
Stable Isotope Systematics of Coalbed Gas during Desorption and Production

Abstract
:1. Introduction
2. Samples and Methods
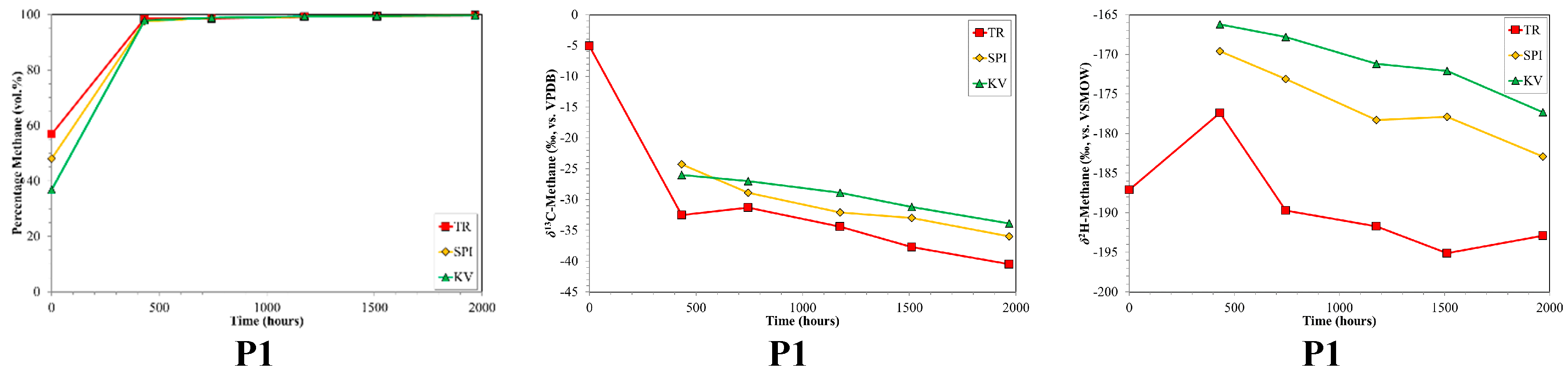
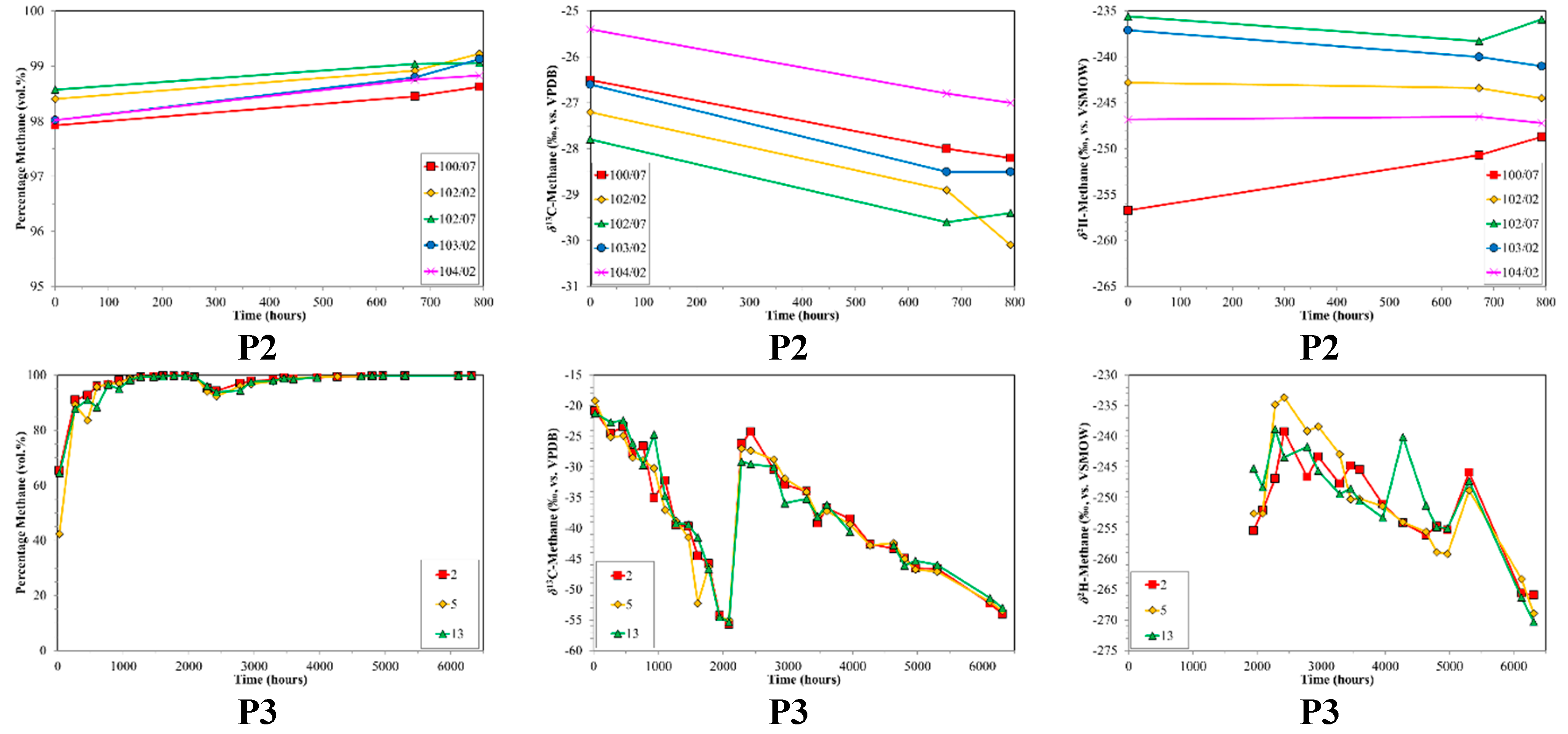
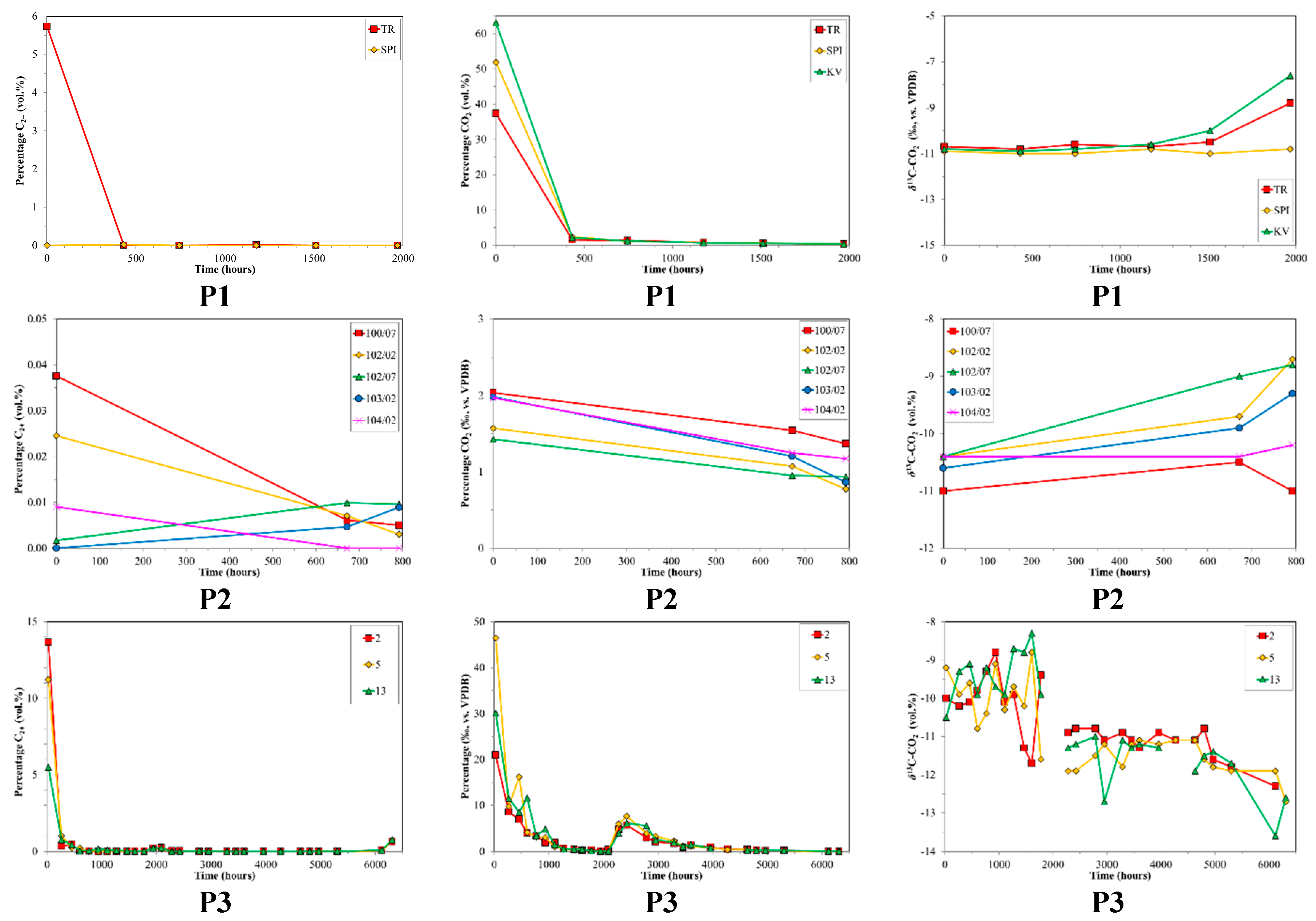
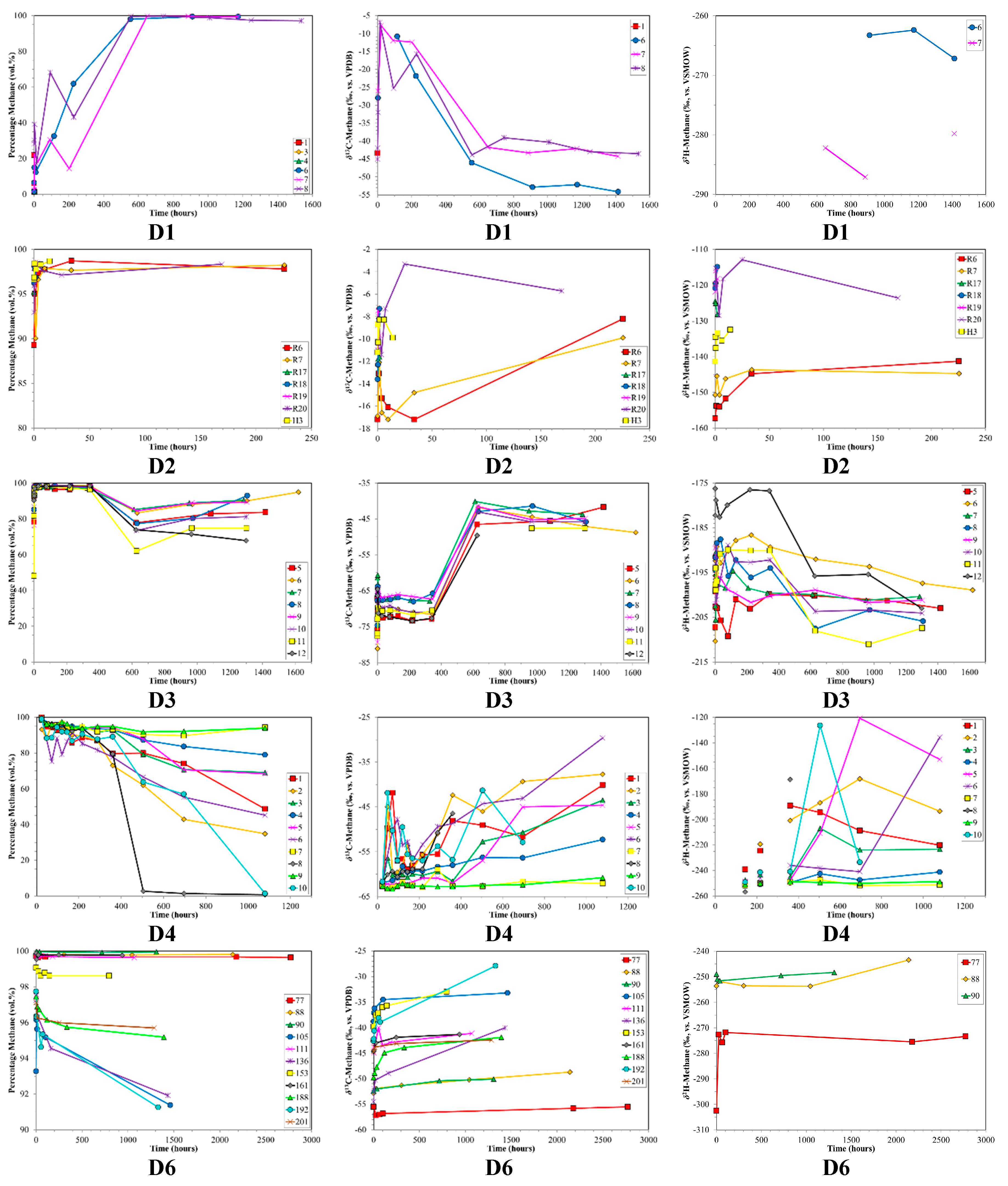
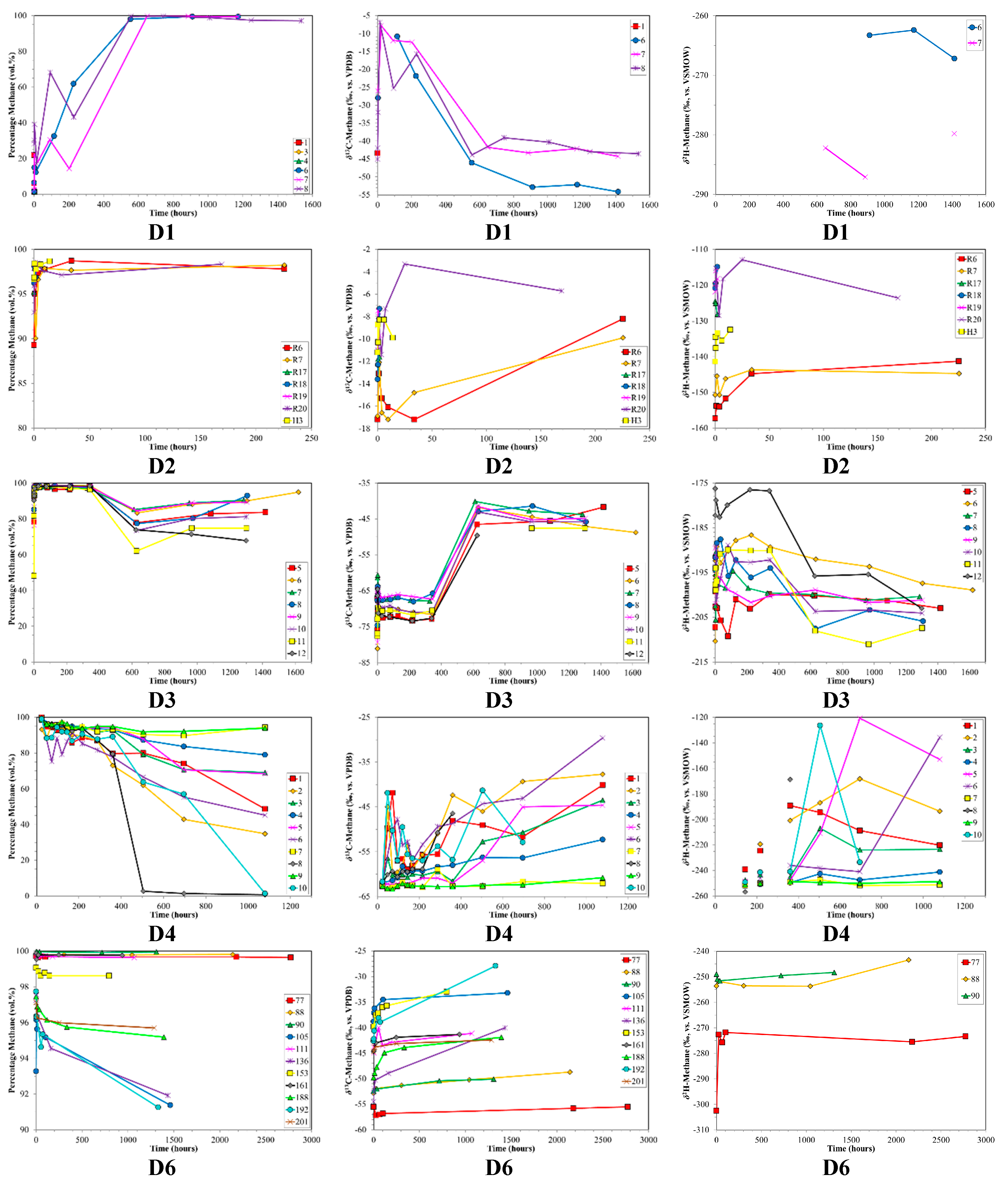
