Genome-Wide Scan for Runs of Homozygosity Identifies Candidate Genes in Three Pig Breeds


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. SNP Genotyping and Quality Control
2.3. Runs of Homozygosity Detection
2.4. Inbreeding Coefficient
2.5. Detection of Common Runs of Homozygosity
3. Results
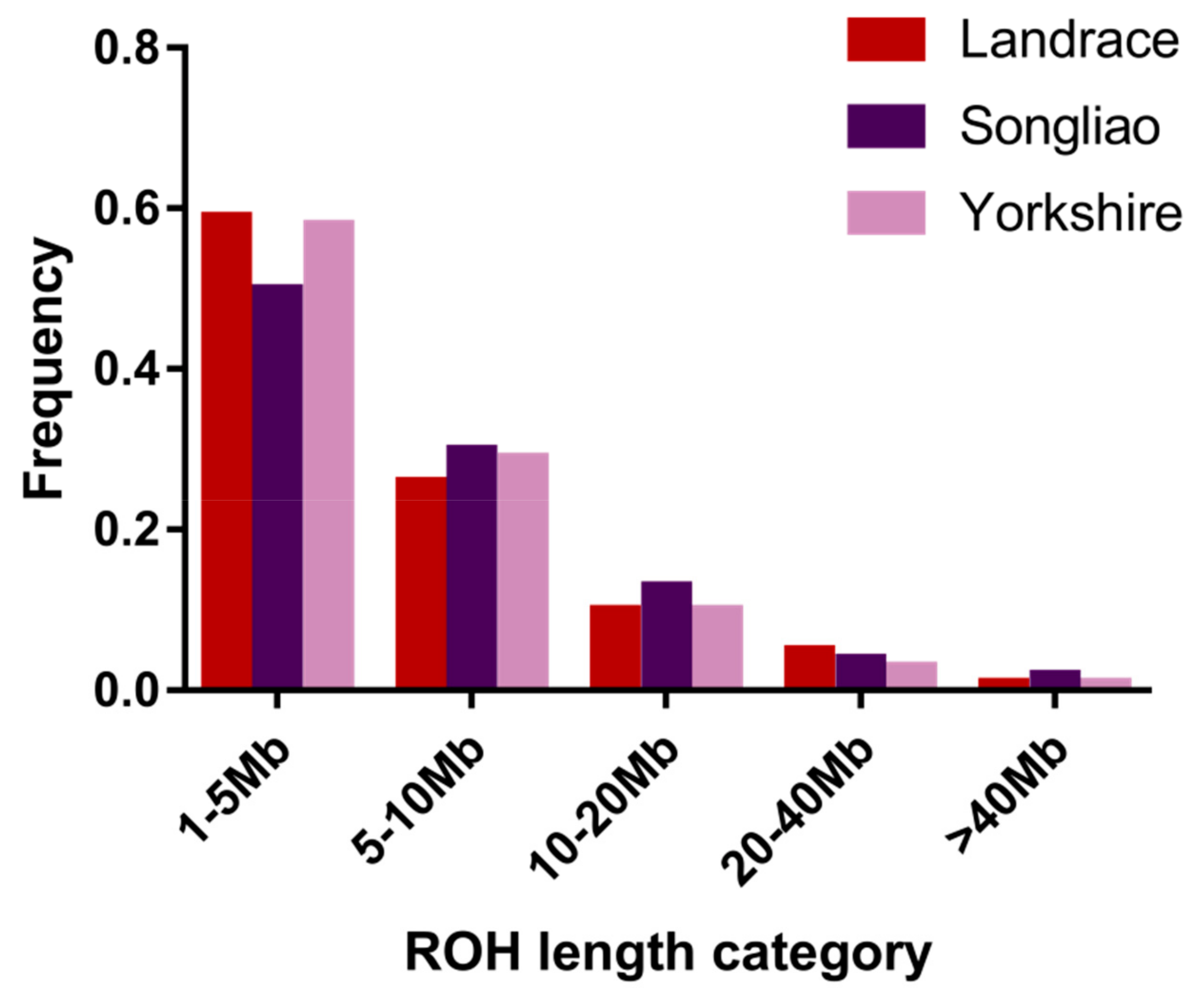
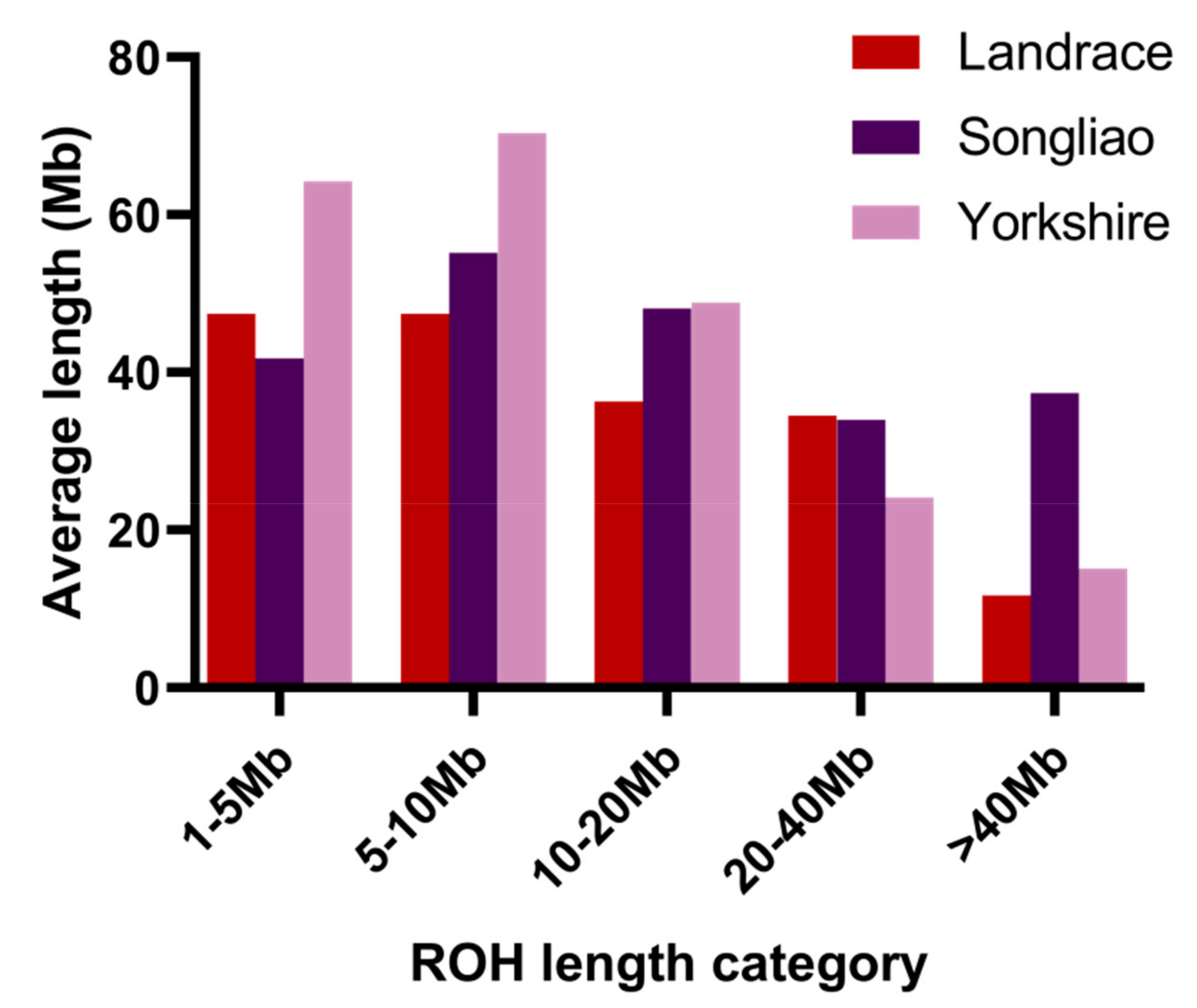
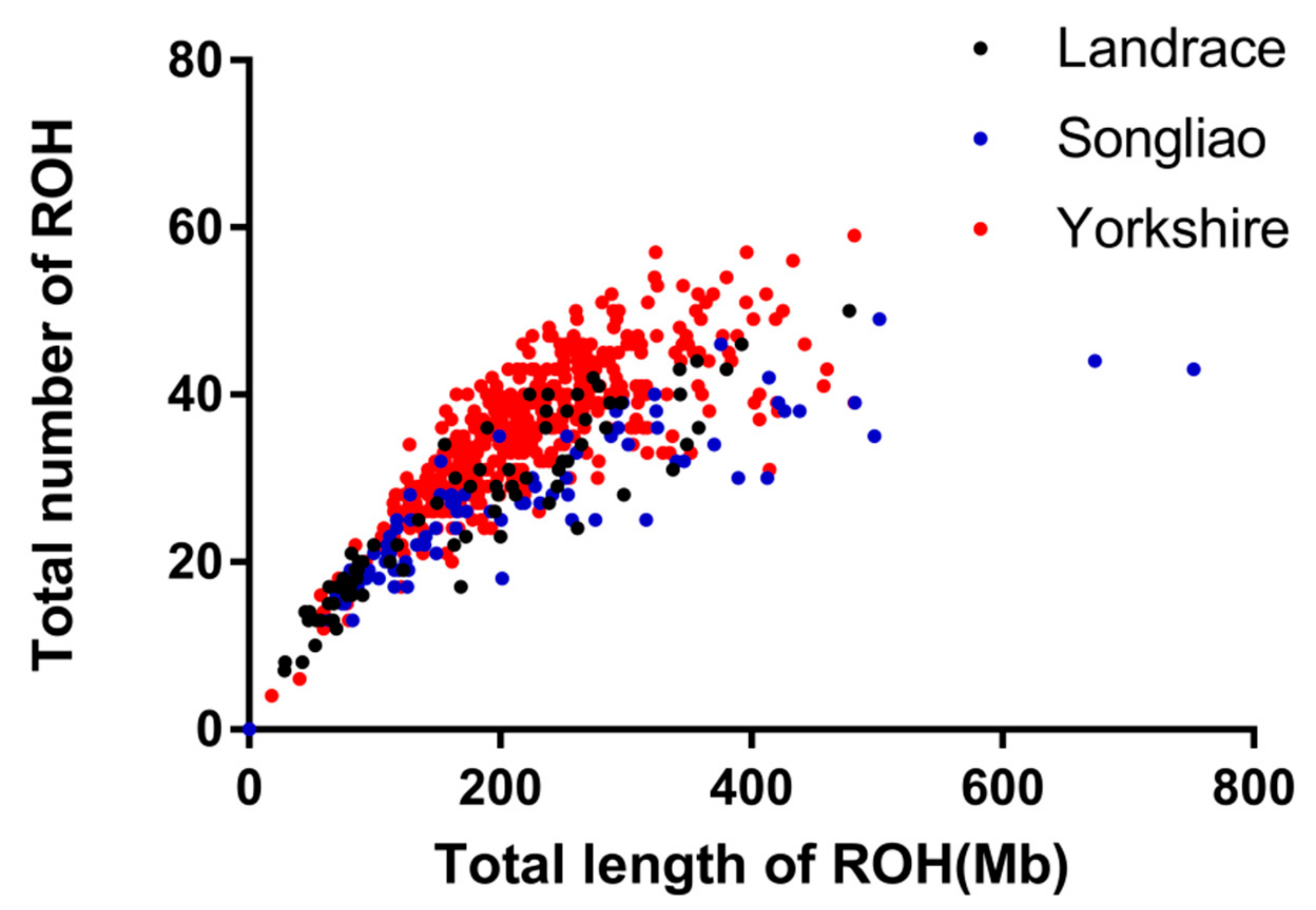
3.1. Distribution of Runs of Homozygosity
3.2. Inbreeding Coefficient of Runs of Homozygosity (FROH)
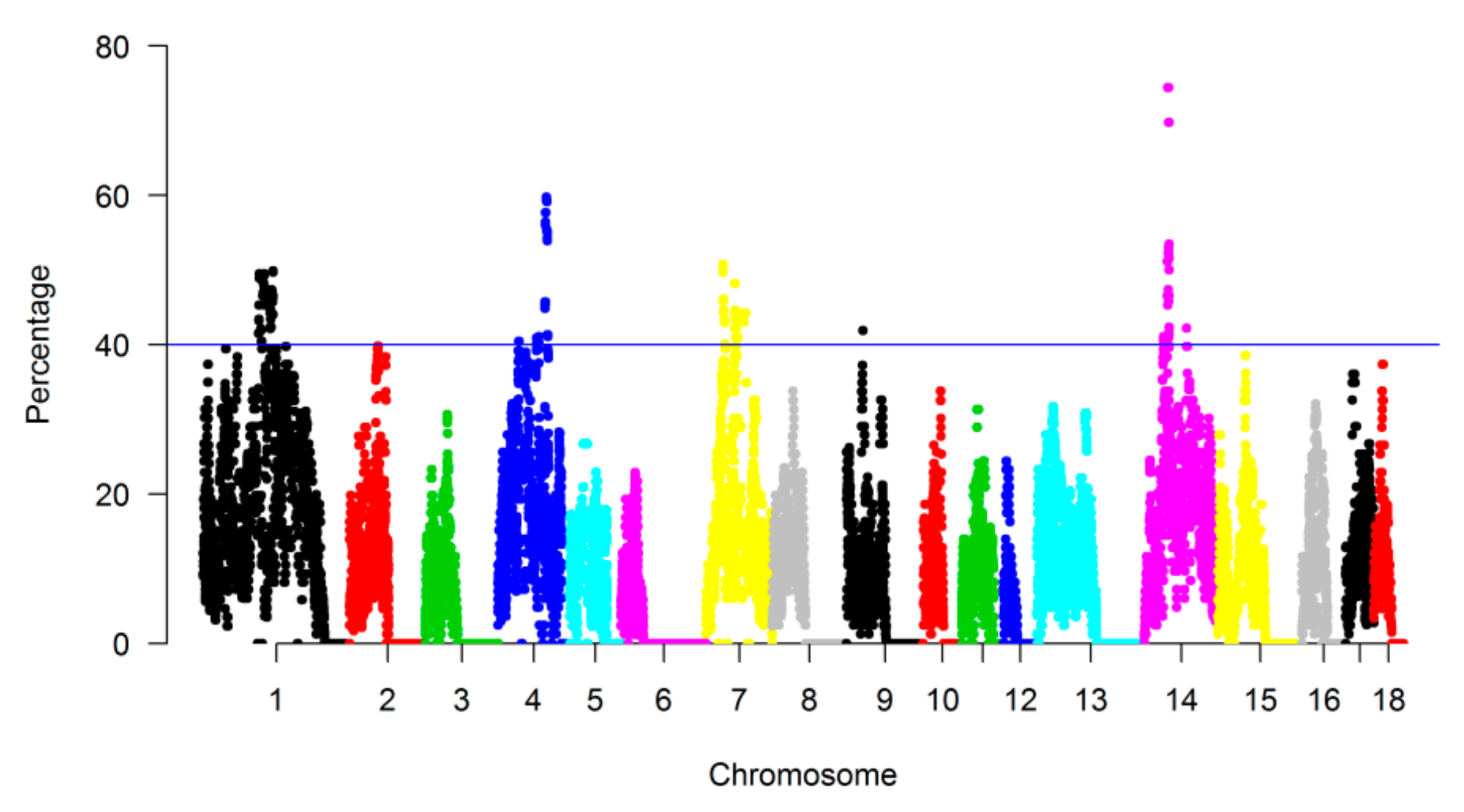
3.3. Genomic Regions with a High Frequency of ROH
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gibson, J.; Morton, N.E.; Collins, A. Extended tracts of homozygosity in outbred human populations. Hum. Mol. Genet. 2006, 15, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Marras, G.; Gaspa, G.; Sorbolini, S.; Dimauro, C.; Ajmone-Marsan, P.; Valentini, A.; Williams, J.L.; Macciotta, N.P. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim. Genet. 2015, 46, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Ferencakovic, M.; Hamzic, E.; Gredler-Grandl, B.; Solberg, T.R.; Klemetsdal, G.; Curik, I.; Sölkner, J. Estimates of autozygosity derived from runs of homozygosity: Empirical evidence from selected cattle populations. J. Anim. Breed. Genet. 2013, 130, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, S.; Tolone, M.; Di Gerlando, R.; Fontanesi, L.; Sardina, M.T.; Portolano, B. Genomic inbreeding estimation in small populations: Evaluation of runs of homozygosity in three local dairy cattle breeds. Animal 2016, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Silió, L.; Rodríguez, M.C.; Fernández, A.; Barragán, C.; Benítez, R.C.Ó.; Fernández, A.I. Measuring inbreeding and inbreeding depression on pig growth from pedigree or SNP-derived metrics. J. Anim. Breed. Genet 2014, 130, 349–360. [Google Scholar]
- Frankham, R. Introduction to quantitative genetics (4th ed.). Am. J. Hum. Genet. 1996, 46, 1231. [Google Scholar]
