

Insights into the Donkey Hindgut Microbiome Using Metagenome-Assembled Genomes

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Selection, Husbandry, and Sample Collection
2.2. Genomic DNA and Genome Sequencing
2.3. Metagenomic Assembly and Binning
2.4. Annotation and Functional Analyses of MAGs
2.5. Analyses of Interaction Networks of MAGs
2.6. Statistical Analysis
2.7. Data Availability
3. Results
3.1. Metagenome-Assembled Genomes
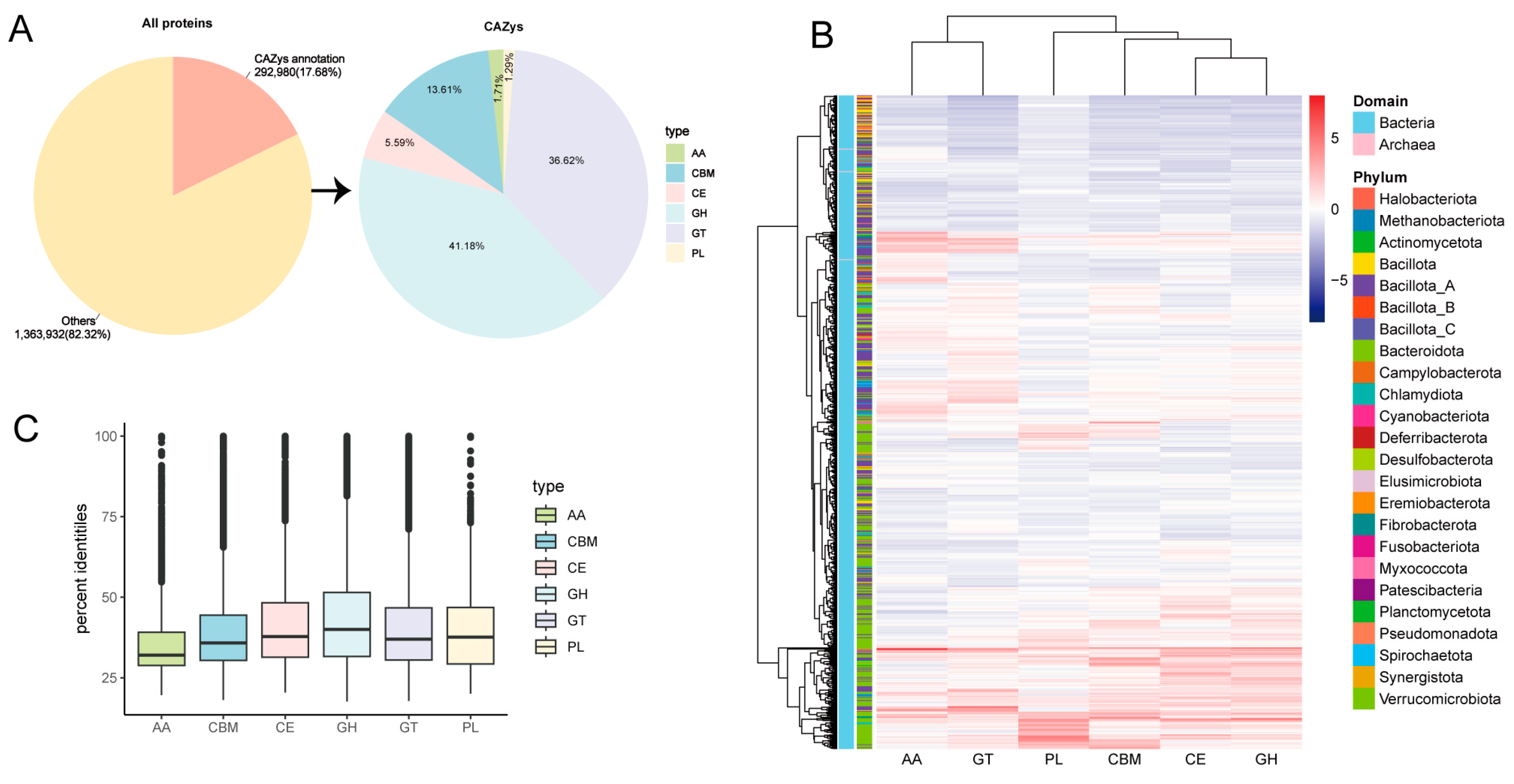
3.2. Novel CAZymes of MAGs
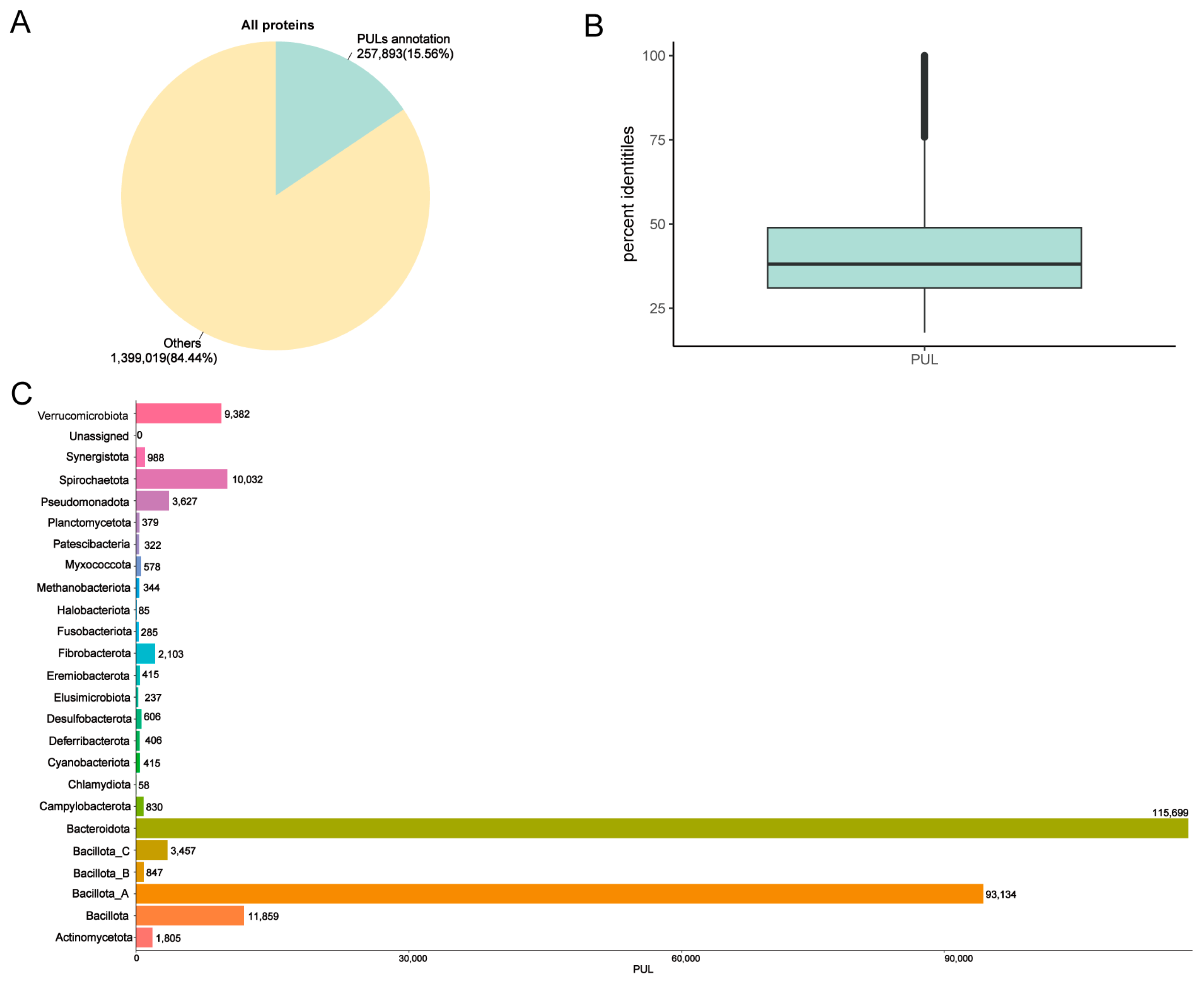
3.3. Annotation PULs of MAGs
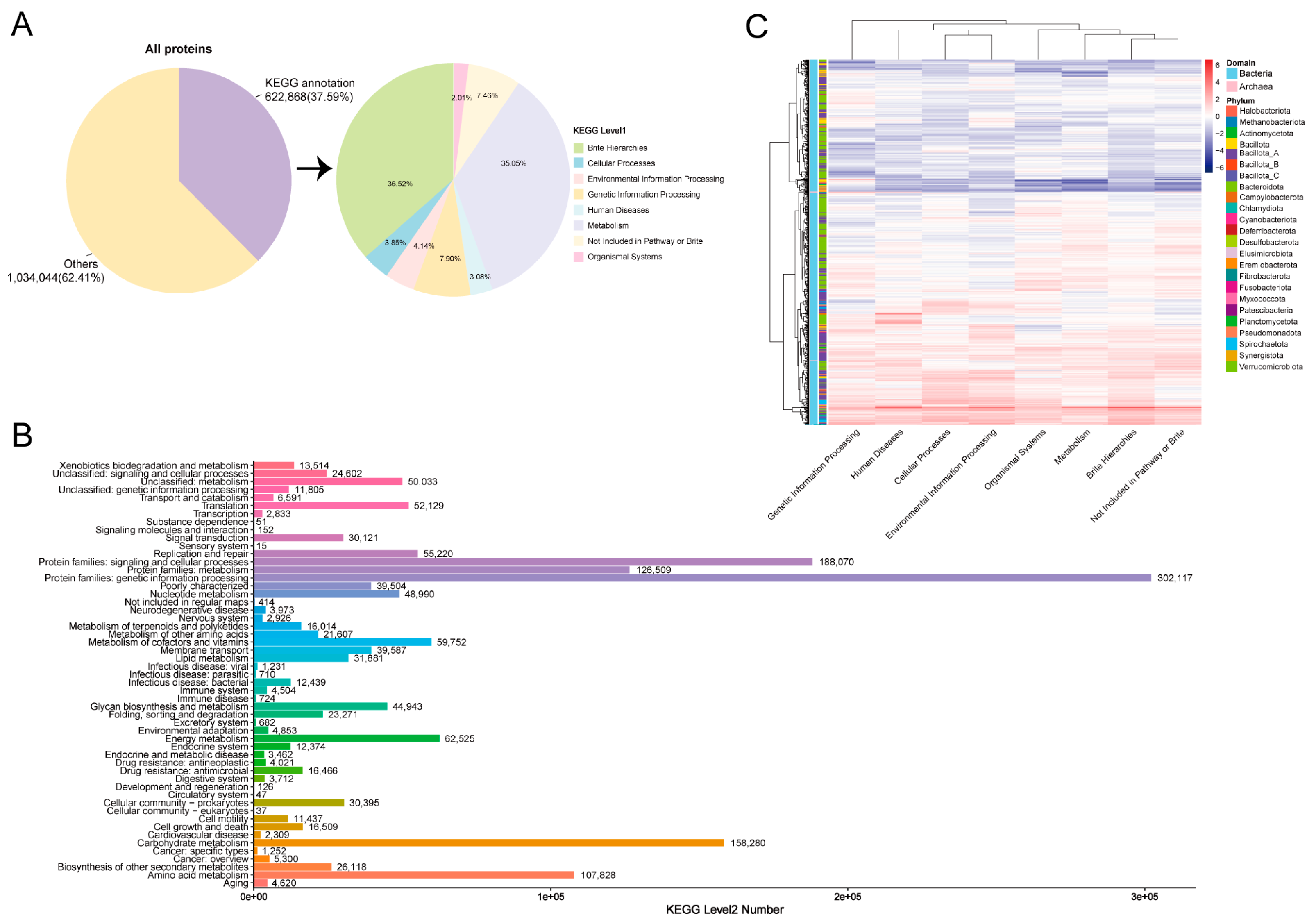
3.4. KEGG and MCI Analyses of Hindgut MAGs
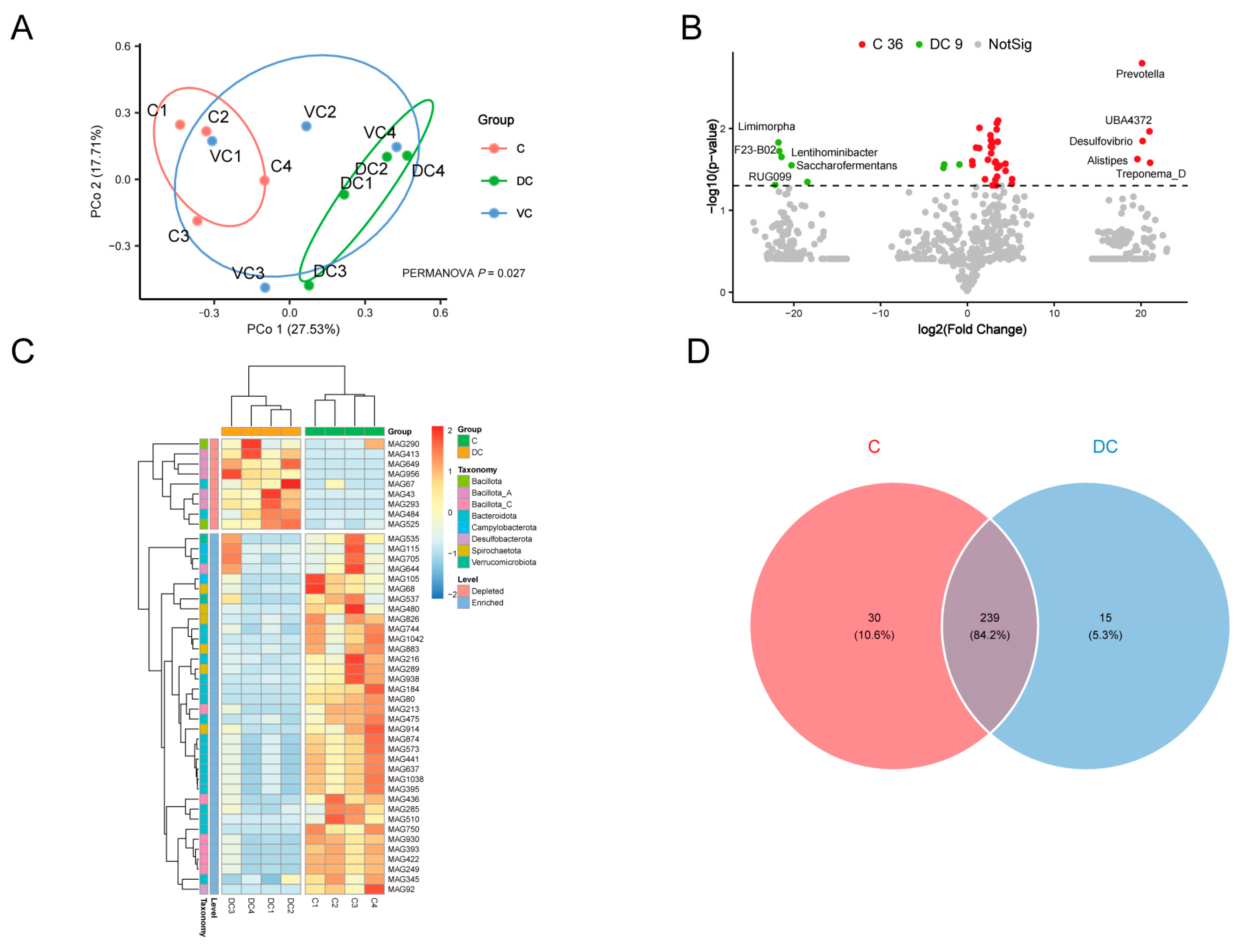
3.5. Analysis of Hindgut Differential MAGs
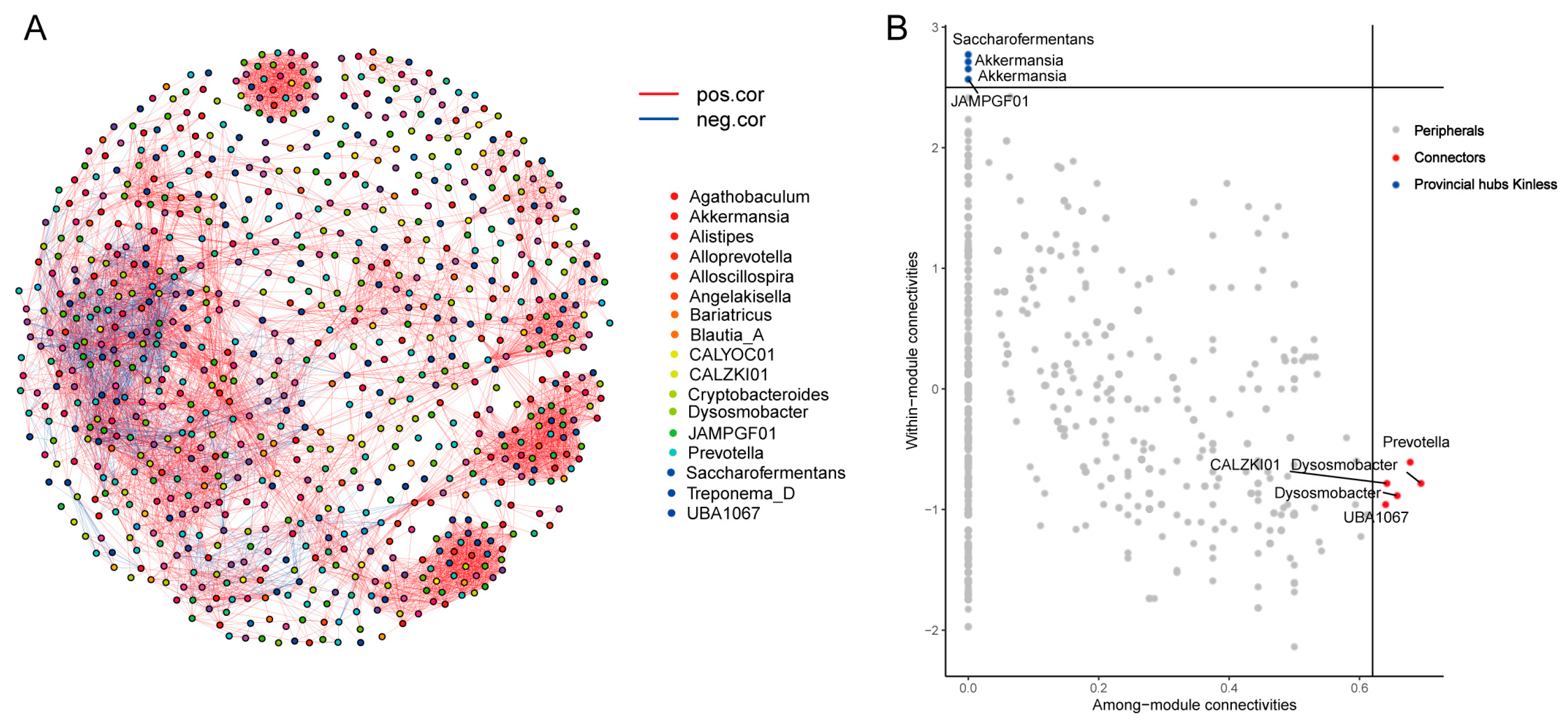
3.6. MAGs Co-Occurrence and Interaction Networks in the Cecum
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Peixoto, R.S.; Harkins, D.M.; Nelson, K.E. Advances in Microbiome Research for Animal Health. Annu. Rev. Anim. Biosci. 2021, 9, 289–311. [Google Scholar] [CrossRef]
- Ezenwa, V.O.; Gerardo, N.M.; Inouye, D.W.; Medina, M.; Xavier, J.B. Animal Behavior and the Microbiome. Science 2012, 338, 198–199. [Google Scholar] [CrossRef]
- Khan, M.Z.; Chen, W.; Wang, X.; Liang, H.; Wei, L.; Huang, B.; Kou, X.; Liu, X.; Zhang, Z.; Chai, W.; et al. A review of genetic resources and trends of omics applications in donkey research: Focus on China. Front. Vet. Sci. 2024, 11, 1366128. [Google Scholar] [CrossRef]
- Hao, Z.; Ding, X.; Wang, J. Chapter Four—Effects of gut bacteria and their metabolites on gut health of animals. In Advances in Applied Microbiology; Gadd, G.M., Sariaslani, S., Eds.; Academic Press: Cambridge, MA, USA, 2024; Volume 127, pp. 223–252. [Google Scholar]
- Balasundaram, D.; Veerasamy, V.; Sylvia Singarayar, M.; Neethirajan, V.; Ananth Devanesan, A.; Thilagar, S. Therapeutic potential of probiotics in gut microbial homeostasis and Rheumatoid arthritis. Int. Immunopharmacol. 2024, 137, 112501. [Google Scholar] [CrossRef]
