
Evolutionary Insights into the Relationship of Frogs, Salamanders, and Caecilians and Their Adaptive Traits, with an Emphasis on Salamander Regeneration and Longevity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection, Transcriptome Sequencing, and De Novo Assembly
2.2. Phylogenetic Analyses and Date Estimation
2.2.1. Construction of the Raw Phylogenomic Datasets
2.2.2. Data Filtration
2.2.3. Phylogenomic Tree Construction
2.2.4. Divergence Time Estimates
2.3. Test for Selections on Lissamphibia
2.3.1. Lineage-Specific dN/dS and dS Estimates
2.3.2. Rapidly Evolving GO Categories
2.3.3. Identification of Genes under Positive Selection
2.3.4. Identification of Fast-Evolving Genes
2.4. Test for Parallel Evolution
2.5. Prediction of Functional Impact of Variants
2.6. Quantitative Real-Time PCR
3. Results and Discussion
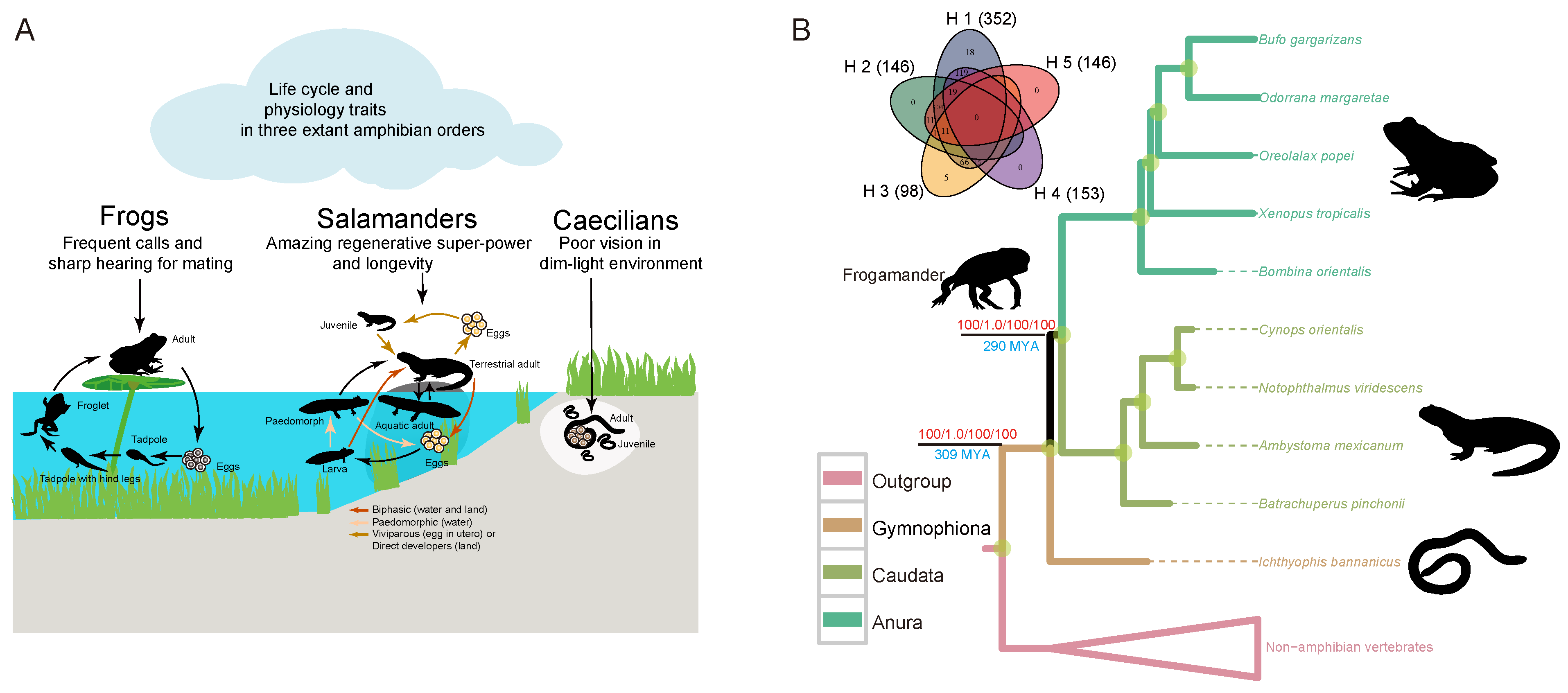
3.1. Robust Support for Frog–Salamander Sister Relationship
3.2. Fossil-Compatible Divergence Time of Lissamphibia
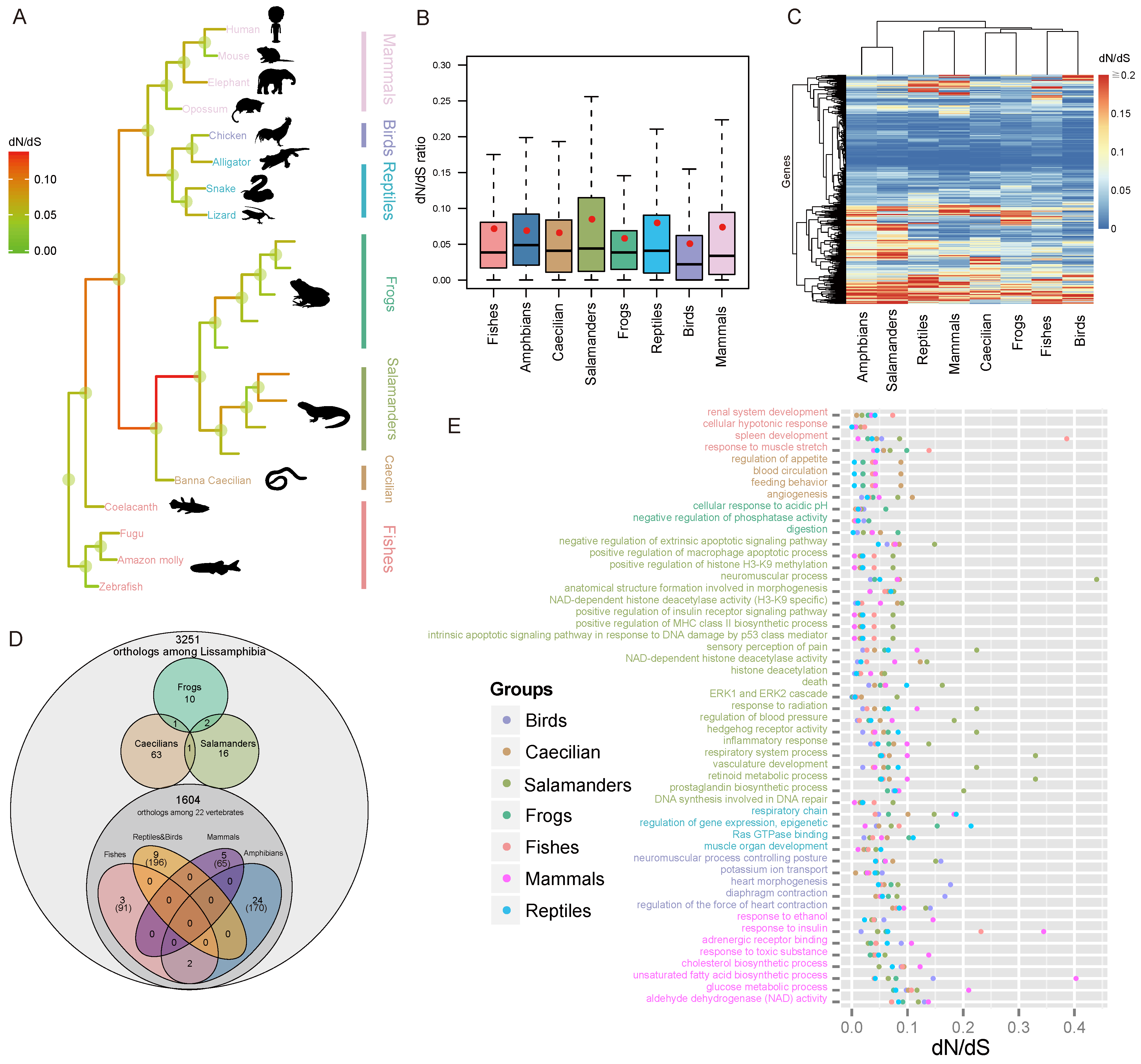
3.3. Rapid Evolution of Salamander Genes
3.4. Detection of Selection
3.5. Regeneration- and Development-Related Genes in Salamanders
3.6. Longevity-Related Genes in Salamanders
3.7. Vocalization- and Hearing-Related Genes in Frogs
3.8. Vision-Related Genes in Caecilians
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 4D | Four-fold degenerate sites |
| BEBPP | Bayes empirical Bayes posterior probabilities |
| BI | Bayesian inference |
| BIC | Bayesian Information Criterion |
| CDS | Coding sequence |
| CH | Compositional heterogeneity |
| ECM | Extracellular matrix |
| FDR | False discovery rate |
| FEGs | Fast-evolving genes |
| GAC | Genes as characters |
| GO | Gene Ontology |
| HaMSTR | Hidden Markov Model-based Search for Orthologs using Reciprocity |
| LRT | Likelihood ratio test |
| ML | Maximum likelihood |
| MPEs | Maximum pseudo-likelihood estimates |
| PSGs | Positively selected genes |
| RCFV | Relative Composition Frequency Variability |
| REGOs | Rapidly evolving GO categories |
| ROL | Rate of living |
| SRH | Stationary, reversible, and homogeneous conditions |
| WRST | Wilcoxon rank sum test |
References
- Brockes, J.P.; Kumar, A. Comparative aspects of animal regeneration. Annu. Rev. Cell. Dev. Biol. 2008, 24, 525–549. [Google Scholar] [CrossRef]
- Tanaka, E.M. The molecular and cellular choreography of appendage regeneration. Cell 2016, 165, 1598–1608. [Google Scholar] [CrossRef] [PubMed]
- Sandoval-Guzmán, T.; Wang, H.; Khattak, S.; Schuez, M.; Roensch, K.; Nacu, E.; Tazaki, A.; Joven, A.; Tanaka, E.M.; Simon, A. Fundamental differences in dedifferentiation and stem cell recruitment during skeletal muscle regeneration in two salamander species. Cell Stem Cell 2014, 14, 174–187. [Google Scholar] [CrossRef]
- Fei, J.-F.; Schuez, M.; Knapp, D.; Taniguchi, Y.; Drechsel, D.N.; Tanaka, E.M. Efficient gene knockin in axolotl and its use to test the role of satellite cells in limb regeneration. Proc. Natl. Acad. Sci. USA 2017, 114, 12501–12506. [Google Scholar] [CrossRef] [PubMed]