- Curik, I.; Ferenčaković, M.; Sölkner, J. Inbreeding and runs of homozygosity: A possible solution to an old problem. Livest. Sci. 2014, 166, 26–34. [Google Scholar] [CrossRef]
- Keller, M.C.; Visscher, P.M.; Goddard, M.E. Quantification of Inbreeding Due to Distant Ancestors and Its Detection Using Dense Single Nucleotide Polymorphism Data. Genetics 2011, 189, 237–249. [Google Scholar] [CrossRef]
- Mastrangelo, S.; Tolone, M.; Sardina, M.T.; Sottile, G.; Sutera, A.M.; Di Gerlando, R.; Portolano, B. Genome-wide scan for runs of homozygosity identifies potential candidate genes associated with local adaptation in Valle del Belice sheep. Genet. Sel. Evol. 2017, 49, 84. [Google Scholar] [CrossRef]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef]
- Broman, K.W.; Weber, J.L. Long Homozygous Chromosomal Segments in Reference Families from the Centre d’Étude du Polymorphisme Humain. Am. J. Hum. Genet. 1999, 65, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Medrano, J.M.; Megens, H.-J.; Groenen, M.A.; Ramis, G.; Bosse, M.; Perez-Enciso, M.; Crooijmans, R.P. Conservation genomic analysis of domestic and wild pig populations from the Iberian Peninsula. BMC Genet. 2013, 14, 106. [Google Scholar] [CrossRef] [PubMed]
- Bosse, M.; Megens, H.J.; Madsen, O.; Paudel, Y.; Frantz, L.A.F.; Schook, L.B.; Crooijmans, R.P.M.A.; Groenen, M.A.M. Regions of Homozygosity in the Porcine Genome: Consequence of Demography and the Recombination Landscape. PLoS Genet. 2012, 8, e1003100. [Google Scholar] [CrossRef] [PubMed]
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef] [PubMed]
- Leutenegger, A.-L.; Prum, B.; Génin, E.; Verny, C.; Lemainque, A.; Clerget-Darpoux, F.; Thompson, E.A. Estimation of the Inbreeding Coefficient through Use of Genomic Data. Am. J. Hum. Genet. 2003, 73, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Woods, C.G.; Valente, E.M.; Bond, J.; Roberts, E. A new method for autozygosity mapping using single nucleotide polymorphisms (SNPs) and EXCLUDEAR. J. Med Genet. 2004, 41, e101. [Google Scholar] [CrossRef] [PubMed]