- Durack, J.; Lynch, S.V. The gut microbiome: Relationships with disease and opportunities for therapy. J. Exp. Med. 2019, 216, 20–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhang, J.; Dang, W.; Irwin, D.; Wang, Z.; Zhang, S. Unveiling the Biogeography and Potential Functions of the Intestinal Digesta- and Mucosa-Associated Microbiome of Donkeys. Front. Microbiol. 2020, 11, 596882. [Google Scholar] [CrossRef]
- Julliand, V.; Grimm, P. HORSE SPECIES SYMPOSIUM: The microbiome of the horse hindgut: History and current knowledge. J. Anim. Sci. 2016, 94, 2262–2274. [Google Scholar] [CrossRef] [PubMed]
- Murru, F.; Fliegerova, K.; Mura, E.; Mrázek, J.; Kopečný, J.; Moniello, G. A comparison of methanogens of different regions of the equine hindgut. Anaerobe 2018, 54, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.C.; Arroyo, L.G.; Allen-Vercoe, E.; Stämpfli, H.R.; Kim, P.T.; Sturgeon, A.; Weese, J.S. Comparison of the fecal microbiota of healthy horses and horses with colitis by high throughput sequencing of the V3-V5 region of the 16S rRNA gene. PLoS ONE 2012, 7, e41484. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.S.; Rodrigues, M.A.M.; Bessa, R.J.B.; Ferreira, L.M.; Martin-Rosset, W. Understanding the equine cecum-colon ecosystem: Current knowledge and future perspectives. Animal 2011, 5, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Lindenberg, F.; Krych, L.; Kot, W.; Fielden, J.; Frøkiær, H.; van Galen, G.; Nielsen, D.S.; Hansen, A.K. Development of the equine gut microbiota. Sci. Rep. 2019, 9, 14427. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Huang, B.; Gao, X.; Shi, X.; Wang, X.; Wang, T.; Wang, Y.; Liu, G.; Wang, C. Dynamic changes in fecal microbiota in donkey foals during weaning: From pre-weaning to post-weaning. Front. Microbiol. 2023, 14, 1105330. [Google Scholar] [CrossRef]
- Liu, G.; Bou, G.; Su, S.; Xing, J.; Qu, H.; Zhang, X.; Wang, X.; Zhao, Y.; Dugarjaviin, M. Microbial diversity within the digestive tract contents of Dezhou donkeys. PLoS ONE 2019, 14, e0226186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Huang, B.; Shi, X.; Wang, T.; Wang, Y.; Zhu, M.; Wang, C. Comparative analysis of bacterial diversity between the liquid phase and adherent fraction within the donkey Caeco-Colic ecosystem. Animals 2022, 12, 1116. [Google Scholar] [CrossRef]
