Selection Signature Analysis of Whole-Genome Sequences to Identify Genome Differences Between Selected and Unselected Holstein Cattle


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population and Sample Collection
2.2. Whole Genome Sequencing and Data Quality Control
2.3. Analysis of Diversity and Population Structure
2.4. Selection Signature Analysis
2.5. Functional Annotation and Enrichment Analysis
2.6. Cattle QTL Annotation and Analysis
3. Results
3.1. Population Analysis Identified Genome-Wide Differences Between Selected and Unselected Cattle
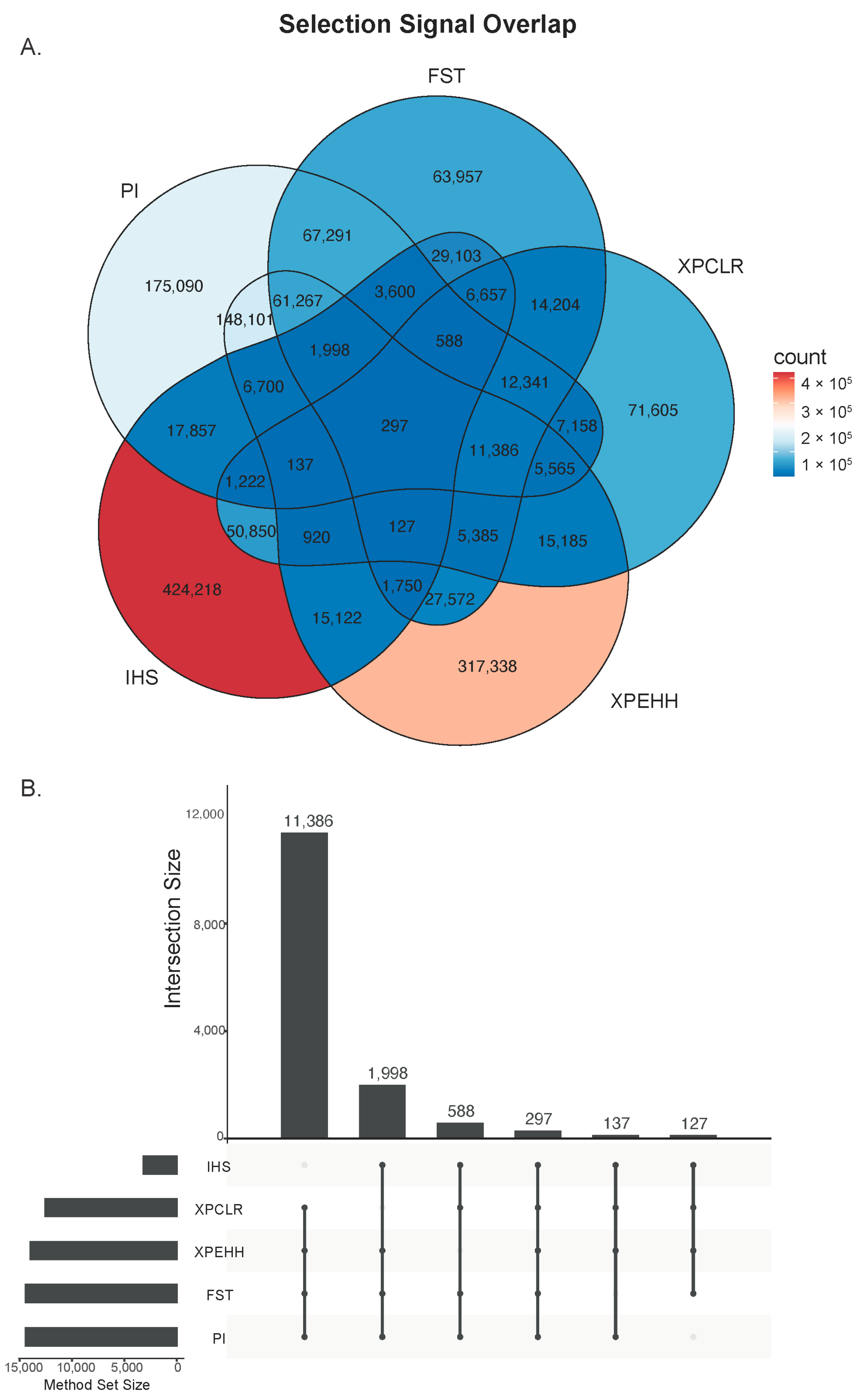
3.2. Selection Signature Analysis with Five Methods Identified Candidate Regions Under Selection
3.3. Gene Annotation and Enrichment Analysis of Selection Signatures
3.4. Cattle QTL Enrichment Analysis in Candidate Selection Signatures
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hayes, B.J.; Lewin, H.A.; Goddard, M.E. The Future of Livestock Breeding: Genomic Selection for Efficiency, Reduced Emissions Intensity, and Adaptation. Trends Genet. 2013, 29, 206–214. [Google Scholar] [CrossRef]
- Wiggans, G.R.; Cole, J.B.; Hubbard, S.M.; Sonstegard, T.S. Genomic Selection in Dairy Cattle: The Usda Experience. Annu. Rev. Anim. Biosci. 2017, 5, 309–327. [Google Scholar] [CrossRef]
- Hansen, L. Consequences of Selection for Milk Yield from a Geneticist’s Viewpoint. J. Dairy Sci. 2000, 83, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Brito, L.; Bédère, N.; Douhard, F.; Oliveira, H.; Arnal, M.; Peñagaricano, F.; Schinckel, A.; Baes, C.; Miglior, F. Genetic Selection of High-Yielding Dairy Cattle Toward Sustainable Farming Systems in a Rapidly Changing World. Animal 2021, 15, 100292. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Li, W.-H. Mathematical Model for Studying Genetic Variation in Terms of Restriction Endonucleases. Proc. Natl. Acad. Sci. USA 1979, 76, 5269–5273. [Google Scholar] [CrossRef] [PubMed]
- Sabeti, P.C.; Varilly, P.; Fry, B.; Lohmueller, J.; Hostetter, E.; Cotsapas, C.; Xie, X.; Byrne, E.H.; McCarroll, S.A.; Gaudet, R. Genome-Wide Detection and Characterization of Positive Selection in Human Populations. Nature 2007, 449, 913–918. [Google Scholar] [CrossRef]
- Voight, B.F.; Kudaravalli, S.; Wen, X.; Pritchard, J.K. A Map of Recent Positive Selection in the Human Genome. PLoS Biol. 2006, 4, e72. [Google Scholar]
- Yu, Y.; Fu, J.; Xu, Y.; Zhang, J.; Ren, F.; Zhao, H.; Tian, S.; Guo, W.; Tu, X.; Zhao, J. Genome Re-Sequencing Reveals the Evolutionary History of Peach Fruit Edibility. Nat. Commun. 2018, 9, 5404. [Google Scholar] [CrossRef]