- Scraggs, E.; Zanella, R.; Wojtowicz, A.; Taylor, J.F.; Gaskins, C.T.; Reeves, J.J.; de Avila, J.M.; Neibergs, H.L. Estimation of inbreeding and effective population size of full-blood Wagyu cattle registered with the American Wagyu Cattle Association. J. Anim. Breed Genet. 2014, 131, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, J.P.; Marmi, J.; Goyache, F.; Jordana, J. Pedigree information reveals moderate to high levels of inbreeding and a weak population structure in the endangered Catalonian donkey breed. J. Anim. Breed. Genet. 2005, 122, 378–386. [Google Scholar]
- Saura, M.; Fernández, A.; Varona, L.; Fernández, A.I.; de Cara, M.A.; Ángeles, R.; Barragán, C.; Villanueva, B. Detecting inbreeding depression for reproductive traits in Iberian pigs using genome-wide data. Genet. Sel. Evol. 2015, 47, 329. [Google Scholar] [CrossRef]
- Banos, G.; Wiggans, G.; Powell, R. Impact of Paternity Errors in Cow Identification on Genetic Evaluations and International Comparisons. J. Dairy Sci. 2001, 84, 2523–2529. [Google Scholar] [CrossRef]
- Gomez-Raya, L.; Rodriguez, C.; Barragán, C.; Silió, L. Genomic inbreeding coefficients based on the distribution of the length of runs of homozygosity in a closed line of Iberian pigs. Genet. Sel. Evol. 2015, 47, 177. [Google Scholar] [CrossRef] [PubMed]
- Forutan, M.; Mahyari, S.A.; Baes, C.; Melzer, N.; Schenkel, F.S.; Sargolzaei, M. Inbreeding and runs of homozygosity before and after genomic selection in North American Holstein cattle. BMC Genom. 2018, 19, 98. [Google Scholar] [CrossRef]
- Zhang, Q.; Calus, M.P.; Guldbrandtsen, B.; Lund, M.S.; Sahana, G. Estimation of inbreeding using pedigree, 50k SNP chip genotypes and full sequence data in three cattle breeds. BMC Genet. 2015, 16, 330. [Google Scholar] [CrossRef] [PubMed]