- Xing, J.; Liu, G.; Zhang, X.; Bai, D.; Yu, J.; Li, L.; Wang, X.; Su, S.; Zhao, Y.; Bou, G.; et al. The Composition and Predictive Function of the Fecal Microbiota Differ Between Young and Adult Donkeys. Front. Microbiol. 2020, 11, 596394. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, Q.; Liu, G.; Zhang, Z.; Zhan, Y.; Zhu, M.; Wang, C. Metabolic Alternations During Gestation in Dezhou Donkeys and the Link to the Gut Microbiota. Front. Microbiol. 2022, 13, 801976. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, Q.; Shi, X.; Liu, G.; Wang, C. Integrated multi-omics reveals novel microbe-host lipid metabolism and immune interactions in the donkey hindgut. Front. Immunol. 2022, 13, 1003247. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.D.; Auffret, M.D.; Warr, A.; Walker, A.W.; Roehe, R.; Watson, M. Compendium of 4,941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat. Biotechnol. 2019, 37, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Glendinning, L.; Genç, B.; Wallace, R.J.; Watson, M. Metagenomic analysis of the cow, sheep, reindeer and red deer rumen. Sci. Rep. 2021, 11, 1990. [Google Scholar] [CrossRef]
- Yutin, N.; Benler, S.; Shmakov, S.A.; Wolf, Y.I.; Tolstoy, I.; Rayko, M.; Antipov, D.; Pevzner, P.A.; Koonin, E.V. Analysis of metagenome-assembled viral genomes from the human gut reveals diverse putative CrAss-like phages with unique genomic features. Nat. Commun. 2021, 12, 1044. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, X.; Guo, R.; Ni, W.; Liu, K.; Liu, Z.; Dai, J.; Xu, Y.; Abduriyim, S.; Wu, Z.; et al. Expanded catalogue of metagenome-assembled genomes reveals resistome characteristics and athletic performance-associated microbes in horse. Microbiome 2023, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, Y.; Huang, B.; Zhu, M.; Wang, C. The Fibrolytic Enzyme Profiles and the Composition of Fungal Communities in Donkey Cecum-Colon Ecosystem. Animals 2022, 12, 412. [Google Scholar] [CrossRef]