- Sousounis, K.; Athippozhy, A.T.; Voss, S.R.; Tsonis, P.A. Plasticity for axolotl lens regeneration is associated with age-related changes in gene expression. Regeneration 2014, 1, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, G.; Eguchi, Y.; Nakamura, K.; Yadav, M.C.; Millán, J.L.; Tsonis, P.A. Regenerative capacity in newts is not altered by repeated regeneration and ageing. Nat. Commun. 2011, 2, 384. [Google Scholar] [CrossRef]
- Torres, M. Regeneration: Limb regrowth takes two. Nature 2016, 533, 328–330. [Google Scholar] [CrossRef][Green Version]
- Godwin, J.W.; Pinto, A.R.; Rosenthal, N.A. Macrophages are required for adult salamander limb regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 9415–9420. [Google Scholar] [CrossRef]
- Yun, M.H.; Gates, P.B.; Brockes, J.P. Sustained ERK activation underlies reprogramming in regeneration-competent salamander cells and distinguishes them from their mammalian counterparts. Stem Cell Rep. 2014, 3, 15–23. [Google Scholar] [CrossRef]
- Calve, S.; Odelberg, S.J.; Simon, H.-G. A transitional extracellular matrix instructs cell behavior during muscle regeneration. Dev. Biol. 2010, 344, 259–271. [Google Scholar] [CrossRef]
- Kumar, A.; Godwin, J.W.; Gates, P.B.; Garza-Garcia, A.A.; Brockes, J.P. Molecular basis for the nerve dependence of limb regeneration in an adult vertebrate. Science 2007, 318, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.L.; Baselga-Garriga, C.; Melton, D.A. Blastemal progenitors modulate immune signaling during early limb regeneration. Development 2019, 146, dev169128. [Google Scholar] [CrossRef]
- Rando, T.A. Stem cells, ageing and the quest for immortality. Nature 2006, 441, 1080–1086. [Google Scholar] [CrossRef] [PubMed]
- Lunghi, E. Doubling the lifespan of European plethodontid salamanders. Ecology 2022, 103, e03581. [Google Scholar] [CrossRef] [PubMed]
- Yun, M.H. Salamander insights into ageing and rejuvenation. Front. Cell Dev. Biol. 2021, 9, 689062. [Google Scholar] [CrossRef]
- Walters, H.E.; Troyanovskiy, K.E.; Graf, A.M.; Yun, M.H. Senescent cells enhance newt limb regeneration by promoting muscle dedifferentiation. Aging Cell 2023, 22, e13826. [Google Scholar] [CrossRef]
- Zhang, P.; Zhou, H.; Chen, Y.-Q.; Liu, Y.-F.; Qu, L.-H. Mitogenomic perspectives on the origin and phylogeny of living amphibians. Syst. Biol. 2005, 54, 391–400. [Google Scholar] [CrossRef]
- Nowoshilow, S.; Schloissnig, S.; Fei, J.-F.; Dahl, A.; Pang, A.W.; Pippel, M.; Winkler, S.; Hastie, A.R.; Young, G.; Roscito, J.G. The axolotl genome and the evolution of key tissue formation regulators. Nature 2018, 554, 50–55. [Google Scholar] [CrossRef]
- Adams, J. Transcriptome: Connecting the genome to gene function. Nat. Educ. 2008, 1, 195. [Google Scholar]
- Ku, C.-S.; Wu, M.; Cooper, D.N.; Naidoo, N.; Pawitan, Y.; Pang, B.; Iacopetta, B.; Soong, R. Exome versus transcriptome sequencing in identifying coding region variants. Expert Rev. Mol. Diagn. 2012, 12, 241–251. [Google Scholar] [CrossRef]