- Pemberton, T.J.; Absher, D.; Feldman, M.W.; Myers, R.M.; Rosenberg, N.A.; Li, J.Z. Genomic Patterns of Homozygosity in Worldwide Human Populations. Am. J. Hum. Genet. 2012, 91, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Szmatoła, T.; Gurgul, A.; Ropka-Molik, K.; Jasielczuk, I.; Ząbek, T.; Bugno-Poniewierska, M. Characteristics of runs of homozygosity in selected cattle breeds maintained in Poland. Livest. Sci. 2016, 188, 72–80. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Q.; Xiao, Q.; Sun, H.; Gao, H.; Yang, Y.; Chen, J.; Li, Z.; Xue, M.; Ma, P.; et al. Distribution of runs of homozygosity in Chinese and Western pig breeds evaluated by reduced-representation sequencing data. Anim. Genet. 2018, 49, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Lencz, T.; Lambert, C.; DeRosse, P.; Burdick, K.E.; Morgan, T.V.; Kane, J.M.; Kucherlapati, R.; Malhotra, A.K. Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc. Natl. Acad. Sci. USA 2007, 104, 19942–19947. [Google Scholar] [CrossRef]
- Kirin, M.; McQuillan, R.; Franklin, C.S.; Campbell, H.; McKeigue, P.M.; Wilson, J.F. Genomic Runs of Homozygosity Record Population History and Consanguinity. PLoS ONE 2010, 5, e13996. [Google Scholar] [CrossRef]
- McQuillan, R.; Leutenegger, A.-L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polašek, O.; Tenesa, A.; et al. Runs of Homozygosity in European Populations. Am. J. Hum. Genet. 2008, 83, 658. [Google Scholar] [CrossRef]
- CNCAGR (China National Commission of Animal Genetic Resources). Animal Genetic Resources in China—Pigs; China Agriculture Press: Beijing, China, 2012. (In Chinese) [Google Scholar]
- Kim, E.S.; Cole, J.B.; Huson, H.; Wiggans, G.R.; Van Tassell, C.P.; Crooker, B.A.; Liu, G.; Da, Y.; Sonstegard, T.S. Effect of artificial selection on runs of homozygosity in u.s. Holstein cattle. PLoS ONE 2013, 8, e80813. [Google Scholar] [CrossRef] [PubMed]
- Traspov, A.; Deng, W.; Kostyunina, O.; Ji, J.; Shatokhin, K.; Lugovoy, S.; Zinovieva, N.; Yang, B.; Huang, L. Population structure and genome characterization of local pig breeds in Russia, Belorussia, Kazakhstan and Ukraine. Genet. Sel. Evol. 2016, 48, 1785. [Google Scholar]