- Ma, L.; Sonstegard, T.S.; Cole, J.B.; VanTassell, C.P.; Wiggans, G.R.; Crooker, B.A.; Tan, C.; Prakapenka, D.; Liu, G.E.; Da, Y. Genome Changes Due to Artificial Selection in Us Holstein Cattle. BMC Genom. 2019, 20, 128. [Google Scholar] [CrossRef]
- Poplin, R.; Chang, P.-C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T. A Universal Snp and Small-Indel Variant Caller Using Deep Neural Networks. Nat. Biotechnol. 2018, 36, 983–987. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-Generation Plink: Rising to the Challenge of Larger and Richer Datasets. GigaScience 2015, 4, s13742-015-0047-8. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, R.A.M.; Chen, Z.J. Ggplot2: Elegant Graphics for Data Analysis; Taylor & Francis: Abingdon, UK, 2019. [Google Scholar]
- Zhang, C.; Dong, S.-S.; Xu, J.-Y.; He, W.-M.; Yang, T.-L. PopLDdecay: A Fast and Effective Tool for Linkage Disequilibrium Decay Analysis Based on Variant Call Format Files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Patterson, N.; Reich, D. Population Differentiation as a Test for Selective Sweeps. Genome Res. 2010, 20, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T. The Variant Call Format and Vcftools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Szpiech, Z.A.; Hernandez, R.D. Selscan: An Efficient Multithreaded Program to Perform Ehh-Based Scans for Positive Selection. Mol. Biol. Evol. 2014, 31, 2824–2827. [Google Scholar] [CrossRef]
- Rosen, B.D.; Bickhart, D.M.; Schnabel, R.D.; Koren, S.; Elsik, C.G.; Tseng, E.; Rowan, T.N.; Low, W.Y.; Zimin, A.; Couldrey, C. De Novo Assembly of the Cattle Reference Genome with Single-Molecule Sequencing. GigaScience 2020, 9, giaa021. [Google Scholar] [CrossRef]
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Matsuura, Y.; Ishiguro-Watanabe, M. KEGG: Biological Systems Database as a Model of the Real World. Nucleic Acids Res. 2025, 53, D672–D677. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape Provides a Biologist-Oriented Resource for the Analysis of Systems-Level Datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Fonseca, P.A.; Suarez-Vega, A.; Marras, G.; Cánovas, Á. GALLO: An R Package for Genomic Annotation and Integration of Multiple Data Sources in Livestock for Positional Candidate Loci. GigaScience 2020, 9, giaa149. [Google Scholar] [CrossRef]
- Hu, Z.-L.; Park, C.A.; Wu, X.-L.; Reecy, J.M. Animal QTLdb: An Improved Database Tool for Livestock Animal Qtl/Association Data Dissemination in the Post-Genome Era. Nucleic Acids Res. 2013, 41, D871–D879. [Google Scholar] [CrossRef] [PubMed]