- Wilkinson, G.S.; Adams, D.M. Recurrent evolution of extreme longevity in bats. Biol. Lett. 2019, 15, 20180860. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.F.; Blanquart, F.; Bataillon, T.; Kassen, R. What drives parallel evolution? How population size and mutational variation contribute to repeated evolution. Bioessays 2017, 39, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fei, L.; Ye, C.; Jiang, J. Colored Atlas of Chinese Amphibians and Their Distributions; Sichuan Publishing Group: Chengdu, China, 2012. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Simpson, J.T.; Wong, K.; Jackman, S.D.; Schein, J.E.; Jones, S.J.M.; Birol, İ. ABySS: A parallel assembler for short read sequence data. Genome Res. 2009, 19, 1117–1123. [Google Scholar] [CrossRef]
- Robertson, G.; Schein, J.; Chiu, R.; Corbett, R.; Field, M.; Jackman, S.D.; Mungall, K.; Lee, S.; Okada, H.M.; Qian, J.Q.; et al. De novo assembly and analysis of RNA-seq data. Nat. Methods 2010, 7, 909–912. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef]
- Qiao, L.; Yang, W.; Fu, J.; Song, Z. Transcriptome Profile of the Green Odorous Frog (Odorrana margaretae). PLoS ONE 2013, 8, e75211. [Google Scholar] [CrossRef]
- Yang, W.; Qi, Y.; Lu, B.; Qiao, L.; Wu, Y.; Fu, J. Gene expression variations in high-altitude adaptation: A case study of the Asiatic toad (Bufo gargarizans). BMC Genet. 2017, 18, 62. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-H.; Tsai, M.-H.; Ho, C.-C.; Chen, C.-Y.; Lee, H.-S. De novo transcriptome sequencing of axolotl blastema for identification of differentially expressed genes during limb regeneration. BMC Genom. 2013, 14, 434. [Google Scholar] [CrossRef] [PubMed]
- Looso, M.; Preussner, J.; Sousounis, K.; Bruckskotten, M.; Michel, C.S.; Lignelli, E.; Reinhardt, R.; Höffner, S.; Krüger, M.; Tsonis, P.A.; et al. A de novo assembly of the newt transcriptome combined with proteomic validation identifies new protein families expressed during tissue regeneration. Genome Biol. 2013, 14, R16. [Google Scholar] [CrossRef]
- Hellsten, U.; Harland, R.M.; Gilchrist, M.J.; Hendrix, D.; Jurka, J.; Kapitonov, V.; Ovcharenko, I.; Putnam, N.H.; Shu, S.; Taher, L.; et al. The genome of the Western clawed frog Xenopus tropicalis. Science 2010, 328, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Ebersberger, I.; Strauss, S.; von Haeseler, A. HaMStR: Profile hidden markov model based search for orthologs in ESTs. BMC Evol. Biol. 2009, 9, 157. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Hartmann, S.; Vision, T.J. Using ESTs for phylogenomics: Can one accurately infer a phylogenetic tree from a gappy alignment? BMC Evol. Biol. 2008, 8, 95. [Google Scholar] [CrossRef]
- Philippe, H.; Snell, E.A.; Bapteste, E.; Lopez, P.; Holland, P.W.H.; Casane, D. Phylogenomics of eukaryotes: Impact of missing data on large alignments. Mol. Biol. Evol. 2004, 21, 1740–1752. [Google Scholar] [CrossRef]
- Misof, B.; Meyer, B.; von Reumont, B.M.; Kück, P.; Misof, K.; Meusemann, K. Selecting informative subsets of sparse supermatrices increases the chance to find correct trees. BMC Bioinform. 2013, 14, 348. [Google Scholar] [CrossRef]