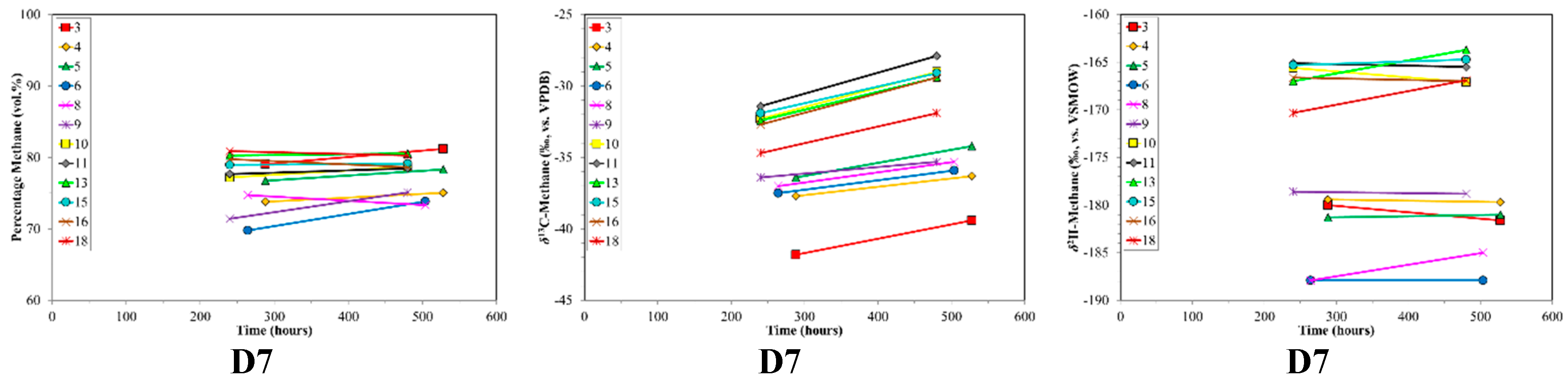
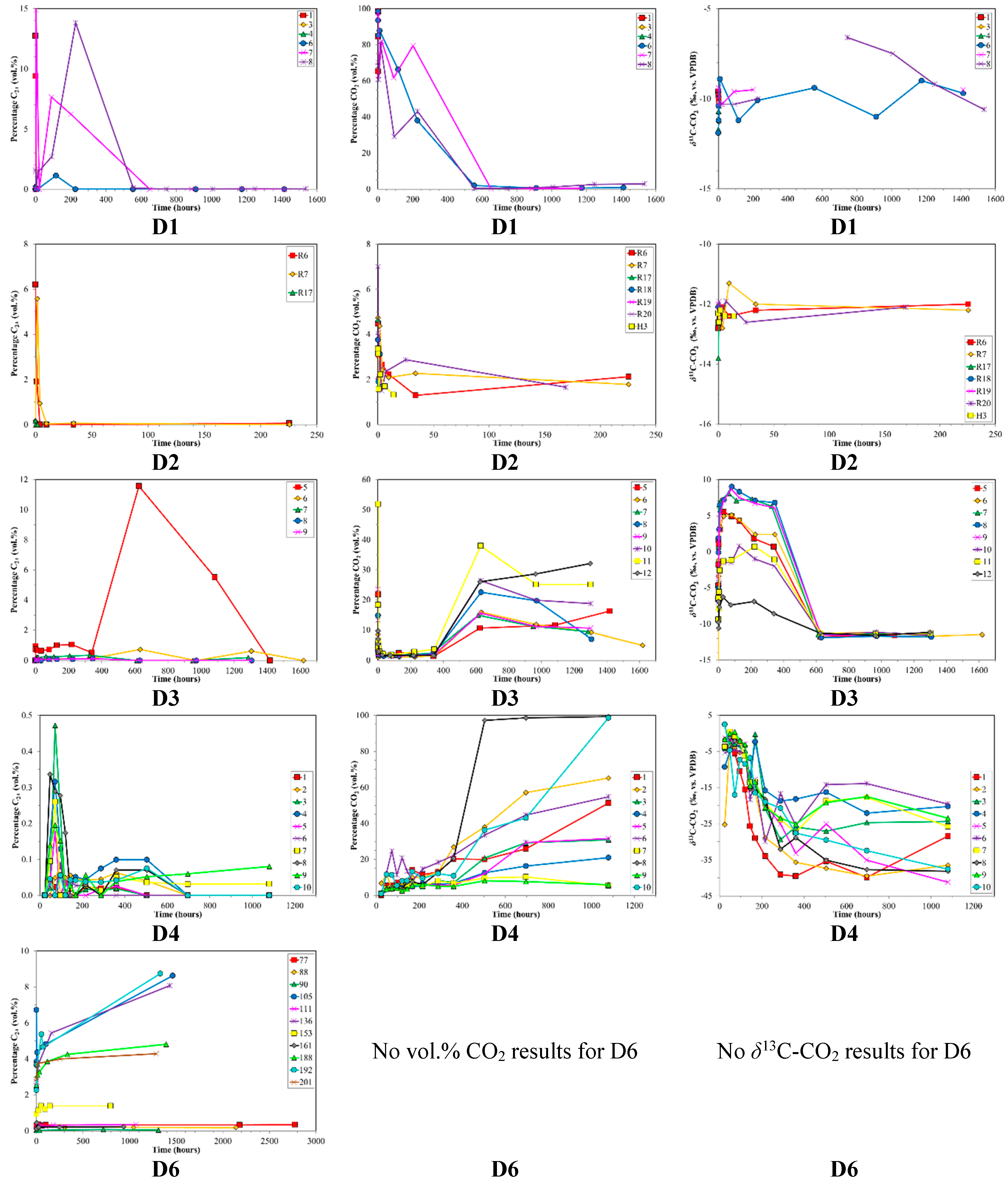
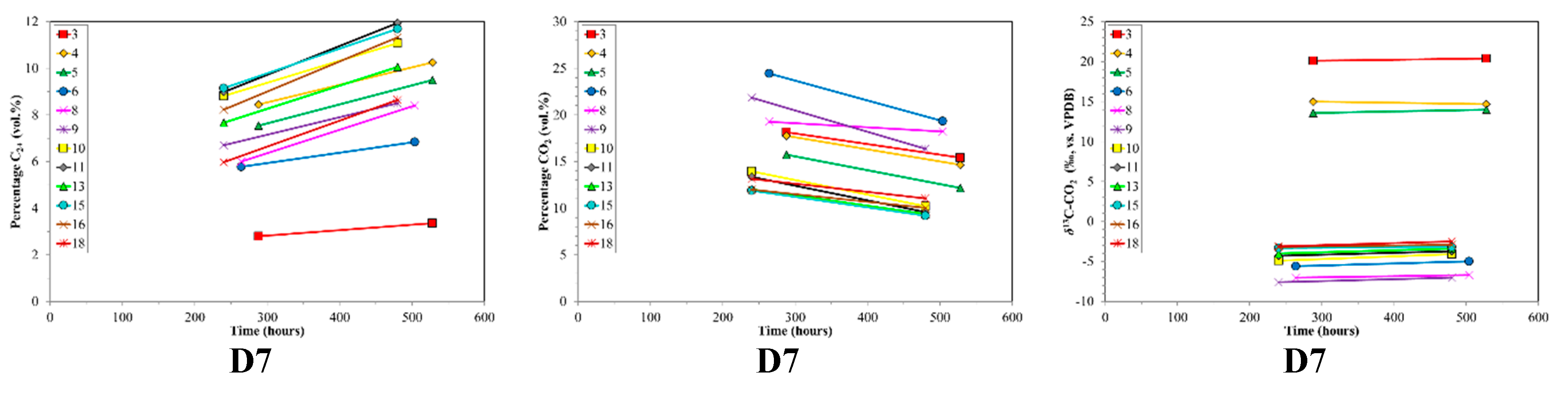
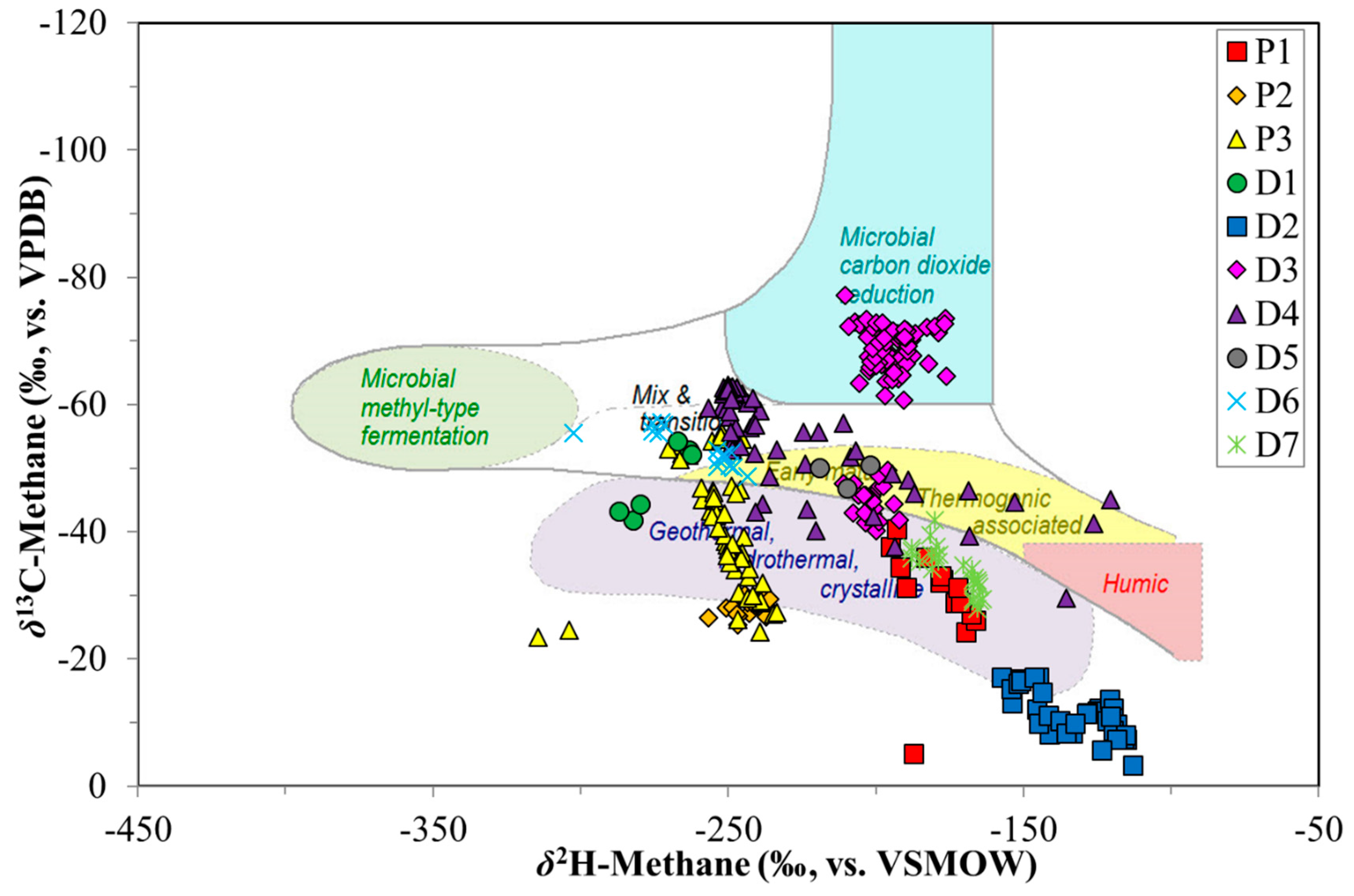
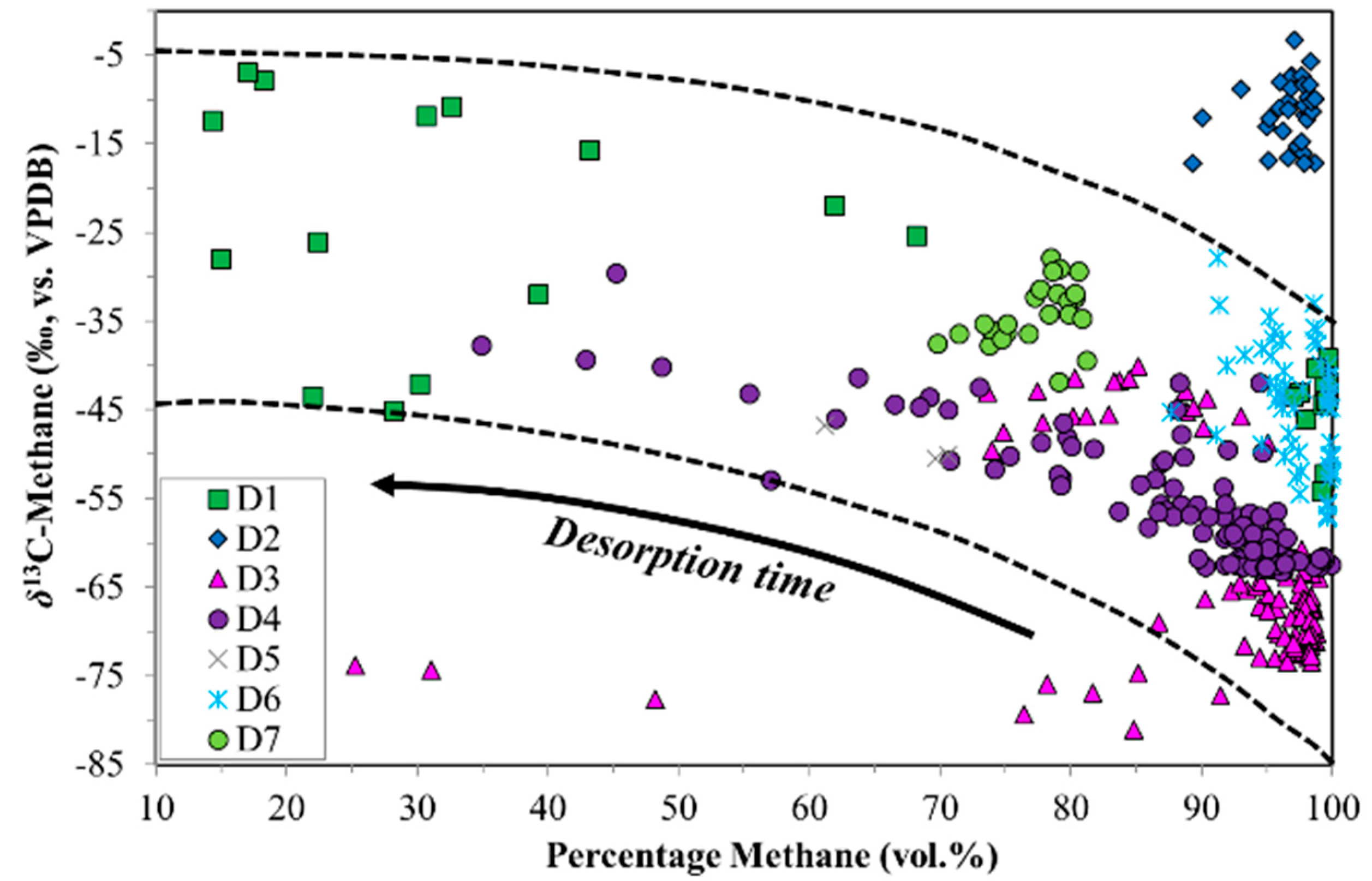
3. Results
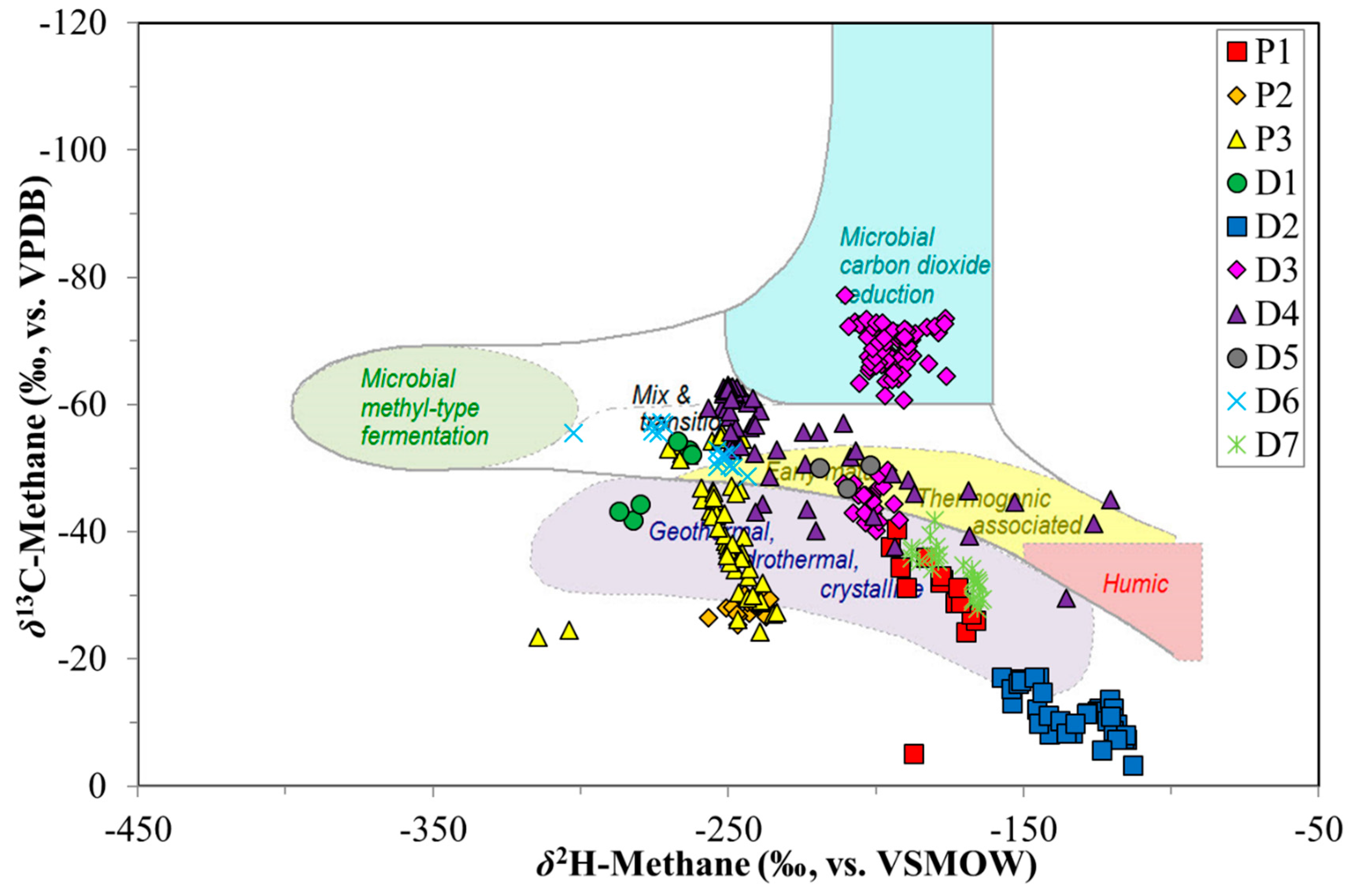
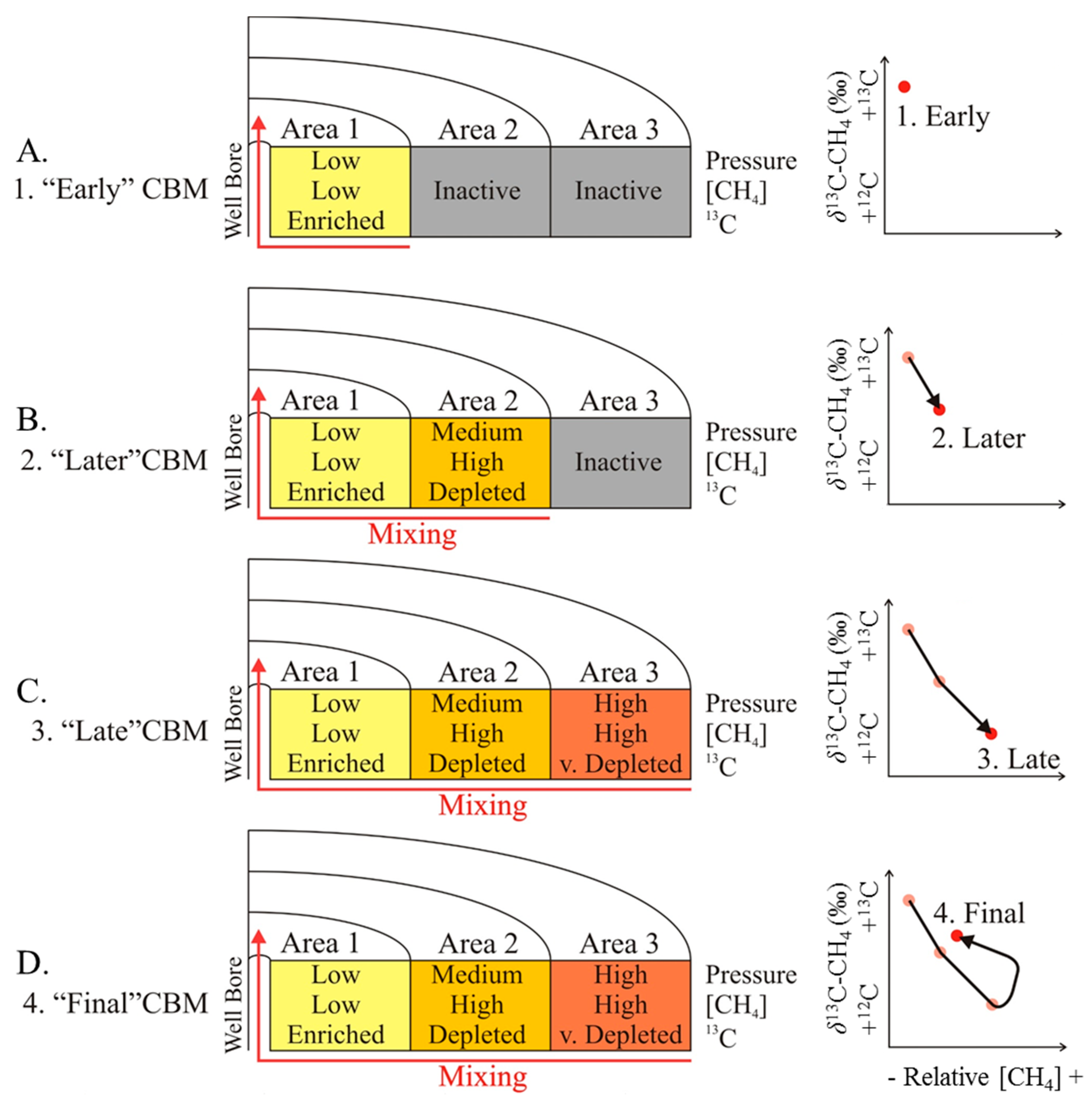