- Ren, Y.; Yu, G.; Shi, C.; Liu, L.; Guo, Q.; Han, C.; Zhang, D.; Zhang, L.; Liu, B.; Gao, H.; et al. Majorbio Cloud: A one-stop, comprehensive bioinformatic platform for multiomics analyses. iMeta 2022, 1, e12. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015, 3, e1165. [Google Scholar] [CrossRef]
- Alneberg, J.; Bjarnason, B.S.; de Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Lahti, L.; Loman, N.J.; Andersson, A.F.; Quince, C. Binning metagenomic contigs by coverage and composition. Nat. Methods 2014, 11, 1144–1146. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2015, 32, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Olm, M.R.; Brown, C.T.; Brooks, B.; Banfield, J.F. dRep: A tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. Isme J. 2017, 11, 2864–2868. [Google Scholar] [CrossRef]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk v2: Memory friendly classification with the genome taxonomy database. Bioinformatics 2022, 38, 5315–5316. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef]
- Ausland, C.; Zheng, J.; Yi, H.; Yang, B.; Li, T.; Feng, X.; Zheng, B.; Yin, Y. dbCAN-PUL: A database of experimentally characterized CAZyme gene clusters and their substrates. Nucleic Acids Res. 2021, 49, D523–D528. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Li, L.; Luo, X.; Chen, M.; Tang, W.; Zhan, L.; Dai, Z.; Lam, T.T.; Guan, Y.; Yu, G. Ggtree: A serialized data object for visualization of a phylogenetic tree and annotation data. iMeta 2022, 1, e56. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Gu, F.; Zhu, S.; Hou, J.; Tang, Y.; Liu, J.-X.; Xu, Q.; Sun, H.-Z. The hindgut microbiome contributes to host oxidative stress in postpartum dairy cows by affecting glutathione synthesis process. Microbiome 2023, 11, 87. [Google Scholar] [CrossRef]