- D’Occhio, M.J.; Campanile, G.; Baruselli, P.S.; Porto Neto, L.R.; Hayes, B.J.; Snr, A.C.; Fortes, M.R. Pleomorphic Adenoma Gene1 in Reproduction and Implication for Embryonic Survival in Cattle: A Review. J. Anim. Sci. 2024, 102, skae103. [Google Scholar] [CrossRef] [PubMed]
- Gai, Z.; Hu, S.; Ma, J.; Wang, Y.; Gong, G.; Zhao, J. Whole Genome-Wide Analysis of Dep Family Members in Sheep (Ovis Aries) Reveals Their Potential Roles in Regulating Lactation. Chem. Biol. Technol. Agric. 2022, 9, 68. [Google Scholar] [CrossRef]
- Magalhaes, A.F.; De Camargo, G.M.; Fernandes, G.A.; Gordo, D.G.; Tonussi, R.L.; Costa, R.B.; Espigolan, R.; Silva, R.M.d.O.; Bresolin, T.; De Andrade, W.B. Genome-Wide Association Study of Meat Quality Traits in Nellore Cattle. PLoS ONE 2016, 11, e0157845. [Google Scholar] [CrossRef]
- Puglisi, R.; Cambuli, C.; Capoferri, R.; Giannino, L.; Lukaj, A.; Duchi, R.; Lazzari, G.; Galli, C.; Feligini, M.; Galli, A. Differential Gene Expression in Cumulus Oocyte Complexes Collected by Ovum Pick up from Repeat Breeder and Normally Fertile Holstein Friesian Heifers. Anim. Reprod. Sci. 2013, 141, 26–33. [Google Scholar] [CrossRef]
- Mukherjee, J.; Das, P.K.; Banerjee, D. Lactation Physiology. In Textbook of Veterinary Physiology; Springer: Berlin/Heidelberg, Germany, 2023; pp. 639–674. [Google Scholar]
- Arnandis, T.; Ferrer-Vicens, I.; García-Trevijano, E.; Miralles, V.; García, C.; Torres, L.; Viña, J.; Zaragozá, R. Calpains Mediate Epithelial-Cell Death During Mammary Gland Involution: Mitochondria and Lysosomal Destabilization. Cell Death Differ. 2012, 19, 1536–1548. [Google Scholar] [CrossRef]
- García-Trevijano, E.R.; Ortiz-Zapater, E.; Gimeno, A.; Viña, J.R.; Zaragozá, R. Calpains, the Proteases of Two Faces Controlling the Epithelial Homeostasis in Mammary Gland. Front. Cell Dev. Biol. 2023, 11, 1249317. [Google Scholar] [CrossRef] [PubMed]
- Tornillo, G.; Knowlson, C.; Kendrick, H.; Cooke, J.; Mirza, H.; Aurrekoetxea-Rodríguez, I.; dM Vivanco, M.; Buckley, N.E.; Grigoriadis, A.; Smalley, M.J. Dual Mechanisms of Lyn Kinase Dysregulation Drive Aggressive Behavior in Breast Cancer Cells. Cell Rep. 2018, 25, 3674–3692.e10. [Google Scholar] [CrossRef]
- Vallone, S.A.; García Solá, M.; Schere-Levy, C.; Meiss, R.P.; Hermida, G.N.; Chodosh, L.A.; Kordon, E.C.; Hynes, N.E.; Gattelli, A. Aberrant Ret Expression Affects Normal Mammary Gland Post-Lactation Transition, Enhancing Cancer Potential. Dis. Models Mech. 2022, 15, dmm049286. [Google Scholar] [CrossRef]
- Li, C.; Sun, D.; Zhang, S.; Wang, S.; Wu, X.; Zhang, Q.; Liu, L.; Li, Y.; Qiao, L. Genome Wide Association Study Identifies 20 Novel Promising Genes Associated with Milk Fatty Acid Traits in Chinese Holstein. PLoS ONE 2014, 9, e96186. [Google Scholar] [CrossRef]