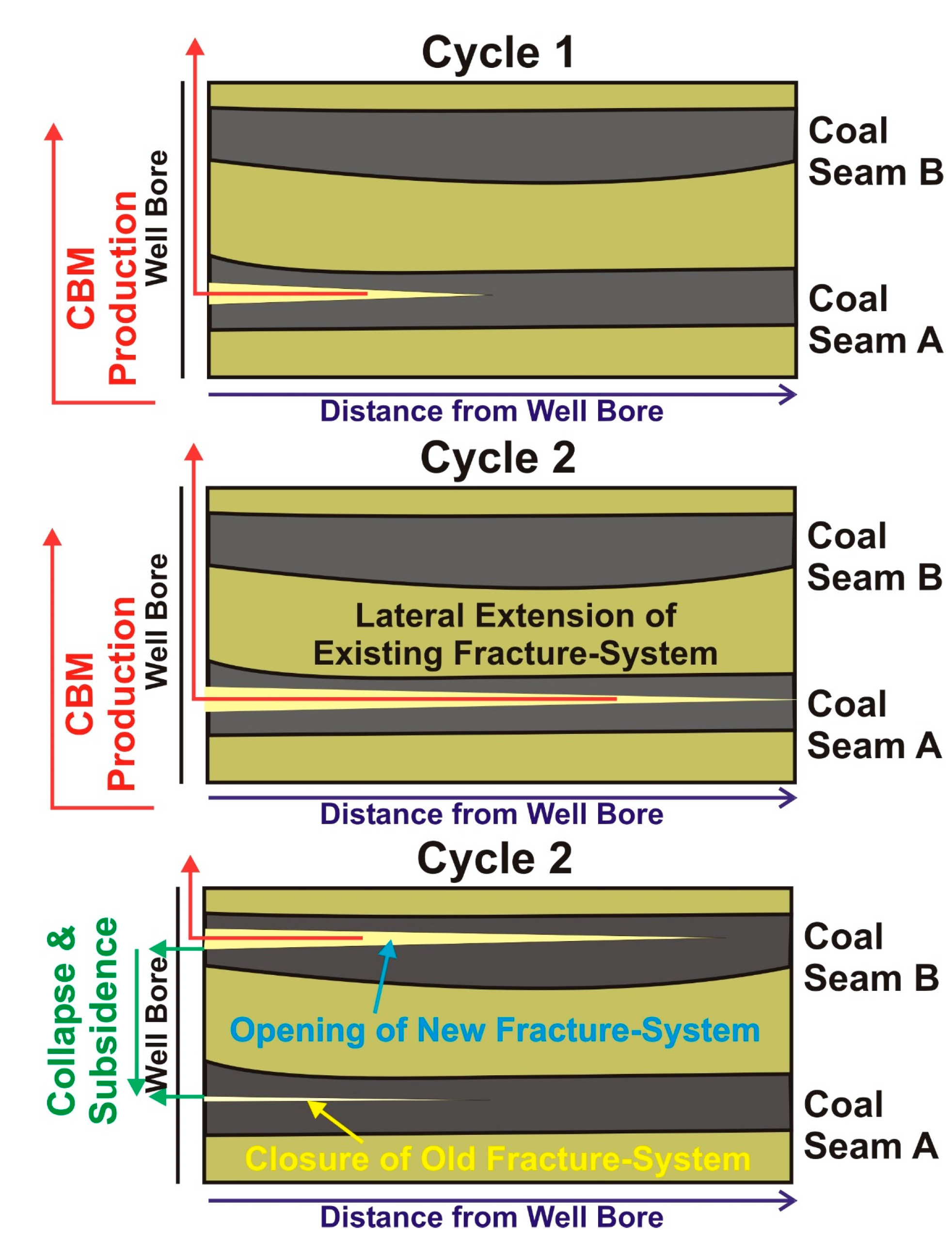
4. Discussion
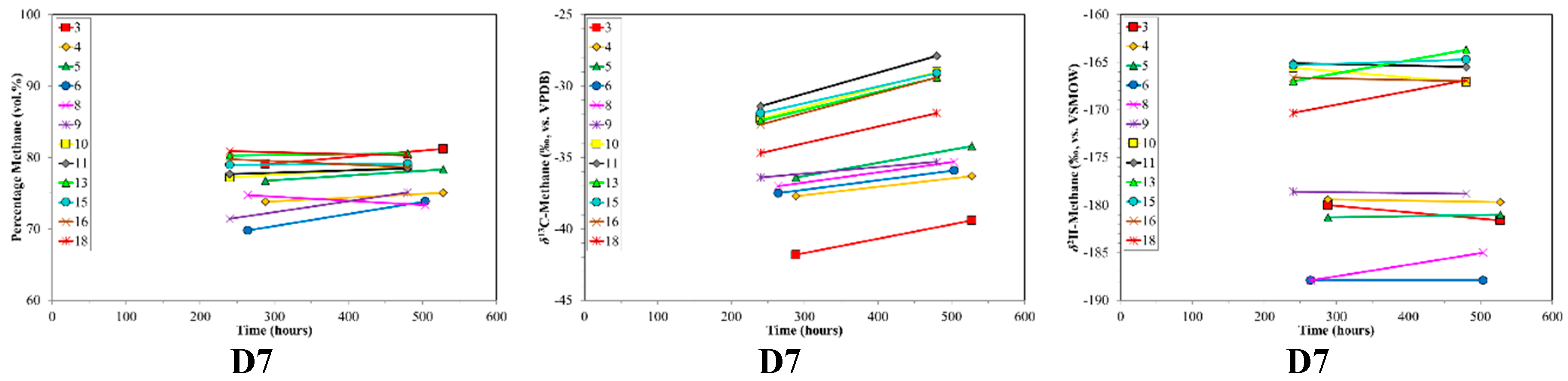
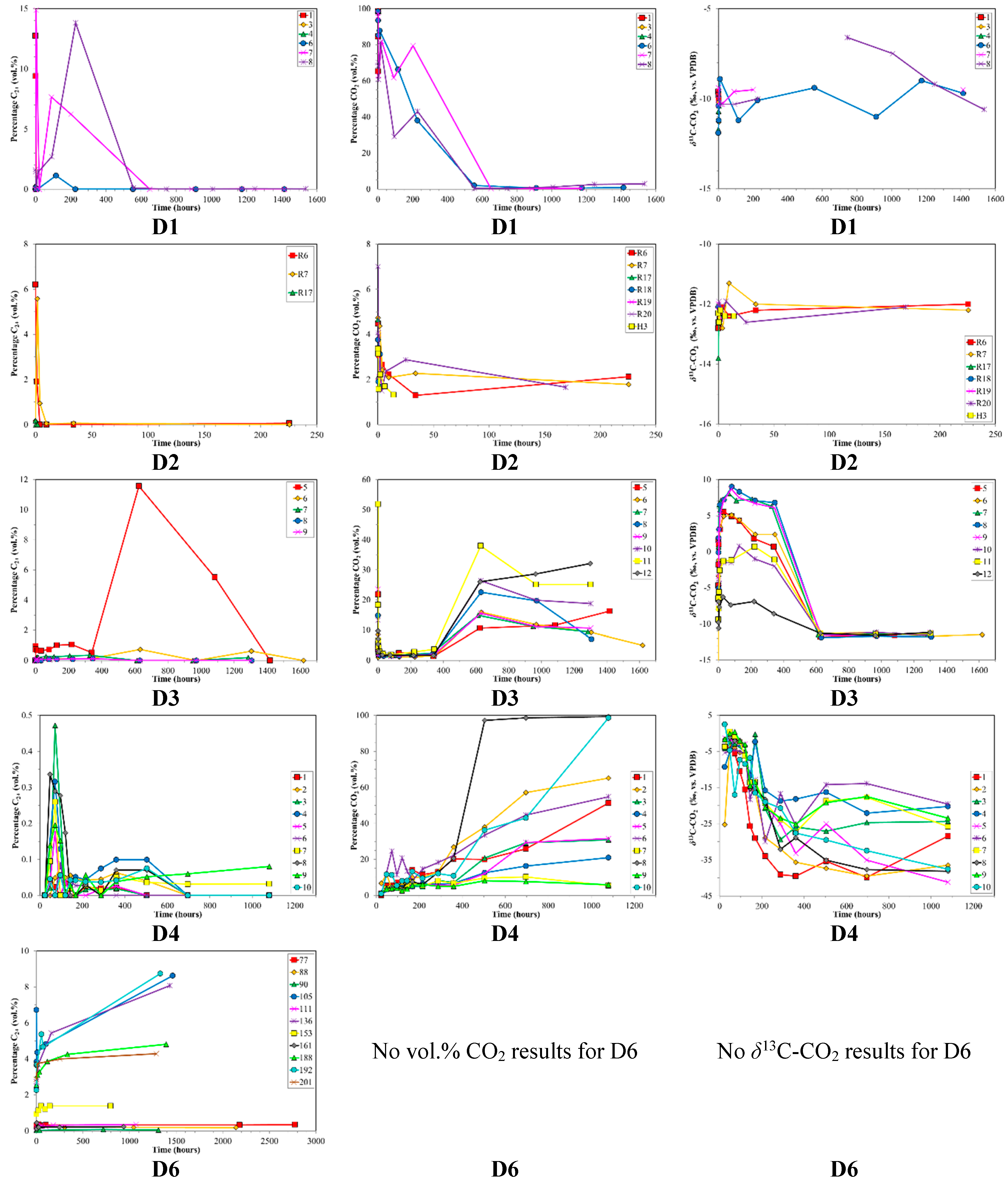
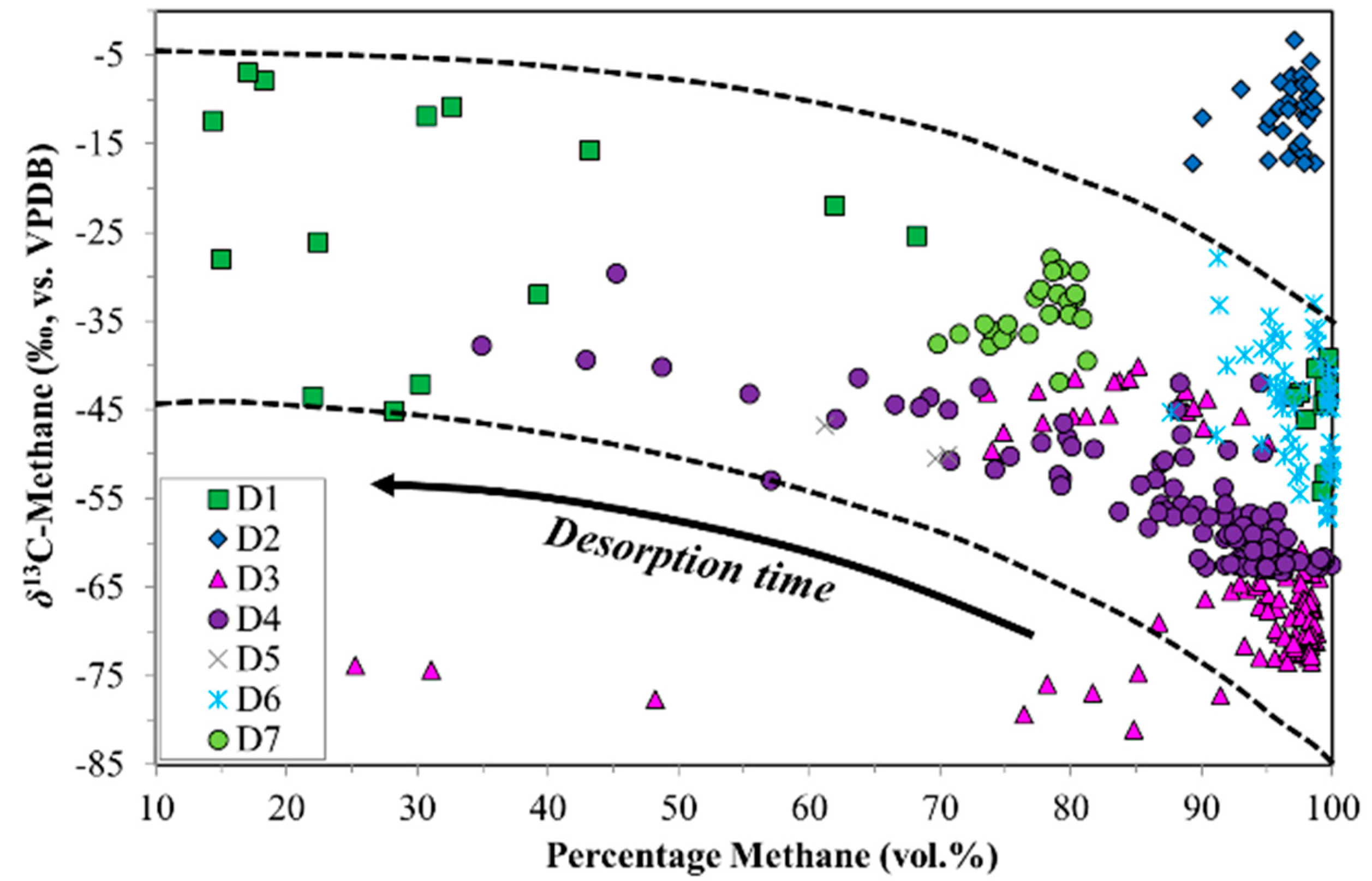
4.1. Canister Desorption Experiments
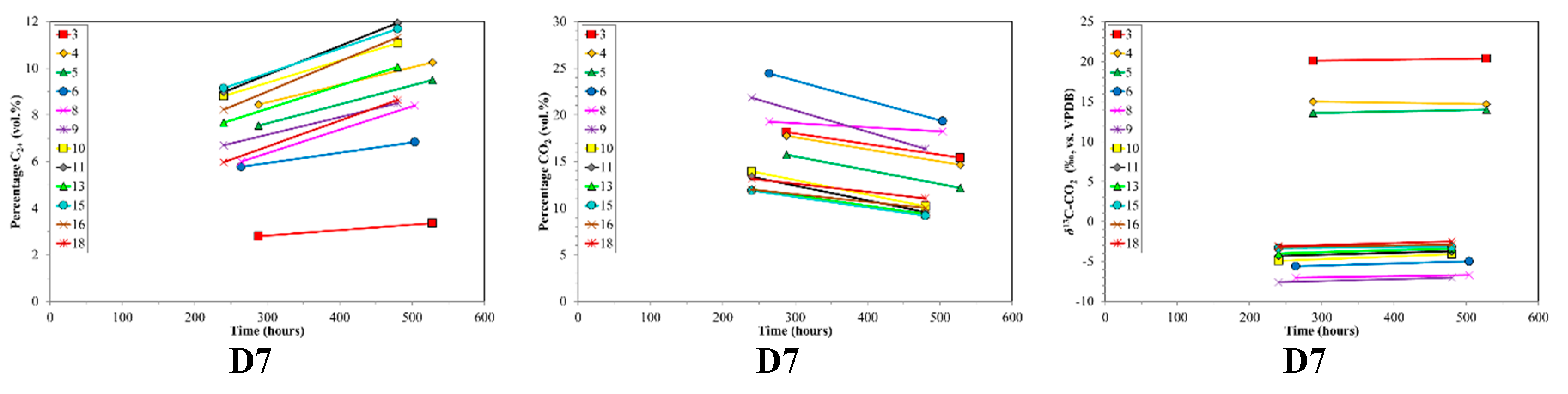
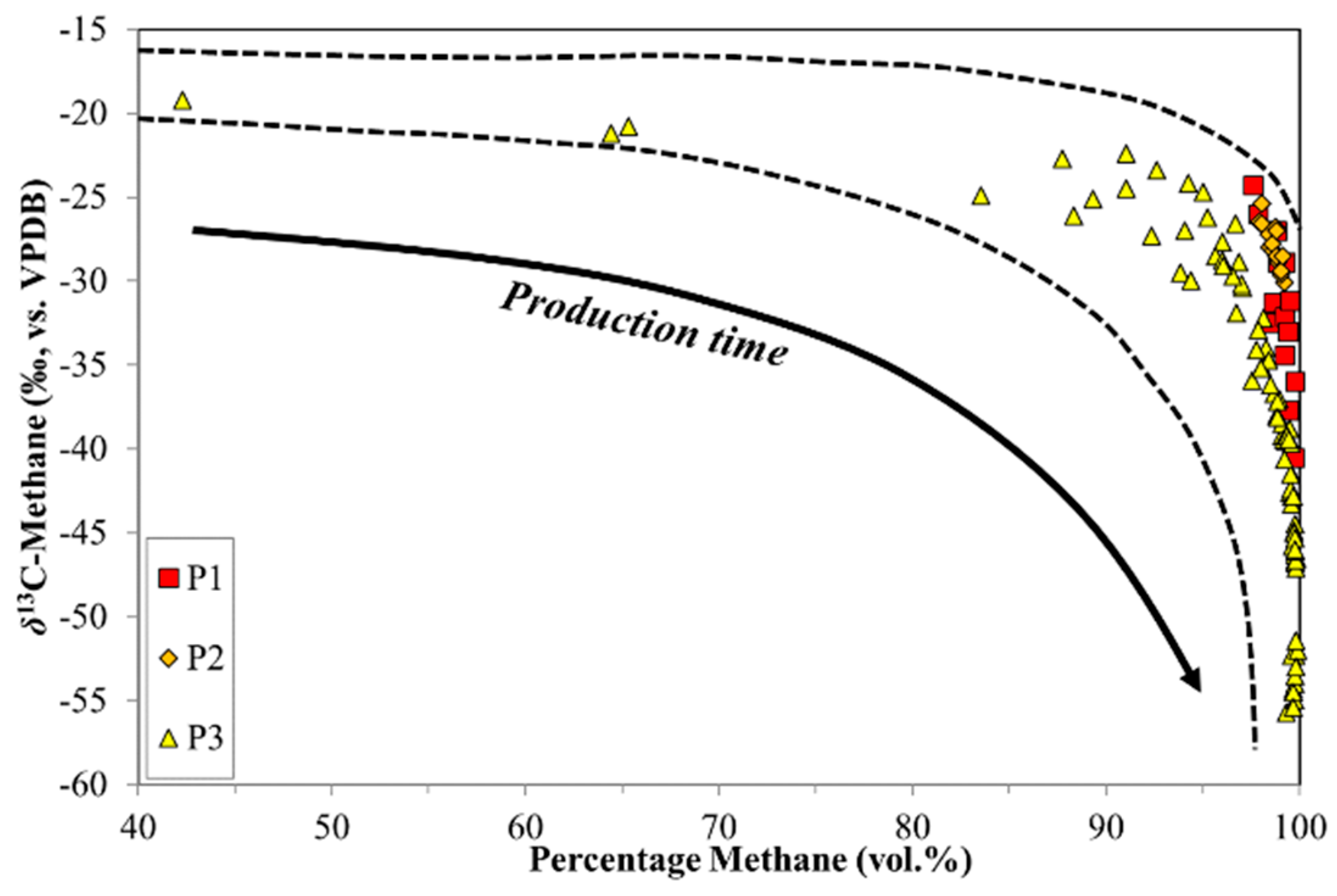