- Kück, P.; Struck, T.H. BaCoCa—A heuristic software tool for the parallel assessment of sequence biases in hundreds of gene and taxon partitions. Mol. Phylogenetics Evol. 2014, 70, 94–98. [Google Scholar] [CrossRef]
- Zhong, M.; Hansen, B.; Nesnidal, M.; Golombek, A.; Halanych, K.M.; Struck, T.H. Detecting the symplesiomorphy trap: A multigene phylogenetic analysis of terebelliform annelids. BMC Evol. Biol. 2011, 11, 369. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.Y.; Jermiin, L. Tracing the decay of the historical signal in biological sequence data. Syst. Biol. 2004, 53, 623–637. [Google Scholar] [CrossRef]
- Jermiin, L.S.; Ho, S.Y.; Ababneh, F.; Robinson, J.; Larkum, A.W. The biasing effect of compositional heterogeneity on phylogenetic estimates may be underestimated. Syst. Biol. 2004, 53, 638–643. [Google Scholar] [CrossRef]
- Jermiin, L.S.; Ho, J.W.K.; Lau, K.W.; Jayaswal, V. SeqVis: A tool for detecting compositional heterogeneity among aligned nucleotide sequences. Bioinform. DNA Seq. Anal. 2009, 537, 65–91. [Google Scholar] [CrossRef]
- Collins, T.M.; Fedrigo, O.; Naylor, G.J. Choosing the best genes for the job: The case for stationary genes in genome-scale phylogenetics. Syst. Biol. 2005, 54, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Rannala, B.; Yang, Z. Phylogenetic inference using whole genomes. Annu. Rev. Genom. Hum. Genet. 2008, 9, 217–231. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Lartillot, N.; Lepage, T.; Blanquart, S. PhyloBayes 3: A Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 2009, 25, 2286–2288. [Google Scholar] [CrossRef]
- Lanfear, R.; Calcott, B.; Ho, S.Y.W.; Guindon, S. PartitionFinder: Combined Selection of Partitioning Schemes and Substitution Models for Phylogenetic Analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef]
- Ruta, M.; Coates, M.I.; Quicke, D.L.J. Early tetrapod relationships revisited. Biol. Rev. 2003, 78, 251–345. [Google Scholar] [CrossRef]
- Zardoya, R.; Meyer, A. On the origin of and phylogenetic relationships among living amphibians. Proc. Natl. Acad. Sci. USA 2001, 98, 7380–7383. [Google Scholar] [CrossRef] [PubMed]
- Hugall, A.F.; Foster, R.; Lee, M.S.Y. Calibration Choice, Rate Smoothing, and the Pattern of Tetrapod Diversification According to the Long Nuclear Gene RAG-1. Syst. Biol. 2007, 56, 543–563. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wake, D.B. Higher-level salamander relationships and divergence dates inferred from complete mitochondrial genomes. Mol. Phylogen. Evol. 2009, 53, 492–508. [Google Scholar] [CrossRef] [PubMed]
- Vallin, G.; Laurin, M. Cranial morphology and affinities of Microbrachis, and a reappraisal of the phylogeny and lifestyle of the first amphibians. J. Vert. Paleontol. 2004, 24, 56–72. [Google Scholar] [CrossRef]
- Feller, A.E.; Hedges, S.B. Molecular Evidence for the Early History of Living Amphibians. Mol. Phylogen. Evol. 1998, 9, 509–516. [Google Scholar] [CrossRef]
- Hedges, S.B.; Maxson, L.R. A Molecular Perspective on Lissamphibian Phylogeny. Herpetol. Monogr. 1993, 7, 27–42. [Google Scholar] [CrossRef]