- Sato, Y.; Sato, R.; Fukui, E.; Yoshizawa, F. Impact of rumen microbiome on cattle carcass traits. Sci. Rep. 2024, 14, 6064. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, N.; Carlsen, B.; Christensen, M.H.; Pertoldi, C.; Nielsen, J.L. The Gut Microbiome of 54 Mammalian Species. Front. Microbiol. 2022, 13, 886252. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhao, X.; Han, X.; Xu, S.; Zhao, L.; Hu, L.; Xu, T.; Zhao, N.; Zhang, X.; Chen, D.; et al. Comparative study of gut microbiota in Tibetan wild asses (Equus kiang) and domestic donkeys (Equus asinus) on the Qinghai-Tibet plateau. PeerJ 2020, 8, e9032. [Google Scholar] [CrossRef]
- Holman, D.B.; Kommadath, A.; Tingley, J.P.; Abbott, D.W. Novel Insights into the Pig Gut Microbiome Using Metagenome-Assembled Genomes. Microbiol. Spectr. 2022, 10, e0238022. [Google Scholar] [CrossRef] [PubMed]
- Burden, F.A.; Bell, N. Donkey Nutrition and Malnutrition. Vet. Clin. N. Am. Equine Pract. 2019, 35, 469–479. [Google Scholar] [CrossRef]
- Orellana, L.H.; Francis, T.B.; Ferraro, M.; Hehemann, J.H.; Fuchs, B.M.; Amann, R.I. Verrucomicrobiota are specialist consumers of sulfated methyl pentoses during diatom blooms. Isme J. 2022, 16, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Grondin, J.M.; Tamura, K.; Déjean, G.; Abbott, D.W.; Brumer, H. Polysaccharide utilization loci: Fueling microbial communities. J. Bacteriol. 2017, 199, e00860-16. [Google Scholar] [CrossRef] [PubMed]
- Dao, T.-K.; Do, T.-H.; Le, N.-G.; Nguyen, H.-D.; Nguyen, T.-Q.; Le, T.-T.-H.; Truong, N.-H. Understanding the Role of Prevotella Genus in the Digestion of Lignocellulose and Other Substrates in Vietnamese Native Goats’ Rumen by Metagenomic Deep Sequencing. Animals 2021, 11, 3257. [Google Scholar] [CrossRef] [PubMed]
- Fehlner-Peach, H.; Magnabosco, C.; Raghavan, V.; Scher, J.U.; Tett, A.; Cox, L.M.; Gottsegen, C.; Watters, A.; Wiltshire-Gordon, J.D.; Segata, N.; et al. Distinct Polysaccharide Utilization Profiles of Human Intestinal Prevotella copri Isolates. Cell Host Microbe 2019, 26, 680–690.e5. [Google Scholar] [CrossRef] [PubMed]
- Accetto, T.; Avguštin, G. Polysaccharide utilization locus and CAZYme genome repertoires reveal diverse ecological adaptation of Prevotella species. Syst. Appl. Microbiol 2015, 38, 453–461. [Google Scholar] [CrossRef]