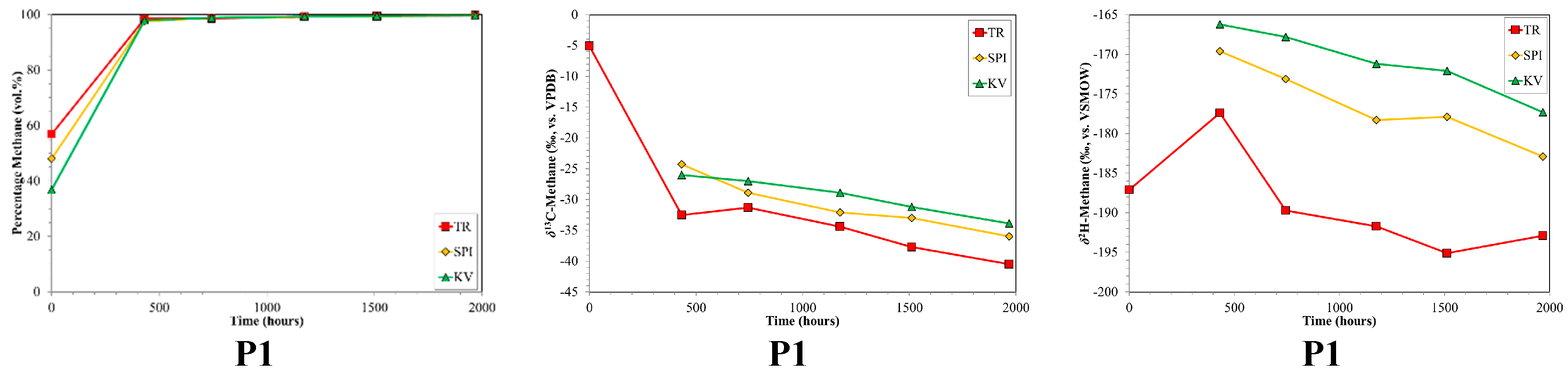
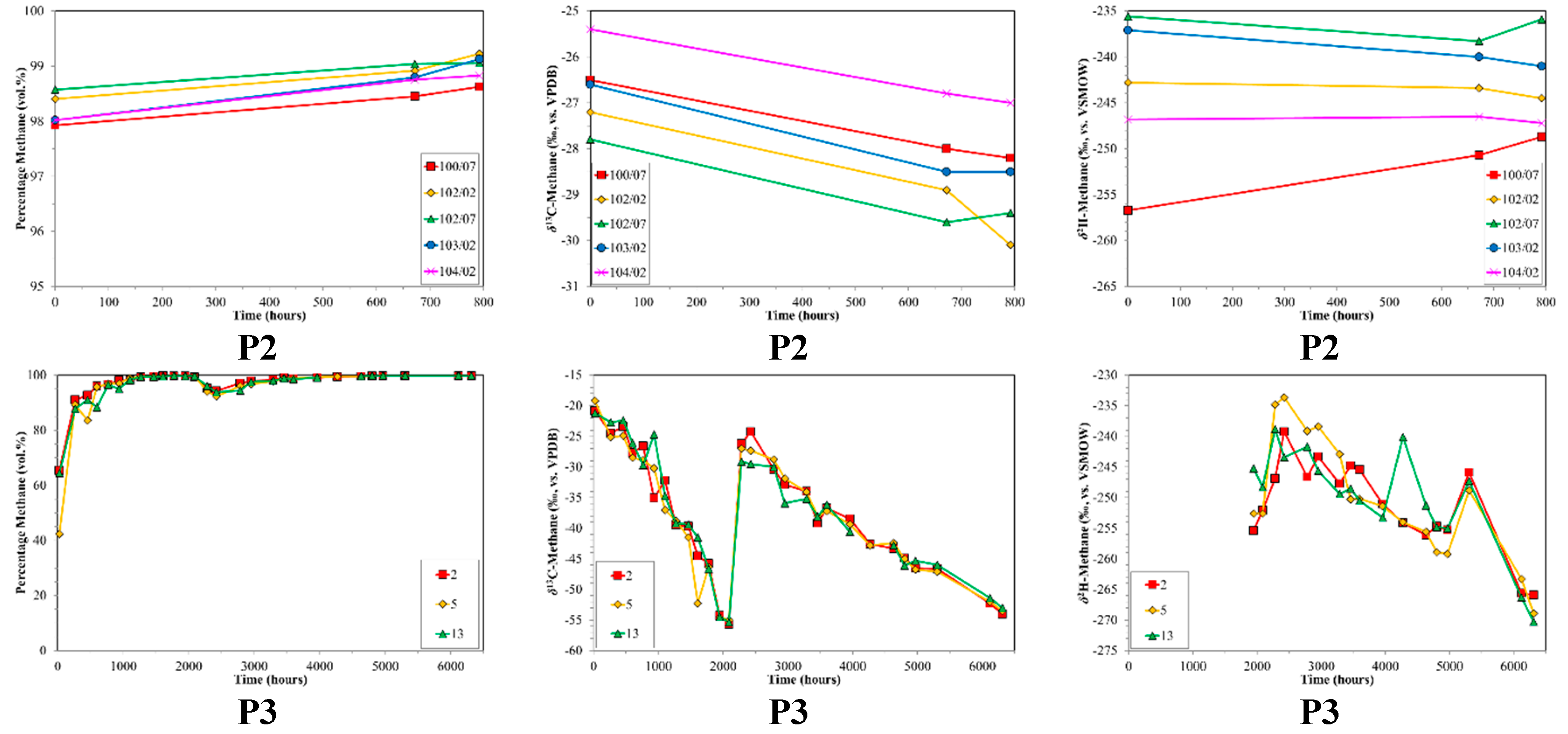
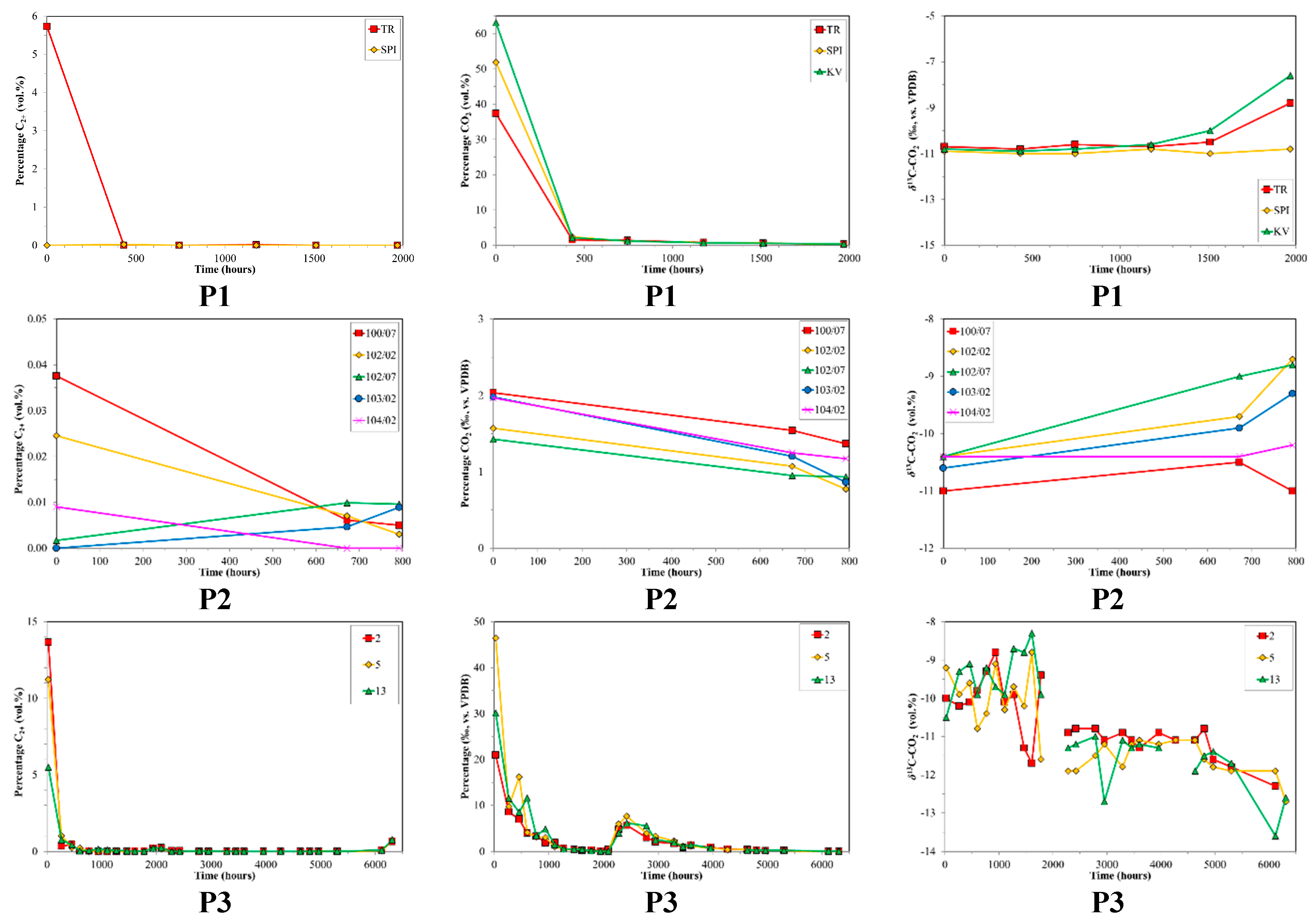
4.2. CBM Production Experiments
- Magnitudes of the molecular and isotope fractionation factors for desorption and diffusion,
- Influence of pressure and temperature on the magnitude of isotope fractionation associated with desorption and diffusion,
- Actual pressure reduction in the coal seam and the pressure gradient throughout the seam,
- Extent (spacing, aperture, length) of natural and artificial fractures,
- Molecular and isotope composition of the original (unaltered) gas, sorbed onto the coal.