- Khan, A.; Tian, L.; Zhang, C.; Yuan, K.; Xu, S. Genetic diversity and natural selection footprints of the glycine amidinotransferase gene in various human populations. Sci. Rep. 2016, 6, 18755. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gu, Y.; Zhang, Z.; Zheng, W.; Yang, L.; Huang, W.; Lin, S.; Li, Y.; Guo, H.; Luo, M. Deficiency of SPATA46, a Novel Nuclear Membrane Protein, Causes Subfertility in Male Mice1. Biol. Reprod. 2016, 95, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Blaha, M.; Nemcova, L.; Kepkova, K.V.; Vodička, P.; Prochazka, R. Gene expression analysis of pig cumulus-oocyte complexes stimulated in vitro with follicle stimulating hormone or epidermal growth factor-like peptides. Reprod. Biol. Endocrinol. 2015, 13, 159. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Latifi, Z.; Kusama, K.; Nakamura, K.; Shimada, M.; Imakawa, K. Induction of immune-related gene expression by seminal exosomes in the porcine endometrium. Biochem. Biophys. Res. Commun. 2017, 495, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, X.; Zhao, Y.; Gao, R.; Geng, Y.; Ding, Y.; Liu, X.; Wang, Y.; He, J. The homologous genes Vangl1 and Vangl2 are required for embryo implantation in the uterus of mice during early pregnancy. Gene 2015, 555, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Shang, P.; Qiangba, Y.; Xu, A.; Wang, Z.; Zhang, H. The association of NR1H3 gene with lipid deposition in the pig. Lipids Health Dis. 2016, 15, 212. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, P.A.; Gomes, R.C.; Santana, M.H.; Silva, S.L.; Leme, P.R.; Mudadu, M.A.; Regitano, L.C.; Meirelles, F.V.; Ferraz, J.B.; Fukumasu, H. Bovine NR1I3 gene polymorphisms and its association with feed efficiency traits in Nellore cattle. Meta Gene 2014, 2, 206–217. [Google Scholar] [CrossRef]
- Ballester, M.; Revilla, M.; Puig-Oliveras, A.; Marchesi, J.A.P.; Castelló, A.; Corominas, J.; Fernández, A.I.; Folch, J.M. Analysis of the porcine APOA2 gene expression in liver, polymorphism identification and association with fatty acid composition traits. Anim. Genet. 2016, 47, 552–559. [Google Scholar] [CrossRef]