- Accetto, T.; Avguštin, G. The diverse and extensive plant polysaccharide degradative apparatuses of the rumen and hindgut Prevotella species: A factor in their ubiquity? Syst. Appl. Microbiol. 2019, 42, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Chang, J.; Zhang, R.; Fang, W.; Chen, L.; Ma, W.; Zhang, Y.; Yang, W.; Li, Y.; Zhang, P.; et al. Metagenomic analysis reveals the efficient digestion mechanism of corn stover in Angus bull rumen: Microbial community succession, CAZyme composition and functional gene expression. Chemosphere 2023, 336, 139242. [Google Scholar] [CrossRef]
- Sun, Z.; Jiang, X.; Wang, B.; Tian, F.; Zhang, H.; Yu, L. Novel Phocaeicola Strain Ameliorates Dextran Sulfate Sodium-induced Colitis in Mice. Curr. Microbiol. 2022, 79, 393. [Google Scholar] [CrossRef]
- Murray, E.; Epstein, K. Case 12.4—Cecal Impaction-Anatomical features in equids. In Comparative Veterinary Anatomy; Orsini, J.A., Grenager, N.S., de Lahunta, A., Eds.; Academic Press: Cambridge, MA, USA, 2022; pp. 744–749. [Google Scholar]
- Chen, C.; Fang, S.; Wei, H.; He, M.; Fu, H.; Xiong, X.; Zhou, Y.; Wu, J.; Gao, J.; Yang, H.; et al. Prevotella copri increases fat accumulation in pigs fed with formula diets. Microbiome 2021, 9, 175. [Google Scholar] [CrossRef]
- Betancur-Murillo, C.L.; Aguilar-Marín, S.B.; Jovel, J. Prevotella: A Key Player in Ruminal Metabolism. Microorganisms 2023, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, B.; Luckey, D.; Bodhke, R.; Chen, J.; Marietta, E.; Jeraldo, P.; Murray, J.; Taneja, V. Prevotella histicola Protects From Arthritis by Expansion of Allobaculum and Augmenting Butyrate Production in Humanized Mice. Front. Immunol. 2021, 12, 609644. [Google Scholar] [CrossRef] [PubMed]