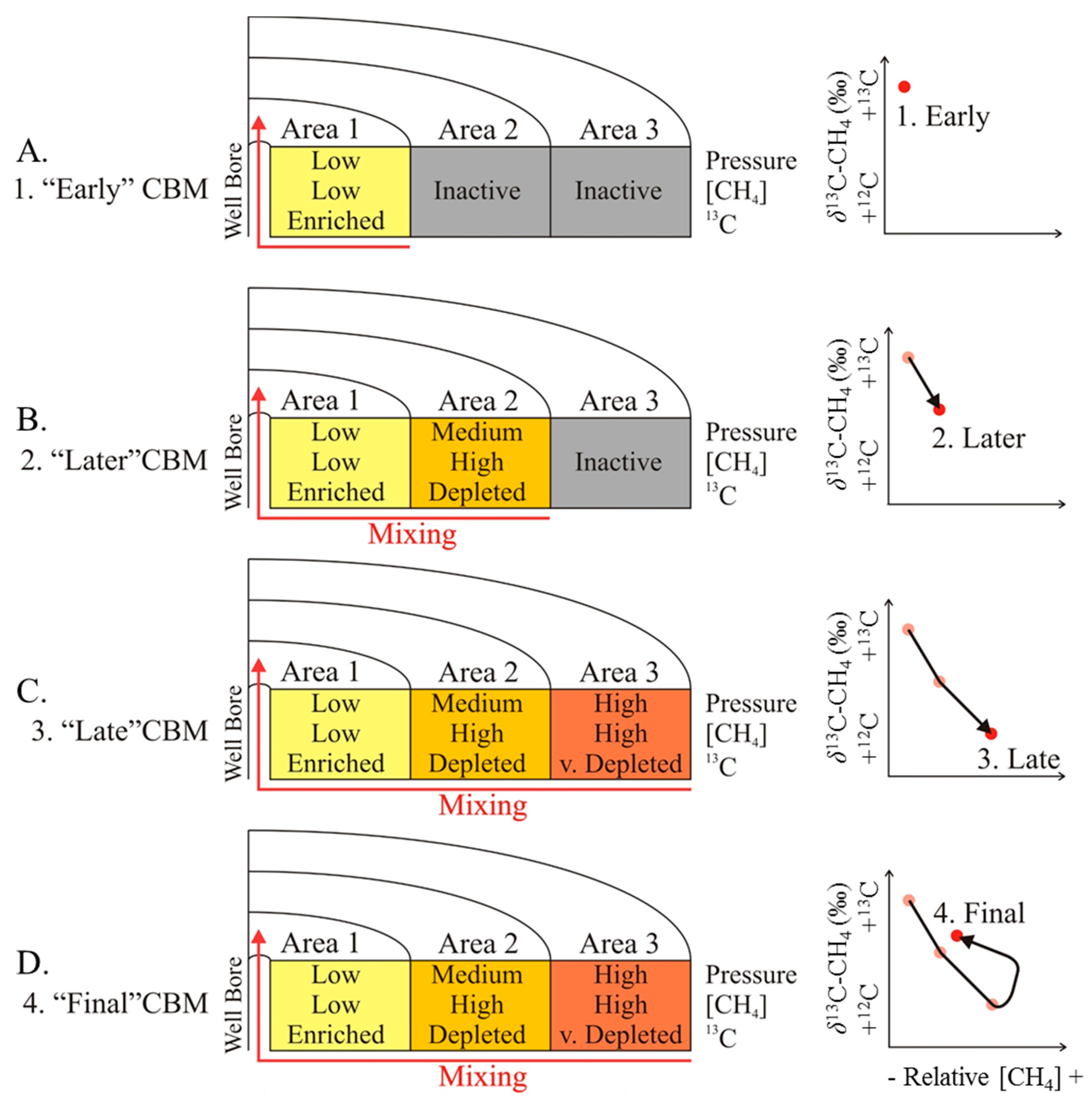
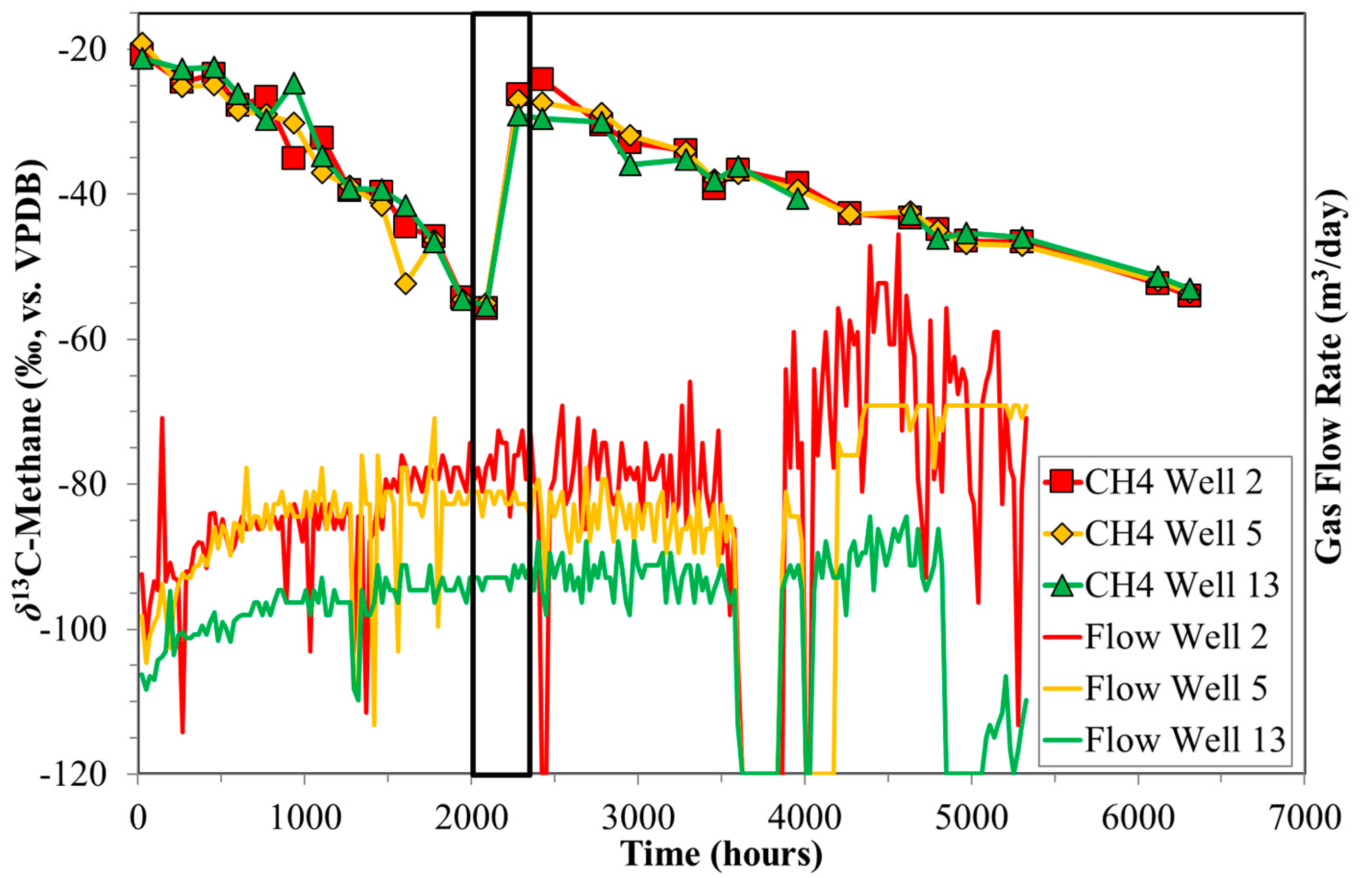
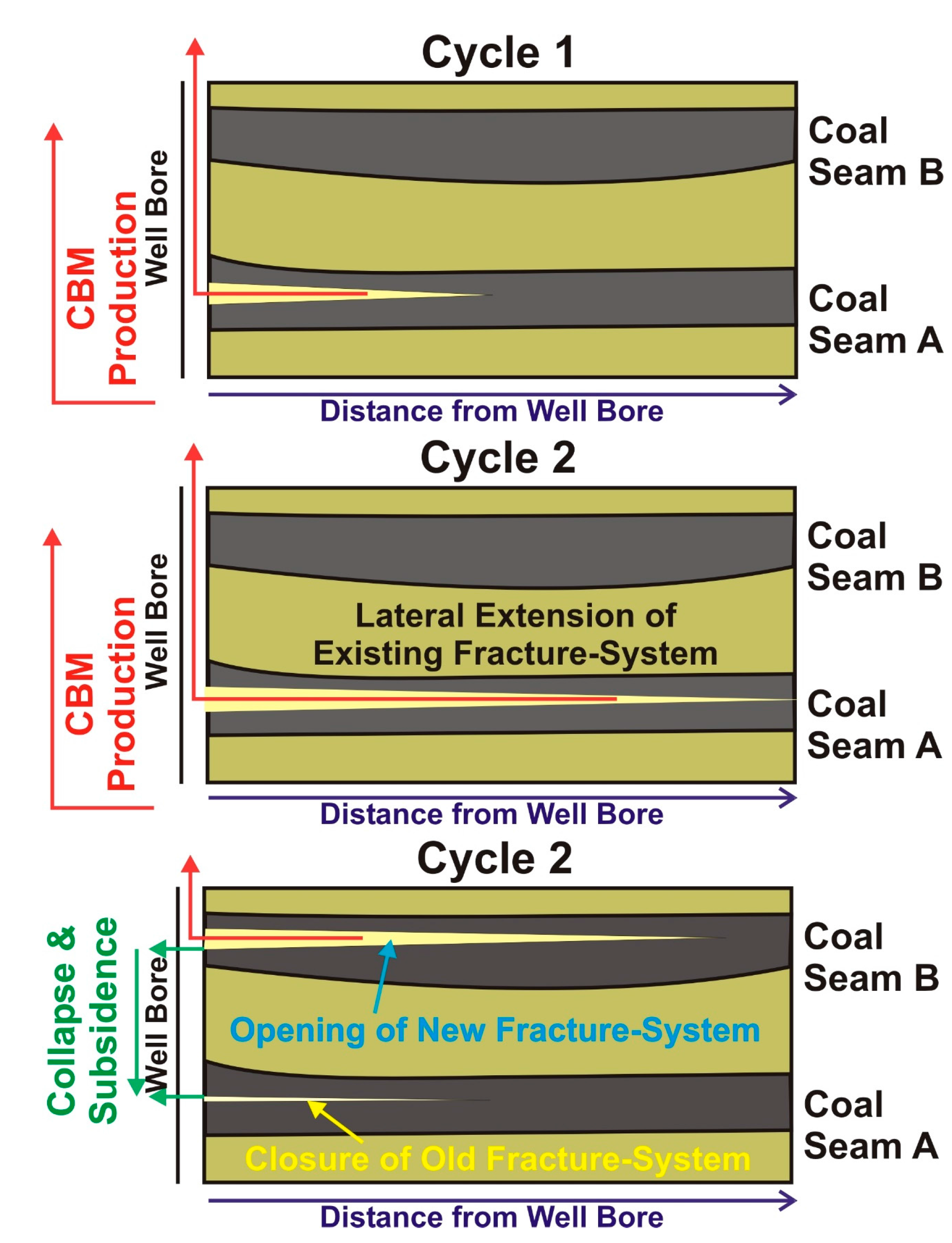
4.3. Indications for Opening of New Fracture Systems
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kawata, Y.; Kazuo, F. Some predictions of possible unconventional hydrocarbons availability until 2100. In Proceedings of the SPE Asia Pacific Oil and Gas Conference and Exhibition, Jakarta, Indonesia, 17–19 April 2001. [Google Scholar]
- Al-Jubori, A.; Johnston, S.; Boyer, C.; Lambert, S.W.; Bustos, O.A.; Pashin, J.C.; Wray, A. Coal bed methane: Clean energy for the world. Oilfield Rev. 2009, 21, 4–13. [Google Scholar]
- U.S. Energy Information Administration. Annual Energy Outlook 2013 with Projections to 2040; EIA: Washington, DC, USA, 2013.
- Xu, C.; Dunnahoe, T.; Slocum, M.T.; Bell, L. Reserves grow modestly as crude oil production climbs. Oil Gas J. 2015, 113, 20–35. [Google Scholar]
- Abas, N.; Kalair, A.; Khan, N. Review of fossil fuels and future energy technologies. Futures 2015, 69, 31–49. [Google Scholar] [CrossRef]
- Environmental Protection Agency. Compilation of Air Pollutant Emission Factors; EPA: Washington, DC, USA, 1998.
- Chamberlin, R.T. Notes on explosive mine gases and dusts, with special reference to the explosions in the Monongah, Darr and Naomi coal mines. U.S. Geol. Surv. Bull. 1909, 383. Available online: https://digital.library.unt.edu/ark:/67531/metadc38812/ (accessed on 6 January 2017).