- Chen, Z.; Yao, Y.; Ma, P.; Wang, Q.; Pan, Y. Haplotype-Based Genome-Wide Association Study Identifies Loci and Candidate Genes for Milk Yield in Holsteins. PLoS ONE 2018, 13, e0192695. [Google Scholar] [CrossRef]
- Pečnik, Ž.; Jevšinek Skok, D. Identification of Genomic Regions Affecting Nitrogen Excretion Intensity in Brown Swiss Dairy Cows. Anim. Biotechnol. 2024, 35, 2434097. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Guldbrandtsen, B.; Lund, M.S.; Sahana, G. Prioritizing Candidate genes Post-Gwas Using Multiple Sources of Data for Mastitis Resistance in Dairy Cattle. BMC Genom. 2018, 19, 656. [Google Scholar] [CrossRef] [PubMed]
- Zare, M.; Atashi, H.; Hostens, M. Genome-Wide Association Study for Lactation Performance in the Early and Peak Stages of Lactation in Holstein Dairy Cows. Animals 2022, 12, 1541. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-W.; Sung, Y.-W.; Hsueh, Y.-W.; Chen, Y.-Y.; Ho, M.; Hsu, H.-C.; Yang, T.-C.; Lin, W.-C.; Chang, H.-M. Growth Hormone in Fertility and Infertility: Mechanisms of Action and Clinical Applications. Front. Endocrinol. 2022, 13, 1040503. [Google Scholar] [CrossRef]
- Li, F.; Feng, X.; Li, R.; Du, B.; Xue, X. Genetic Bases and Molecular Breeding of Key Economic Traits in China Dairy Cattle: A Progress Report. Int. J. Clin. Case Rep. Rev. 2022, 12. [Google Scholar] [CrossRef]
- Chen, S.; Paul, M.R.; Sterner, C.J.; Belka, G.K.; Wang, D.; Xu, P.; Sreekumar, A.; Pan, T.-c.; Pant, D.K.; Makhlin, I. PAQR8 Promotes Breast Cancer Recurrence and Confers Resistance to Multiple Therapies. Breast Cancer Res. 2023, 25, 1. [Google Scholar] [CrossRef]
- Zhang, Y.T.; Hong, W.S.; Liu, D.T.; Qiu, H.T.; Zhu, Y.; Chen, S.X. Involvement of Membrane Progestin Receptor Beta (Mprβ/Paqr8) in Sex Pheromone Progestin-Induced Expression of Luteinizing Hormone in the Pituitary of Male Chinese Black Sleeper (Bostrychus sinensis). Front. Endocrinol. 2018, 9, 397. [Google Scholar] [CrossRef]
- Laodim, T.; Koonawootrittriron, S.; Elzo, M.A.; Suwanasopee, T.; Jattawa, D.; Sarakul, M. Genetic Factors Influencing Milk and Fat Yields in Tropically Adapted Dairy Cattle: Insights from Quantitative Trait Loci Analysis and Gene Associations. Anim. Biosci. 2023, 37, 576. [Google Scholar] [CrossRef]
- Daldaban, F.; Kıyıcı, J.M.; Akyüz, B.; Aksel, E.G.; Kaliber, M.; Çınar, M.U.; Arslan, K. Association of the Cacna2d1 Gene with Milk Yield and Milk Quality Traits in Holstein Cattle. J. Dairy Res. 2024, 91, 373–377. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, H.; Zhao, X.; Zhang, J.; Zhao, Z.; Lian, X.; Zhang, J.; Kong, F.; Hu, T.; Wang, T. Targeting the Immunoglobulin Igsf9 Enhances Antitumor T-Cell Activity and Sensitivity to Anti–Pd-1 Immunotherapy. Cancer Res. 2023, 83, 3385–3399. [Google Scholar] [CrossRef]