- Sawin, E.A.; De Wolfe, T.J.; Aktas, B.; Stroup, B.M.; Murali, S.G.; Steele, J.L.; Ney, D.M. Glycomacropeptide is a prebiotic that reduces Desulfovibrio bacteria, increases cecal short-chain fatty acids, and is anti-inflammatory in mice. Am. J. Physiol. Gastrointest Liver Physiol. 2015, 309, G590–G601. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.J.; Wearsch, P.A.; Veloo, A.C.M.; Rodriguez-Palacios, A. The Genus Alistipes: Gut Bacteria with Emerging Implications to Inflammation, Cancer, and Mental Health. Front. Immunol. 2020, 11, 906. [Google Scholar] [CrossRef]
- Xu, R.; Li, Q.; Wang, H.; Su, Y.; Zhu, W. Reduction of Redox Potential Exerts a Key Role in Modulating Gut Microbial Taxa and Function by Dietary Supplementation of Pectin in a Pig Model. Microbiol. Spectr. 2023, 11, e0328322. [Google Scholar] [CrossRef] [PubMed]
- Ramayo-Caldas, Y.; Mach, N.; Lepage, P.; Levenez, F.; Denis, C.; Lemonnier, G.; Leplat, J.J.; Billon, Y.; Berri, M.; Doré, J.; et al. Phylogenetic network analysis applied to pig gut microbiota identifies an ecosystem structure linked with growth traits. Isme J. 2016, 10, 2973–2977. [Google Scholar] [CrossRef]
- Tokuda, G.; Mikaelyan, A.; Fukui, C.; Matsuura, Y.; Watanabe, H.; Fujishima, M.; Brune, A. Fiber-associated spirochetes are major agents of hemicellulose degradation in the hindgut of wood-feeding higher termites. Proc. Natl. Acad. Sci. USA 2018, 115, E11996–E12004. [Google Scholar] [CrossRef]
- Zhang, J.; Loh, K.-C.; Lee, J.; Wang, C.-H.; Dai, Y.; Wah Tong, Y. Three-stage anaerobic co-digestion of food waste and horse manure. Sci. Rep. 2017, 7, 1269. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Luo, X.; Lin, H.; Han, F.; Qin, J.G.; Chen, L.; Xu, C.; Li, E. Growth, Health, and Gut Microbiota of Female Pacific White Shrimp, Litopenaeus vannamei Broodstock Fed Different Phospholipid Sources. Antioxidants 2022, 11, 1143. [Google Scholar] [CrossRef] [PubMed]
- Amat, S.; Lantz, H.; Munyaka, P.M.; Willing, B.P. Prevotella in Pigs: The Positive and Negative Associations with Production and Health. Microorganisms 2020, 8, 1584. [Google Scholar] [CrossRef]
- Le Roy, T.; Moens de Hase, E.; Van Hul, M.; Paquot, A.; Pelicaen, R.; Régnier, M.; Depommier, C.; Druart, C.; Everard, A.; Maiter, D.; et al. Dysosmobacter welbionis is a newly isolated human commensal bacterium preventing diet-induced obesity and metabolic disorders in mice. Gut 2022, 71, 534–543. [Google Scholar] [CrossRef]