- Bertard, C.; Bruyet, B.; Gunther, J. Determination of desorbable gas concentration of coal (direct method). Int. J. Rock Mech. Min. 1970, 7, 43–65. [Google Scholar] [CrossRef]
- Joubert, J.L.; Grein, C.T.; Bienstock, D. Effect of moisture on the methane capacity of American coals. Fuel 1974, 53, 186–191. [Google Scholar] [CrossRef]
- McCulloch, C.M.; Diamond, W.P. Inexpensive method helps predict methane content of coalbeds. Coal Age 1976, 81, 102–106. [Google Scholar]
- Kim, A.G. Estimating methane content of bituminous coalbeds from adsorption data. U.S. Bur. Mines Rep. Investig. 1977, 8245, 22. [Google Scholar]
- Diamond, W.P.; Levine, J.R. Direct method determination of the gas content of coal: Procedures and results. U.S. Bur. Mines Rep. Investig. 1981, 8515, 35. [Google Scholar]
- Rigby, D.; Smith, J.W. An isotopic study of gases and hydrocarbons in the Cooper basin. Aust. Pet. Explor. Assoc. J. 1981, 21, 222–229. [Google Scholar]
- Meissner, F.F. Cretaceous and Lower Tertiary coals as sources for gas accumulations in the Rocky Mountain area. In Hydrocarbon Source Rocks of the Greater Rocky Mountain Region; Woodward, J., Meissner, F.F., Clayton, J.L., Eds.; Rocky Mountain Association of Geologists: Denver, CO, USA, 1984; pp. 401–431. ISBN 9780933979062. [Google Scholar]
- Creedy, D.P. Geological controls on the formation and distribution of gas in British coal measure strata. Int. J. Coal Geol. 1988, 10, 1–31. [Google Scholar] [CrossRef]
- Levine, J.R. Coalification: The evolution of coal as a source rock and reservoir rock for oil and gas. In Hydrocarbons from Coal; Law, B.E., Rice, D.D., Eds.; American Association of Petroleum Geologists: Tulsa, OK, USA, 1993; Volume 38, pp. 39–77. ISBN 9781629811048. [Google Scholar]
- Rice, D.D. Compositions and origins of coalbed gas. In Hydrocarbons from Coal; Law, B.E., Rice, D.D., Eds.; American Association of Petroleum Geologists: Tulsa, OK, USA, 1993; Volume 34, pp. 159–184. ISBN 9780891810469. [Google Scholar]
- Yee, D.; Seidle, J.P.; Hanson, W.B. Gas sorption on coal and measurement of gas content. In Hydrocarbons from Coal; Law, B.E., Rice, D.D., Eds.; American Association of Petroleum Geologists: Tulsa, OK, USA, 1993; Volume 38, pp. 203–218. ISBN 9780891810469. [Google Scholar]
- Ryan, B.D.; Dawson, F.M. Coalbed methane canister desorption techniques. In Geological Fieldwork 1993; A Summary of Field Activities and Current Research; Grant, B., Newell, J.M., Eds.; British Columbia Geological Survey: Victoria, BC, Canada, 1994; pp. 245–256. [Google Scholar]
- Beamish, B.B.; Crosdale, P.J. The influence of maceral content on the sorption of gases by coal and the association with outbursting. In Management and Control of High Gas Outbursts in Underground Coal Mines; Lama, R.D., Ed.; Westonprint: Kiama, NSW, Australia, 1995; pp. 353–362. [Google Scholar]
- Bustin, R.M.; Clarkson, C.; Levy, J. Coalbed methane adsorption of coals of the Bulli and Wongawilli seams, southern Sydney basin: Effect of maceral composition. In Proceedings of the 29th Newcastle Symposium on Advances in the Study of the Sydney Basin, Newcastle, NSW, Australia, 6–9 April 1995; pp. 22–28. [Google Scholar]
- McLennan, J.D.; Schafer, P.S.; Pratt, T.J. A Guide to Determining Coalbed Gas Content; Gas Research Institute: Des Plaines, IL, USA, 1995. [Google Scholar]
- Diamond, W.P.; Schatzel, S.J. Measuring the gas content of coal: A review. Int. J. Coal Geol. 1998, 35, 311–331. [Google Scholar] [CrossRef]
- Crosdale, P.J.; Beamish, B.B.; Valix, M. Coalbed methane sorption related to coal composition. Int. J. Coal Geol. 1998, 35, 147–158. [Google Scholar] [CrossRef]
- Laubach, S.E.; Marrett, R.A.; Olson, J.E.; Scott, A.R. Characteristics and origins of coal cleat: A review. Int. J. Coal Geol. 1998, 35, 175–207. [Google Scholar] [CrossRef]
- Kopp, O.C.; Bennett, M.E.; Clark, C.E. Volatiles lost during coalification. Int. J. Coal Geol. 2000, 44, 69–84. [Google Scholar] [CrossRef]
- Radlinski, A.P.; Mastalerz, M.; Hinde, A.L.; Hainbuchner, M.; Rauch, H.; Maron, M.; Lin, J.-S.; Fan, L.; Thiyagarajan, P. Application of SAXS and SANS in evaluation of porosity, pore size distribution and surface area of coals. Int. J. Coal Geol. 2004, 59, 245–271. [Google Scholar] [CrossRef]
- Mastalerz, M.; Drobniak, A.; Strąpoć, D.; Solana Acosta, W.; Rupp, J. Variations in pore characteristics in high-volatile bituminous coals: Implications for coal bed gas content. Int. J. Coal Geol. 2008, 76, 205–216. [Google Scholar] [CrossRef]
- Melnichenko, Y.B.; Radlinski, A.P.; Mastalerz, M.; Cheng, G.; Rupp, J. Characterization of the CO2 fluid adsorption in coal as a function of pressure using neutron scattering techniques (SANS and USANS). Int. J. Coal Geol. 2009, 77, 69–79. [Google Scholar] [CrossRef]
- Colombo, U.; Gazzarrini, F.; Gonfiantini, R.; Kneuper, G.; Teichmüller, M.; Teichmüller, R. Carbon isotope study on methane from German coal deposits. In Advances in Organic Geochemistry 1966, Proceedings of the 3rd International Congress on Organic Geochemistry, London, UK, 26–28 September 1966; Hobson, G.D., Speers, G.C., Eds.; Pergamon Press: Oxford, UK, 1970; pp. 1–26. [Google Scholar]
- Whiticar, M.J. Stable isotope geochemistry of coals, humic kerogens and related natural gases. Int. J. Coal Geol. 1996, 32, 191–215. [Google Scholar] [CrossRef]
- Clayton, J.L. Geochemistry of coalbed gas—A review. Int. J. Coal Geol. 1998, 35, 159–173. [Google Scholar] [CrossRef]
- Strąpoć, D.; Schimmelmann, A.; Mastalerz, M. Carbon isotopic fractionation of CH4 and CO2 during canister desorption of coal. Org. Geochem. 2006, 37, 152–164. [Google Scholar] [CrossRef]
- Dai, J.; Ni, Y.; Zou, C.; Tao, S.; Hu, G.; Hu, A.; Yang, C.; Tao, X. Stable carbon isotopes of alkane gases from the Xujiahe coal measures and implication for gas-source correlation in the Sichuan Basin, SW China. Org. Geochem. 2009, 40, 638–646. [Google Scholar] [CrossRef]
- Golding, S.D.; Boreham, C.J.; Esterle, J.S. Stable isotope geochemistry of coal bed and shale gas and related production waters: A review. Int. J. Coal Geol. 2013, 120, 24–40. [Google Scholar] [CrossRef]
- Gaschnitz, R.; Kroos, B.M.; Gerling, P.; Faber, E.; Littke, R. On-line pyrolysis-GC-IRMS: Isotope fractionation of thermally generated gases from coal. Fuel 2001, 80, 2139–2153. [Google Scholar] [CrossRef]
- Cramer, B. Methane generation from coal during open system pyrolysis investigated by isotope specific, Gaussian distributed reaction kinetics. Org. Geochem. 2004, 35, 379–392. [Google Scholar] [CrossRef]
- Kotarba, M.J.; Lewan, M.D. Characterizing thermogenic coalbed gas from Polish coals of different ranks by hydrous pyrolysis. Org. Geochem. 2004, 35, 615–646. [Google Scholar] [CrossRef]
- Friedrich, H.U.; Jüntgen, H. Some measurements of the 12C/13C ratio in methane or ethane desorbed from hard coal or released by pyrolysis. In Advances in Organic Geochemistry 1971, Proceedings of the 5th International Congress on Organic Geochemistry, Hannover, Germany, 7–10 September 1971; Gärtner, H.R., Wehner, H., Eds.; Pergamon Press: Oxford, UK, 1972; pp. 639–646. [Google Scholar]
- Friedrich, H.U.; Jüntgen, H. Aussagen zum 13C/12C-Verhältnis des bei der Inkohlung gebildeten Methans aufgrund von Pyrolyse-Versuchen. Erdöl Kohle-Erdgas Petrochem. 1973, 26, 636–639. [Google Scholar]
- Smith, J.W.; Rigby, D.; Gould, K.W.; Hart, G.; Hargraves, A.J. An isotopic study of hydrocarbon generation processes. Org. Geochem. 1985, 8, 341–347. [Google Scholar] [CrossRef]
- Gould, K.W.; Hargraves, A.J.; Smith, J.W. Variation in the composition of seam gases issuing from coal. Bull. Aust. Inst. Min. Metall. 1987, 292, 69–73. [Google Scholar]
- Hosgörmez, H.; Yalcin, M.N.; Cramer, B.; Gerling, P.; Faber, E.; Schäfer, R.G.; Mann, U. Isotopic and molecular composition of coal-bed gas in the Amasre region (Zonguldak basin–Western Black Sea). Org. Geochem. 2002, 33, 1429–1439. [Google Scholar] [CrossRef]
- Strąpoć, D.; Mastalerz, M.; Eble, C.; Schimmelmann, A. Characterization of the origin of coalbed gases in southeastern Illinois Basin by compound-specific carbon and hydrogen stable isotope ratios. Org. Geochem. 2007, 38, 267–287. [Google Scholar] [CrossRef]
- Strąpoć, D.; Mastalerz, M.; Schimmelmann, A.; Drobniak, A.; Hedges, S. Variability of geochemical properties in a microbially dominated coalbed gas system from the eastern margin of the Illinois Basin, USA. Int. J. Coal Geol. 2008, 76, 98–110. [Google Scholar] [CrossRef]
- Strąpoć, D.; Picardal, F.W.; Turich, C.; Schaperdoth, I.; Macalady, J.L.; Lipp, J.S.; Lin, Y.; Ertefai, T.F.; Schubotz, F.; Hinrichs, K.; et al. Methane-producing microbial community in a coal bed of the Illinois Basin. Appl. Environ. Microbiol. 2008, 74, 2424–2432. [Google Scholar] [CrossRef] [PubMed]
- Meier-Augenstein, W. Applied gas chromatography coupled to isotope ratio mass spectrometry. J. Chromatogr. 1999, 842, 351–371. [Google Scholar] [CrossRef]
- Niemann, M. Stable Isotope Systematics of Coalbed Methane. Ph.D. Thesis, University of Victoria, Victoria, BC, Canada, 2006. [Google Scholar]
- Coplen, T.B. Guidelines and recommended terms for expression of stable-isotope-ratio and gas-ratio measurement results. Rapid Commun. Mass Spectorm. 2011, 25, 2538–2560. [Google Scholar] [CrossRef]
- Henning, M.; Strąpoć, D.; Lis, G.P.; Sauer, P.; Fong, J.; Schimmelmann, A.; Pratt, L.M. Versatile inlet system for on-line compound-specific δD and δ13C gas chromatography–oxidation/reduction–isotope ratio mass spectrometry analysis of gaseous mixtures. Rapid Commun. Mass Spectorm. 2007, 21, 2269–2272. [Google Scholar] [CrossRef] [PubMed]
- Niemann, M.; Clarke, P.R.; Cornelius, C.T.; Ryan, B.; Whiticar, M.J. Gas origin of the Blackhawk formation, Castlegate coalbed methane field, Utah. In Proceedings of the Rocky Mountain Section AAPG Annual Meeting, Jackson, WY, USA, 24–26 September 2005; AAPG: Tulsa, OK, USA, 2005. [Google Scholar]
- Hunt, J.M. Generation of gas and oil from coal and other terrestrial organic matter. Org. Geochem. 1991, 17, 673–680. [Google Scholar] [CrossRef]
- Jüntgen, H.; Karweil, J. Gasbildung und gasspeicherung in steinkohlenflözen. Part 1. gasbildung. Erdöl Kohle-Erdgas Petrochem. 1966, 19, 251–258. [Google Scholar]
- Jüntgen, H.; Klein, J. Entstehung von Erdgas aus kohligen Sedimenten: Erdol and Kohle. Erdgas Petrochem. Erganz. 1975, 1, 52–68. [Google Scholar]
- Das, B.M.; Nikols, D.J.; Das, Z.U.; Hucka, V.J. Factors affecting rate and total volume of methane desorbed from coalbeds. In Coalbed Methane of Western North America-Guidebook for the Rocky Mountain Association of Geologists Fall Conference and Field Trip; Schwochow, S.D., Murray, D.K., Fahy, M.F., Eds.; Rocky Mountain Association of Geologists: Denver, CO, USA, 1991; pp. 69–76. [Google Scholar]
- Flores, R.M. Coalbed methane: From hazard to resource. Int. J. Coal Geol. 1998, 35, 3–26. [Google Scholar] [CrossRef]