- Wu, H.; Qiao, M.; Peng, X.; Wu, J.; Liu, G.; Sun, H.; Li, L.; Mei, S. Molecular Characterization, Expression Patterns, and Association Analysis with Carcass Traits of Porcine USF1 Gene. Appl. Biochem. Biotechnol. 2013, 170, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Yong, L.I.; Wang, H.; Tang, Z.L.; Zhu, Z.M.; Yang, S.L.; Jin, E.H.; Gong, W.H.; Kui, L.I. Mapping, Cloning and Expression Profile Analysis of Porcine NDUFS2 Gene. Chin. J. Anim. Vet. Sci. 2007, 38, 1137–1142. [Google Scholar]
- Zhang, Z.; Deng, Q.; Wu, Y.; Huang, X.B.; Yao, L.; Jiang, Z.M.; Gui, Y.T. Expression characteristics of the Daxx gene in the mouse testis during spermatogenesis. Zhonghua Nan Ke Xue 2017, 23, 103–109. [Google Scholar] [PubMed]
- Nishimura, Y.; Kano, K.; Naito, K. Porcine CPEB1 is involved in Cyclin B translation and meiotic resumption in porcine oocytes. Anim. Sci. J. 2010, 81, 444–452. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1–5 Mb | 5–10 Mb | 10–20 Mb | 20–40 Mb | >40 Mb | |
|---|---|---|---|---|---|
| Landrace | 1269 | 564 | 216 | 104 | 16 |
| Songliao | 1152 | 676 | 306 | 101 | 53 |
| Yorkshire | 9771 | 4837 | 1691 | 430 | 126 |
| Breed | Sample Size | SNP Number * | Average Length(Mb) | Average Number | FROH | FHOM | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean ± SE | Range | Mean ± SE | Range | Mean ± SE | Range | |||||
| Landrace | 83 | 44~1853 | 6.33 ± 0.21 | 28.11~478.17 | 26.13 ± 1.14 | 7~50 | 0.073 ± 0.0047 | 0.012~0.20 | −0.0013 ± 0.0094 | 0.95 |
| Songliao | 86 | 46~2100 | 7.49 ± 0.31 | 0~752.25 | 26.60 ± 0.96 | 0~49 | 0.089 ± 0.0063 | 0~0.31 | 0.0050 ± 0.010 | 0.98 |
| Yorkshire | 477 | 43~2318 | 6.21 ± 0.063 | 0~482.18 | 35.34 ± 0.39 | 0~59 | 0.092 ± 0.0015 | 0~0.20 | 0.034 ± 0.0031 | 0.93 |
| Average | 215.33 | 43~2318 | 6.40 ± 0.070 | 0~752.25 | 32.99 ± 0.38 | 0~59 | 0.089 ± 0.0015 | 0~0.31 | 0.026 ± 0.0031 | 0.94 |
| CHR | Length (Mb) * | Landrace | SongLiao | Yorkshire | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1–5 Mb | 5–10 Mb | 10–20 Mb | 20–40 Mb | >40 Mb | Total | 1–5 Mb | 5–10 Mb | 10–20 Mb | 20–40 Mb | >40 Mb | Total | 1–5 Mb | 5–10 Mb | 10–20 Mb | 20–40 Mb | >40 Mb | Total | ||