- Pellegrino, A.; Coppola, G.; Santopaolo, F.; Gasbarrini, A.; Ponziani, F.R. Role of Akkermansia in human diseases: From causation to therapeutic properties. Nutrients 2023, 15, 1815. [Google Scholar] [CrossRef] [PubMed]
- Kalia, V.C.; Gong, C.; Shanmugam, R.; Lin, H.; Zhang, L.; Lee, J.K. The emerging biotherapeutic agent: Akkermansia. Indian J. Microbiol. 2022, 62, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Derrien, M.; Belzer, C.; de Vos, W.M. Akkermansia muciniphila and its role in regulating host functions. Microb. Pathog. 2017, 106, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Rabee, A.E.; Younan, B.R.; Kewan, K.Z.; Sabra, E.A.; Lamara, M. Modulation of rumen bacterial community and feed utilization in camel and sheep using combined supplementation of live yeast and microalgae. Sci. Rep. 2022, 12, 12990. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kou, X.; Liu, Y.; Xiang, F.; Zhang, X.; Khan, M.Z.; Wu, B.; Wang, H.; Gong, Y.; Wang, C.; Ma, Q.; et al. Insights into the Donkey Hindgut Microbiome Using Metagenome-Assembled Genomes. Animals 2024, 14, 3625. https://doi.org/10.3390/ani14243625
Kou X, Liu Y, Xiang F, Zhang X, Khan MZ, Wu B, Wang H, Gong Y, Wang C, Ma Q, et al. Insights into the Donkey Hindgut Microbiome Using Metagenome-Assembled Genomes. Animals. 2024; 14(24):3625. https://doi.org/10.3390/ani14243625
Chicago/Turabian StyleKou, Xiyan, Yihong Liu, Fokun Xiang, Xinyue Zhang, Muhammad Zahoor Khan, Boxian Wu, Hua Wang, Yanlin Gong, Changfa Wang, Qingshan Ma, and et al. 2024. "Insights into the Donkey Hindgut Microbiome Using Metagenome-Assembled Genomes" Animals 14, no. 24: 3625. https://doi.org/10.3390/ani14243625
APA StyleKou, X., Liu, Y., Xiang, F., Zhang, X., Khan, M. Z., Wu, B., Wang, H., Gong, Y., Wang, C., Ma, Q., & Li, Y. (2024). Insights into the Donkey Hindgut Microbiome Using Metagenome-Assembled Genomes. Animals, 14(24), 3625. https://doi.org/10.3390/ani14243625