- Carroll, R.L. The origin and early radiation of terrestrial vertebrates. J. Paleontol. 2001, 75, 1202–1213. [Google Scholar] [CrossRef]
- Anderson, J.S.; Reisz, R.R.; Scott, D.; Frobisch, N.B.; Sumida, S.S. A stem batrachian from the Early Permian of Texas and the origin of frogs and salamanders. Nature 2008, 453, 515–518. [Google Scholar] [CrossRef]
- Anderson, J.S. Focal review: The origin (s) of modern amphibians. Evol. Biol. 2008, 35, 231–247. [Google Scholar] [CrossRef]
- Carroll, R.L. The Palaeozoic Ancestry of Salamanders, Frogs and Caecilians. Zool. J. Linn. Soc. 2007, 150, 1–140. [Google Scholar] [CrossRef]
- Carroll, R.L.; Holmes, R. The skull and jaw musculature as guides to the ancestry of salamanders. Zool. J. Linn. Soc. 1980, 68, 1–40. [Google Scholar] [CrossRef]
- Carroll, R.L.; Boisvert, C.; Bolt, J.; Green, D.M.; Philip, N.; Rolian, C.; Schoch, R.; Tarenko, A. Changing patterns of ontogeny from osteolepiform fish through Permian tetrapods as a guide to the early evolution of land vertebrates. In Recent Advances in the Origin and Early Radiation of Vertebrates; Pfeil: Munich, Germany, 2004; pp. 321–343. [Google Scholar]
- Shimodaira, H. An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 2002, 51, 492–508. [Google Scholar] [CrossRef] [PubMed]
- Shimodaira, H.; Hasegawa, M. CONSEL: For assessing the confidence of phylogenetic tree selection. Bioinformatics 2001, 17, 1246–1247. [Google Scholar] [CrossRef] [PubMed]
- Susko, E.; Leigh, J.; Doolittle, W.; Bapteste, E. Visualizing and assessing phylogenetic congruence of core gene sets: A case study of the γ-Proteobacteria. Mol. Biol. Evol. 2006, 23, 1019–1030. [Google Scholar] [CrossRef]
- Liu, L.; Yu, L.; Edwards, S.V. A maximum pseudo-likelihood approach for estimating species trees under the coalescent model. BMC Evol. Biol. 2010, 10, 302. [Google Scholar] [CrossRef]
- Lu, B.; Yang, W.; Dai, Q.; Fu, J. Using genes as characters and a parsimony analysis to explore the phylogenetic position of turtles. PLoS ONE 2013, 8, e79348. [Google Scholar] [CrossRef]
- Lu, B.; Wang, X.; Fu, J.; Shi, J.; Wu, Y.; Qi, Y. Genetic adaptations of an island pit-viper to a unique sedentary life with extreme seasonal food availability. G3 Genes Genomes Genet. 2020, 10, 1639–1646. [Google Scholar] [CrossRef]
- Birney, E.; Andrews, T.D.; Bevan, P.; Caccamo, M.; Chen, Y.; Clarke, L.; Coates, G.; Cuff, J.; Curwen, V.; Cutts, T. An overview of Ensembl. Genome Res. 2004, 14, 925–928. [Google Scholar] [CrossRef]
- Benton, M.J.; Donoghue, P.C. Paleontological evidence to date the tree of life. Mol. Biol. Evol. 2007, 24, 26–53. [Google Scholar] [CrossRef]
- Yang, Z.; Rannala, B. Bayesian estimation of species divergence times under a molecular clock using multiple fossil calibrations with soft bounds. Mol. Biol. Evol. 2006, 23, 212–226. [Google Scholar] [CrossRef]
- Dos Reis, M.; Yang, Z. The unbearable uncertainty of Bayesian divergence time estimation. J. Syst. Evol. 2013, 51, 30–43. [Google Scholar] [CrossRef]