| 1 | 315.29 | 1.35 | 2.85 | 2.23 | 3.75 | 2.06 | 12.24 | 1.25 | 3.27 | 3.08 | 2.24 | 1.75 | 11.59 | 2.47 | 5.01 | 4.43 | 3.36 | 1.74 | 17.01 |
| 2 | 162.55 | 1.73 | 2.73 | 1.44 | 0.56 | 0 | 6.46 | 1.69 | 2.07 | 1.40 | 0.71 | 1.02 | 6.89 | 2.07 | 1.99 | 1.23 | 0.31 | 0.32 | 5.92 |
| 3 | 144.77 | 1.93 | 1.22 | 0.39 | 0 | 0 | 3.54 | 1.54 | 1.25 | 0.64 | 0.23 | 0.46 | 4.12 | 2.24 | 1.38 | 0.57 | 0.20 | 0.14 | 4.53 |
| 4 | 143.44 | 3.47 | 2.44 | 0.88 | 2.98 | 0 | 9.77 | 2.53 | 4.31 | 3.89 | 3.55 | 1.56 | 15.84 | 4.25 | 7.90 | 3.97 | 1.50 | 0.98 | 18.60 |
| 5 | 111.49 | 1.42 | 1.42 | 3.33 | 0.58 | 0 | 6.75 | 1.47 | 3.78 | 2.25 | 0.28 | 0 | 7.78 | 2.86 | 2.26 | 1.02 | 0.05 | 0 | 6.19 |
| 6 | 157.75 | 1.73 | 0.60 | 0.09 | 0 | 0 | 2.42 | 0.78 | 0.66 | 0.22 | 0 | 0 | 1.66 | 1.63 | 0.49 | 0.20 | 0.09 | 0.06 | 2.47 |
| 7 | 134.75 | 2.69 | 3.63 | 4.62 | 1.71 | 0 | 12.65 | 3.56 | 5.33 | 5.76 | 1.74 | 0.70 | 17.09 | 3.95 | 3.38 | 3.79 | 0.89 | 0.13 | 12.14 |
| 8 | 148.48 | 0.97 | 1.43 | 1.50 | 2.01 | 0 | 5.91 | 1.08 | 0.82 | 0.35 | 1.57 | 0.75 | 4.57 | 1.75 | 1.91 | 0.96 | 0.56 | 0.51 | 5.69 |
| 9 | 153.65 | 1.81 | 1.24 | 0.58 | 0.22 | 0 | 3.85 | 2.62 | 2.87 | 1.28 | 0.16 | 0.80 | 7.73 | 2.59 | 1.69 | 0.64 | 0.28 | 0.20 | 5.40 |
| 10 | 79.38 | 2.89 | 1.31 | 1.30 | 0.40 | 0 | 5.90 | 1.94 | 1.92 | 1.44 | 0.98 | 0 | 6.28 | 2.88 | 0.86 | 0.70 | 0.28 | 0 | 4.72 |
| 11 | 87.68 | 2.70 | 1.14 | 0.34 | 0.75 | 0 | 4.93 | 0.70 | 1.71 | 1.41 | 3.18 | 2.08 | 9.08 | 2.19 | 2.53 | 2.45 | 0.20 | 0.72 | 8.09 |
| 12 | 63.58 | 1.03 | 0.45 | 0.43 | 0 | 0 | 1.91 | 2.26 | 1.65 | 0.78 | 0 | 0 | 4.69 | 1.42 | 0.42 | 0.25 | 0.07 | 0 | 2.16 |
| 13 | 218.61 | 1.20 | 1.25 | 0.67 | 0.66 | 0.82 | 4.60 | 1.33 | 1.37 | 1.05 | 1.23 | 2.31 | 7.29 | 1.90 | 2.61 | 1.85 | 0.95 | 0.47 | 7.78 |
| 14 | 153.83 | 3.24 | 4.07 | 2.47 | 2.69 | 2.21 | 14.68 | 2.59 | 1.84 | 4.00 | 2.96 | 6.79 | 18.18 | 4.36 | 4.86 | 3.79 | 2.11 | 2.56 | 17.68 |
| 15 | 157.67 | 1.98 | 1.60 | 1.49 | 0.74 | 0 | 5.81 | 1.41 | 0.97 | 1.43 | 1.00 | 1.75 | 6.56 | 1.55 | 2.15 | 1.41 | 0.46 | 0.20 | 5.77 |