- Smith, J.W.; Pallasser, R.J. Microbial origin of Australian coalbed methane. Am. Assoc. Pet. Geol. Bull. 1996, 80, 891–897. [Google Scholar]
- Aravena, R.; Harrison, S.M.; Barker, J.F.; Abercombie, H.; Rudolph, D. Origin of methane in the Elk Valley coalfield, southeastern British Columbia, Canada. Chem. Geol. 2003, 195, 219–227. [Google Scholar] [CrossRef]
- Faiz, M.M.; Stalker, L.; Sherwood, N.; Saghafi, A.; Wold, M.; Barclay, S.; Choudhury, J.; Barker, W.; Wang, I. Bio-enhancement of coalbed methane resources in the southern Sydney Basin. APPEA J. 2003, 43, 595–610. [Google Scholar]
- Pashin, J.C. Hydrodynamics of coalbed methane reservoirs in the Black Warrior Basin: Key to understanding reservoir performance and environmental issues. Appl. Geochem. 2007, 22, 2257–2272. [Google Scholar] [CrossRef]
- Strąpoć, D.; Mastalerz, M.; Dawson, K.; Macalady, J.; Callaghan, A.V.; Wawrik, B.; Turich, C.; Ashby, M. Biogeochemistry of microbial coal-bed methane. Annu. Earth Planet. Rev. 2011, 39, 617–656. [Google Scholar] [CrossRef]
- Flores, R.M.; Rice, C.A.; Stricker, G.D.; Warden, A.; Ellis, M.S. Methanogenic pathways of coal-bed gas in the Powder River Basin, United States: The geologic factor. Int. J. Coal Geol. 2008, 76, 52–75. [Google Scholar] [CrossRef]
- Scott, A.R. Improving coal gas recovery with microbially enhanced coalbed methane. In Coalbed Methane: Scientific, Environmental and Economic Evaluation; Mastalerz, M., Glikson, M., Golding, S.D., Eds.; Kluwer: Dordrecht, The Netherlands, 1999; pp. 89–110. ISBN 9780792356981. [Google Scholar]
- Budwill, K.; Beaton, A.; Bustin, M.; Muehlenbachs, K.; Gunter, W.D. Methanogenic activity on coal and sequestered CO2 for enhanced coalbed methane recovery. In Greenhouse Gas Control Technologies, Proceedings of the 6th International Conference on Greenhouse Gas Control Technologies, Kyoto, Japan, 1–4 October 2002; Gale, J., Kaya, Y., Eds.; Elsevier Science: Amsterdam, The Netherlands, 2003; pp. 697–702. [Google Scholar]
- Gao, L.; Mastalerz, M.; Schimmelmann, A. The origin of coalbed methane. In Coal Bed Methane-from Prospect to Pipeline; Thakur, P., Schatzel, S., Aminian, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 7–29. ISBN 9780128008805. [Google Scholar]
- Ashby, M.; Wood, L.; Lidstrom, U.; Clarke, C.; Gould, A.; Strąpoć, D.; Lambo, A.J.; Huizinga, B.J. Compositions and Methods for Identifying and Modifying Carbonaceous Compositions. U.S. Patent 9,206,682 B2, 9 May 2013. [Google Scholar]
- Zhang, J.; Liang, Y.; Harpalani, S. Optimization of methane production from bituminous coal through biogasification. Appl. Energy 2016, 183, 31–42. [Google Scholar] [CrossRef]
- Mastalerz, M.; Drobniak, A.; Schimmelmann, A. Characteristics of microbial coalbed gas during production; example from Pennsylvanian coals in Indiana, USA. Geosciences 2017, 7, 26. [Google Scholar] [CrossRef]
- Fuex, A.N. Experimental evidence against an appreciable isotopic fractionation of methane during migration. In Advances in Organic Geochemistry 1979, Proceedings of the 9th International Meeting on Organic Geochemistry, Newcastle-Upon-Tyne, UK, September 1979; Douglas, A.G., Maxwell, J.R., Eds.; Pergamon Press: Oxford, UK, 1980; pp. 725–732. [Google Scholar]
- Smith, D.M.; Williams, F.L. Diffusion models for gas production from coals: Application to methane content determination. Fuel 1984, 63, 251–255. [Google Scholar] [CrossRef]
- Lebedev, V.S.; Syngayevskiy, Ye. D. The isotopic effect during sorption processes. Geokhimiya 1971, 5, 615–619. [Google Scholar]
- Faber, E. Zur Isotopengeochemie gasförmiger Kohlenwasserstoffe. Erdöl Erdgas und Kohle 1987, 103, 210–218. [Google Scholar]
- Ettinger, I.; Eremin, I.; Zimakov, B.; Yanovska, M. Natural factors influencing coal sorption properties. I. Petrography and sorption properties of coals. Fuel 1966, 45, 267. [Google Scholar]
- Sevenster, P.G. Diffusion of gases through coal. Fuel 1959, 38, 403–418. [Google Scholar]
- Thimons, E.D.; Kissell, F.N. Diffusion of methane through coal. Fuel 1973, 52, 274–280. [Google Scholar] [CrossRef]
- Airey, E.M. Gas emission from broken coal. An experimental and theoretical investigation. Int. J. Rock Mech. Min. 1968, 5, 475–494. [Google Scholar] [CrossRef]
- Clarkson, C.R.; Bustin, R.M. The effect of pore structure and gas pressure upon the transport properties of coal: A laboratory and modeling study. 2. Adsorption rate modeling. Fuel 1999, 78, 1345–1362. [Google Scholar] [CrossRef]
- He, W.; Lv, W.; Dickerson, J.H. Gas diffusion mechanisms and models. In Gas Transport in Solid Oxide Fuel Cells; Springer: Berlin, Germany, 2014; pp. 9–17. [Google Scholar]
- Cui, X.; Bustin, M. Controls of coal fabric and coalbed gas production and compositional shift in both field production and canister desorption tests. SPE J. 2006, 11, 111–119. [Google Scholar] [CrossRef]
- Leythäuser, D.; Schäfer, R.G.; Yükler, A. Diffusion of light hydrocarbons through near-surface rocks. Nature 1980, 284, 522–525. [Google Scholar] [CrossRef]
- Kroos, B.M. Diffusion of C1 to C5 hydrocarbons in water-saturated sedimentary rocks. Erdöl Kohle-Erdgas Petrochem. 1986, 39, 399–402. [Google Scholar]
- Levine, J.R.; Johnson, P.; Beamish, B.B. High pressure microbalance sorption studies. In Proceedings of the International Coalbed Methane Symposium, Tuscaloosa, AL, USA, 17–21 May 1993; pp. 187–195. [Google Scholar]
- Laxminarayana, C.; Crosdale, P.J. Role of coal type and rank on methane sorption characteristics of Bowen Basin, Australia coals. Int. J. Coal Geol. 1999, 40, 309–325. [Google Scholar] [CrossRef]
- Clarkson, C.R.; Bustin, R.M. Binary gas adsorption/desorption isotherms: Effect of moisture and coal composition upon carbon dioxide selectivity over methane. Int. J. Coal Geol. 2000, 42, 241–271. [Google Scholar] [CrossRef]
- Busch, A.; Gensterblum, Y.; Krooss, B.M. Methane and CO2 sorption and desorption measurements on dry Argonne premium coals: Pure components and mixtures. Int. J. Coal Geol. 2003, 55, 205–224. [Google Scholar] [CrossRef]
- Faiz, M.M.; Barclay, S.; Saghafi, A.; Stalker, L.; Wold, M.; Esterle, J.; Sherwood, N. Coal Bed Methane Reservoir Characterization–KP-1 and WG-1, PEL-2, Southern Sydney Basin; CSIRO Petroleum Confidential Report 7; CSIRO: Clayton South, Victoria, Australia, 2002. [Google Scholar]
- Chanton, J.P. The effect of gas transport on the isotope signature of methane in wetlands. Org. Geochem. 2005, 36, 753–768. [Google Scholar] [CrossRef]
- Xia, X.; Tang, Y. Isotope fractionation of methane during natural gas flow with coupled diffusion and adsorption/desorption. Geochim. Cosmochim. Acta 2012, 77, 489–503. [Google Scholar] [CrossRef]
- Colombo, U.; Gazzarrini, F.; Sironi, G. Carbon isotope composition of individual hydrocarbons from Italian natural gases. Nature 1965, 205, 1303–1304. [Google Scholar] [CrossRef]
- Pernaton, E.; Prinzhofer, A.; Schneider, F. Reconsideration of methane isotope signature as a criterion for the genesis of natural gas: Influence of migration on isotopic signatures. Rev. Inst. Fr. Pét. 1996, 51, 635–651. [Google Scholar] [CrossRef]
- Prinzhofer, A.; Pernaton, É. Isotopically light methane in natural gas: Bacterial imprint or diffusive fractionation? Chem. Geol. 1997, 142, 193–200. [Google Scholar] [CrossRef]
- Zhang, T.; Krooss, B.M. Experimental investigation on the carbon isotope fractionation of methane during gas migration by diffusion through sedimentary rocks at elevated temperature and pressure. Geochim. Cosmochim. Acta 2001, 65, 2723–2742. [Google Scholar] [CrossRef]
- Schloemer, S.; Krooss, B.M. Molecular transport of methane, ethane and nitrogen and the influence of diffusion on the chemical and isotopic composition of natural gas accumulations. Geofluids 2004, 4, 81–108. [Google Scholar] [CrossRef]
- Hamilton, S.K.; Golding, S.D.; Baublys, K.A.; Esterle, J.S. Stable isotopic and molecular composition of desorbed coal seam gases from the Walloon Subgroup, eastern Surat Basin, Australia. Int. J. Coal Geol. 2014, 122, 21–36. [Google Scholar] [CrossRef]
- Harpalani, S.; Schraufnagel, R.A. Measurement of parameters impacting methane recovery from coal seams. Int. J. Min. Geol. Eng. 1990, 8, 369–384. [Google Scholar] [CrossRef]
- St. George, J.D.; Barakat, M.A. The change in effective stress associated with shrinkage from gas desorption in coal. Int. J. Coal Geol. 2001, 45, 105–113. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Set | Sample Type | Coal Rank | %RO (Avg.) | Maceral Content (Average %) | Depth Range (m) | Quantity Canister/Wells | Time Range (Hours) | ||
|---|---|---|---|---|---|---|---|---|---|
| Vit. | Inert. | Lipt. | |||||||
| P1 | Prod. | n/a | n/a | n/a | n/a | n/a | n/a | 3 | 1968 |
| P2 | Prod. | HV Bit.A-MV Bit. | 1.20 | n/a | n/a | n/a | 136–172 | 5 | 792 |
| P3 | Prod. | Sub Bit.A-HV Bit.C | 0.57 | 82.6 | 16.2 | 1.2 | 172–188 | 3 | 6312 |
| D1 | Des. | n/a | n/a | n/a | n/a | n/a | 618–650 | 6 | 1.5–1535 |
| D2 | Des. | Anthracite | 4.41 | n/a | n/a | n/a | 123–473 | 7 | 1.8–225 |
| D3 | Des. | Sub Bit.A-HV Bit.C | 0.67 | 75.9 | 17.2 | 6.9 | 134–156 | 8 | 1285–1619 |
| D4 | Des. | Sub Bit.A-HV Bit.C | 0.67 | 75.9 | 17.2 | 6.9 | 10 * | 10 | 1080 |
| D5 | Des. | Sub Bit.A-HV Bit.C | 0.67 | 75.9 | 17.2 | 6.9 | 20 * | 2 | 168–504 |
| D6 | Des. | MV Bit.-LV Bit. | 1.46 | 68.9 | 31.0 | 0.1 | n/a | 14 | 796–2773 |
| D7 | Des. | Sub Bit.A-HV Bit.B | 0.61 | 76.4 | 19.4 | 4.2 | 1268–1312 | 13 | 480–528 |
| Sample Set | Average Relative Abundance (vol. %, Normalized) | |||||
|---|---|---|---|---|---|---|
| Methane | Ethane | Propane | n-Butane | i-Butane | CO2 | |
| P1 | 90.4 | 0.32 | 0.001 | 0 | 0 | 9.28 |
| P2 | 98.7 | 0.01 | 0.003 | 0 | 0 | 1.34 |
| P3 | 96.1 | 0.15 | 0.127 | 0.172 | 0.0002 | 3.41 |
| D1 | 44.3 | 0.5 | 0.554 | 0.801 | 0 | 53.9 |
| D2 | 96.8 | 0.43 | 0.001 | 0 | 0 | 2.8 |
| D3 | 92.3 | 0.26 | 0.005 | 0 | 0 | 7.47 |
| D4 | 86.6 | 0.05 | 0.036 | 0.003 | 0.003 | 13.3 |
| D5 | 67.2 | 0 | 0 | 1.092 | 0 | 31.8 |
| D6 | 97.5 | 2.31 | 0.152 | 0.009 | 0.001 | n/a |
| D7 | 77.3 | 6.98 | 1.15 | 0.051 | 0 | 14.5 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niemann, M.; Whiticar, M.J. Stable Isotope Systematics of Coalbed Gas during Desorption and Production. Geosciences 2017, 7, 43. https://doi.org/10.3390/geosciences7020043
Niemann M, Whiticar MJ. Stable Isotope Systematics of Coalbed Gas during Desorption and Production. Geosciences. 2017; 7(2):43. https://doi.org/10.3390/geosciences7020043
Chicago/Turabian StyleNiemann, Martin, and Michael J. Whiticar. 2017. "Stable Isotope Systematics of Coalbed Gas during Desorption and Production" Geosciences 7, no. 2: 43. https://doi.org/10.3390/geosciences7020043
APA StyleNiemann, M., & Whiticar, M. J. (2017). Stable Isotope Systematics of Coalbed Gas during Desorption and Production. Geosciences, 7(2), 43. https://doi.org/10.3390/geosciences7020043