- Dai, L.; Zhao, J.; Yin, J.; Fu, W.; Chen, G. Cell Adhesion Molecule 2 (Cadm2) Promotes Brain Metastasis by Inducing Epithelial-Mesenchymal Transition (Emt) in Human Non-Small Cell Lung Cancer. Ann. Transl. Med. 2020, 8, 465. [Google Scholar] [CrossRef]
- Cai, W.; Cole, J.B.; Goddard, M.E.; Li, J.; Zhang, S.; Song, J. Mammary Gland Multi-Omics Data Reveals New Genetic Insights into Milk Production Traits in Dairy Cattle. PLoS Genet. 2025, 21, e1011675. [Google Scholar] [CrossRef]
- Arshad, U.; Kennedy, K.M.; Cid de la Paz, M.; Kendall, S.J.; Cangiano, L.R.; White, H.M. Immune Cells Phenotype and Bioenergetic Measures in Cd4+ T Cells Differ between High and Low Feed Efficient Dairy Cows. Sci. Rep. 2024, 14, 15993. [Google Scholar] [CrossRef] [PubMed]
- Peng, V.; Trsan, T.; Sudan, R.; Bhattarai, B.; Cortez, V.S.; Molgora, M.; Vacher, J.; Colonna, M. Inositol Phosphatase Inpp4b Sustains Ilc1s and Intratumoral NK Cells through an Akt-Driven Pathway. J. Exp. Med. 2024, 221, e20230124. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; Mortha, A. Inpp4b Ensures That Ilc1s and Nk Cells Set up a Productive Home Office. J. Exp. Med. 2024, 221, e20232375. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Observed Heterozygosity (Ho) | Expected Heterozygosity (He) | Inbreeding coefficient (F) |
|---|---|---|---|
| Unselected | 0.291 | 0.316 | 0.078 |
| Selected | 0.305 | 0.321 | 0.049 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, J.; Yang, L.; Gao, Y.; Liu, G.E.; Da, Y.; Ma, L. Selection Signature Analysis of Whole-Genome Sequences to Identify Genome Differences Between Selected and Unselected Holstein Cattle. Animals 2025, 15, 2247. https://doi.org/10.3390/ani15152247
Cai J, Yang L, Gao Y, Liu GE, Da Y, Ma L. Selection Signature Analysis of Whole-Genome Sequences to Identify Genome Differences Between Selected and Unselected Holstein Cattle. Animals. 2025; 15(15):2247. https://doi.org/10.3390/ani15152247
Chicago/Turabian StyleCai, Jiarui, Liu Yang, Yahui Gao, George E. Liu, Yang Da, and Li Ma. 2025. "Selection Signature Analysis of Whole-Genome Sequences to Identify Genome Differences Between Selected and Unselected Holstein Cattle" Animals 15, no. 15: 2247. https://doi.org/10.3390/ani15152247
APA StyleCai, J., Yang, L., Gao, Y., Liu, G. E., Da, Y., & Ma, L. (2025). Selection Signature Analysis of Whole-Genome Sequences to Identify Genome Differences Between Selected and Unselected Holstein Cattle. Animals, 15(15), 2247. https://doi.org/10.3390/ani15152247