- Nielsen, R. Molecular signatures of natural selection. Annu. Rev. Genet. 2005, 39, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Yang, Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 1998, 148, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Hubisz, M.J.; Pollard, K.S.; Siepel, A. PHAST and RPHAST: Phylogenetic analysis with space/time models. Brief. Bioinform. 2011, 12, 41–51. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Seim, I.; Fang, X.; Xiong, Z.; Lobanov, A.V.; Huang, Z.; Ma, S.; Feng, Y.; Turanov, A.A.; Zhu, Y.; Lenz, T.L. Genome analysis reveals insights into physiology and longevity of the Brandt’s bat Myotis brandtii. Nat. Commun. 2013, 4, 2212. [Google Scholar] [CrossRef]
- Wilkinson, G.S.; South, J.M. Life history, ecology and longevity in bats. Aging cell 2002, 1, 124–131. [Google Scholar] [CrossRef]
- Kim, E.B.; Fang, X.; Fushan, A.A.; Huang, Z.; Lobanov, A.V.; Han, L.; Marino, S.M.; Sun, X.; Turanov, A.A.; Yang, P.; et al. Genome sequencing reveals insights into physiology and longevity of the naked mole rat. Nature 2011, 479, 223–227. [Google Scholar] [CrossRef]
- Larson, J.; Park, T.J. Extreme hypoxia tolerance of naked mole-rat brain. Neuroreport 2009, 20, 1634–1637. [Google Scholar] [CrossRef]
- Park, T.J.; Lu, Y.; Jüttner, R.; Smith, E.S.J.; Hu, J.; Brand, A.; Wetzel, C.; Milenkovic, N.; Erdmann, B.; Heppenstall, P.A. Selective inflammatory pain insensitivity in the African naked mole-rat (Heterocephalus glaber). PLoS Biol. 2008, 6, e13. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Mele, J.; Wu, Y.; Buffenstein, R.; Hornsby, P.J. Resistance to experimental tumorigenesis in cells of a long-lived mammal, the naked mole-rat (Heterocephalus glaber). Aging cell 2010, 9, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Seluanov, A.; Hine, C.; Azpurua, J.; Feigenson, M.; Bozzella, M.; Mao, Z.; Catania, K.C.; Gorbunova, V. Hypersensitivity to contact inhibition provides a clue to cancer resistance of naked mole-rat. Proc. Natl. Acad. Sci. USA 2009, 106, 19352–19357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Nei, M. Accuracies of ancestral amino acid sequences inferred by the parsimony, likelihood, and distance methods. J. Mol. Evol. 1997, 44, S139–S146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kumar, S. Detection of convergent and parallel evolution at the amino acid sequence level. Mol. Biol. Evol. 1997, 14, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Mitchell, A.; Chang, H.-Y.; Daugherty, L.; Fraser, M.; Hunter, S.; Lopez, R.; McAnulla, C.; McMenamin, C.; Nuka, G.; Pesseat, S. The InterPro protein families database: The classification resource after 15 years. Nucleic Acids Res. 2015, 43, D213–D221. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Dutta, S.; Zardecki, C.; Voigt, M.; Berman, H.M.; Burley, S.K. The RCSB PDB “Molecule of the Month”: Inspiring a Molecular View of Biology. PLoS Biol. 2015, 13, e1002140. [Google Scholar] [CrossRef]
- Mayrose, I.; Graur, D.; Ben-Tal, N.; Pupko, T. Comparison of site-specific rate-inference methods for protein sequences: Empirical Bayesian methods are superior. Mol. Biol. Evol. 2004, 21, 1781–1791. [Google Scholar] [CrossRef] [PubMed]
- Pupko, T.; Bell, R.E.; Mayrose, I.; Glaser, F.; Ben-Tal, N. Rate4Site: An algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics 2002, 18, S71–S77. [Google Scholar] [CrossRef]
- Celniker, G.; Nimrod, G.; Ashkenazy, H.; Glaser, F.; Martz, E.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf: Using evolutionary data to raise testable hypotheses about protein function. Isr. J. Chem. 2013, 53, 199–206. [Google Scholar] [CrossRef]
- Yu, Y.; Nakhleh, L. A maximum pseudo-likelihood approach for phylogenetic networks. BMC Genomics 2015, 16, S10. [Google Scholar] [CrossRef] [PubMed]
- Misof, B.; Liu, S.; Meusemann, K.; Peters, R.S.; Donath, A.; Mayer, C.; Frandsen, P.B.; Ware, J.; Flouri, T.; Beutel, R.G. Phylogenomics resolves the timing and pattern of insect evolution. Science 2014, 346, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.W.; Hejnol, A.; Matus, D.Q.; Pang, K.; Browne, W.E.; Smith, S.A.; Seaver, E.; Rouse, G.W.; Obst, M.; Edgecombe, G.D.; et al. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature 2008, 452, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Hime, P.M.; Lemmon, A.R.; Lemmon, E.C.M.; Prendini, E.; Brown, J.M.; Thomson, R.C.; Kratovil, J.D.; Noonan, B.P.; Pyron, R.A.; Peloso, P.L. Phylogenomics reveals ancient gene tree discordance in the amphibian tree of life. Syst. Biol. 2021, 70, 49–66. [Google Scholar] [CrossRef]
- Chen, M.-Y.; Liang, D.; Zhang, P. Selecting Question-specific Genes to Reduce Incongruence in Phylogenomics: A Case Study of Jawed Vertebrate Backbone Phylogeny. Syst. Biol. 2015, 64, 1104–1120. [Google Scholar] [CrossRef]
- Alexander Pyron, R.; Wiens, J.J. A large-scale phylogeny of Amphibia including over 2800 species, and a revised classification of extant frogs, salamanders, and caecilians. Mol. Phylogen. Evol. 2011, 61, 543–583. [Google Scholar] [CrossRef]
- Werneburg, R.; Witzmann, F.; Schneider, J.W. The oldest known tetrapod (Temnospondyli) from Germany (early Carboniferous, Visean). PalZ 2019, 93, 679–690. [Google Scholar] [CrossRef]
- Marjanović, D.; Laurin, M. Fossils, molecules, divergence times, and the origin of Lissamphibians. Syst. Biol. 2007, 56, 369–388. [Google Scholar] [CrossRef] [PubMed]
- San Mauro, D.; Vences, M.; Alcobendas, M.; Zardoya, R.; Meyer, A. Initial diversification of living amphibians predated the breakup of Pangaea. Am. Nat. 2005, 165, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Wyckoff, G.J.; Malcom, C.M.; Vallender, E.J.; Lahn, B.T. A highly unexpected strong correlation between fixation probability of nonsynonymous mutations and mutation rate. Trends Genet. 2005, 21, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Chong, R.A.; Mueller, R.L. Low metabolic rates in salamanders are correlated with weak selective constraints on mitochondrial genes. Evolution 2013, 67, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Rao, N.; Jhamb, D.; Milner, D.J.; Li, B.; Song, F.; Wang, M.; Voss, S.R.; Palakal, M.; King, M.W.; Saranjami, B. Proteomic analysis of blastema formation in regenerating axolotl limbs. BMC Biol. 2009, 7, 83. [Google Scholar] [CrossRef]
- Wang, H.; Lööf, S.; Borg, P.; Nader, G.A.; Blau, H.M.; Simon, A. Turning terminally differentiated skeletal muscle cells into regenerative progenitors. Nat. Commun. 2015, 6, 7916. [Google Scholar] [CrossRef]
- Yun, M.H.; Gates, P.B.; Brockes, J.P. Regulation of p53 is critical for vertebrate limb regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 17392–17397. [Google Scholar] [CrossRef]
- Hirata, A.; Gardiner, D.M.; Satoh, A. Dermal fibroblasts contribute to multiple tissues in the accessory limb model. Dev. Growth Differ. 2010, 52, 343–350. [Google Scholar] [CrossRef]
- Dujardin, D.L.; Barnhart, L.E.; Stehman, S.A.; Gomes, E.R.; Gundersen, G.G.; Vallee, R.B. A role for cytoplasmic dynein and LIS1 in directed cell movement. J. Cell Biol. 2003, 163, 1205–1211. [Google Scholar] [CrossRef]
- Lee, E.; Ju, B.-G.; Kim, W.-S. Endogenous retinoic acid mediates the early events in salamander limb regeneration. Anim. Cells Syst. 2012, 16, 462–468. [Google Scholar] [CrossRef]
- Zhang, Y.; Desai, A.; Yang, S.Y.; Bae, K.B.; Antczak, M.I.; Fink, S.P.; Tiwari, S.; Willis, J.E.; Williams, N.S.; Dawson, D.M. Inhibition of the prostaglandin-degrading enzyme 15-PGDH potentiates tissue regeneration. Science 2015, 348, aaa2340. [Google Scholar] [PubMed]
- Liu, Y.; Zhou, Q.; Wang, Y.; Luo, L.; Yang, J.; Yang, L.; Liu, M.; Li, Y.; Qian, T.; Zheng, Y. Gekko japonicus genome reveals evolution of adhesive toe pads and tail regeneration. Nat. Commun. 2015, 6, 10033. [Google Scholar]
- Daneau, G.; Boidot, R.; Martinive, P.; Feron, O. Identification of cyclooxygenase-2 as a major actor of the transcriptomic adaptation of endothelial and tumor cells to cyclic hypoxia: Effect on angiogenesis and metastases. Clin. Cancer. Res. 2010, 16, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Zagon, I.S.; Verderame, M.F.; Allen, S.S.; McLaughlin, P.J. Cloning, sequencing, chromosomal location, and function of cDNAs encoding an opioid growth factor receptor (OGFr) in humans. Brain Res. 2000, 856, 75–83. [Google Scholar] [PubMed]
- Zagon, I.S.; Donahue, R.N.; Rogosnitzky, M.; Mclaughlin, P.J. Imiquimod upregulates the opioid growth factor receptor to inhibit cell proliferation independent of immune function. Exp. Biol. Med. 2008, 233, 968–979. [Google Scholar]
- Roet, K.C.; Franssen, E.H.; de Bree, F.M.; Essing, A.H.; Zijlstra, S.-J.J.; Fagoe, N.D.; Eggink, H.M.; Eggers, R.; Smit, A.B.; van Kesteren, R.E. A multilevel screening strategy defines a molecular fingerprint of proregenerative olfactory ensheathing cells and identifies SCARB2, a protein that improves regenerative sprouting of injured sensory spinal axons. J. Neurosci. 2013, 33, 11116–11135. [Google Scholar] [CrossRef] [PubMed]
- Varga, M.; Sass, M.; Papp, D.; Takacs-Vellai, K.; Kobolak, J.; Dinnyes, A.; Klionsky, D.J.; Vellai, T. Autophagy is required for zebrafish caudal fin regeneration. Cell Death Differ. 2014, 21, 547–556. [Google Scholar]
- Gómez, C.M.A.; Echeverri, K. Salamanders: The molecular basis of tissue regeneration and its relevance to human disease. Curr. Top. Dev. Biol. 2021, 145, 235–275. [Google Scholar]
- Gatten, R.; Miller, K.; Full, R. