 | 10 | |||
| C | 0.0006 | 67.0 | −21.3 | 9.2 | −21 | 10.5 | −23 | 7.8 | ||
| C | 0.0125 | 95.0 | −27.9 | 2.0 | −22 | 1 | 0.8 | |||
| C | 0.0078 | 86.0 | −26.0 | 8.9 | 1.5 | 1.8 | ||||
| Ex | 0.0239 | 44 | −24.2 | 26 | −25.2 | 12 | −25 | 10 | −24 |
| Formation | δ13C Rock (‰) | δ13C10 (‰) | ε C1 (‰) | δ13C20 (‰) | ε C2 (‰) | V0 Gas (m3∙t−1) |
|---|---|---|---|---|---|---|
| B | −24.1 | −25.7 | −2.9 | −22 | −3.7 | 0.00165 |
| A | −26.9 | −25.5 | −3 | −21.6 | −2.7 | 0.0017 |
| C | −30.6 | −28.3 | −2.7 | −22.5 | −3.1 | 0.0132 |
| Ex * | −29 | −31 | −3 | −26.8 | 0.200 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frantisek, B.; Eva, G.; Milan, G.; Bohuslava, C.; Ivana, J.; Zdena, L. Evaluation of the Gas Content in Archived Shale Samples: A Carbon Isotope Study. Geosciences 2019, 9, 481. https://doi.org/10.3390/geosciences9110481
Frantisek B, Eva G, Milan G, Bohuslava C, Ivana J, Zdena L. Evaluation of the Gas Content in Archived Shale Samples: A Carbon Isotope Study. Geosciences. 2019; 9(11):481. https://doi.org/10.3390/geosciences9110481
Chicago/Turabian StyleFrantisek, Buzek, Gerslova Eva, Gersl Milan, Cejkova Bohuslava, Jackova Ivana, and Lnenickova Zdena. 2019. "Evaluation of the Gas Content in Archived Shale Samples: A Carbon Isotope Study" Geosciences 9, no. 11: 481. https://doi.org/10.3390/geosciences9110481
APA StyleFrantisek, B., Eva, G., Milan, G., Bohuslava, C., Ivana, J., & Zdena, L. (2019). Evaluation of the Gas Content in Archived Shale Samples: A Carbon Isotope Study. Geosciences, 9(11), 481. https://doi.org/10.3390/geosciences9110481