| 16 | 86.89 | 1.08 | 1.28 | 0.17 | 0 | 0 | 2.53 | 1.02 | 1.06 | 1.16 | 1.44 | 1.19 | 5.87 | 3.48 | 3.10 | 1.14 | 0.47 | 0.31 | 8.50 |
| 17 | 69.69 | 2.93 | 1.94 | 2.80 | 1.88 | 0 | 9.55 | 1.79 | 3.70 | 1.29 | 1.52 | 4.13 | 12.43 | 4.60 | 2.05 | 1.02 | 0.53 | 0 | 8.20 |
| 18 | 61.21 | 2.34 | 1.65 | 0.97 | 3.61 | 0 | 8.57 | 1.74 | 0.85 | 1.73 | 0.64 | 0 | 4.96 | 2.39 | 1.43 | 1.08 | 1.10 | 0 | 6.00 |
| Population | CHR | Start (bp) | End (bp) | Gene Symbol | Distance between ROH Region and Gene * | Function |
|---|---|---|---|---|---|---|
| Landrace | 7 | 57617069 | 58768368 | CPEB1 | 0.17 Mb | Reproduction |
| Songliao | 7 | 57554035 | 58768368 | CPEB1 | 0.24 Mb | Reproduction |
| Yorkshire | 1 | 135628006 | 141358502 | GATM | 5.21 Mb | Reproduction |
| 4 | 94734328 | 100297082 | HSD17B7 | 0.84 Mb | Reproduction | |
| SPATA46 | 1.17 Mb | Reproduction | ||||
| VANGL2 | 3.21 Mb | Reproduction | ||||
| NDUFS2 | 2.37 Mb | Energy conversion | ||||
| NR1I3 | 2.35 Mb | Meat quality | ||||
| APOA2 | 2.36 Mb | Meat quality | ||||
| USF1 | 2.64 Mb | Meat quality | ||||
| 7 | 33743648 | 35729978 | DAXX | 0.44 Mb | Reproduction |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, R.; Shi, L.; Liu, J.; Deng, T.; Wang, L.; Liu, Y.; Zhao, F. Genome-Wide Scan for Runs of Homozygosity Identifies Candidate Genes in Three Pig Breeds. Animals 2019, 9, 518. https://doi.org/10.3390/ani9080518
Xie R, Shi L, Liu J, Deng T, Wang L, Liu Y, Zhao F. Genome-Wide Scan for Runs of Homozygosity Identifies Candidate Genes in Three Pig Breeds. Animals. 2019; 9(8):518. https://doi.org/10.3390/ani9080518
Chicago/Turabian StyleXie, Rui, Liangyu Shi, Jiaxin Liu, Tianyu Deng, Lixian Wang, Yang Liu, and Fuping Zhao. 2019. "Genome-Wide Scan for Runs of Homozygosity Identifies Candidate Genes in Three Pig Breeds" Animals 9, no. 8: 518. https://doi.org/10.3390/ani9080518
APA StyleXie, R., Shi, L., Liu, J., Deng, T., Wang, L., Liu, Y., & Zhao, F. (2019). Genome-Wide Scan for Runs of Homozygosity Identifies Candidate Genes in Three Pig Breeds. Animals, 9(8), 518. https://doi.org/10.3390/ani9080518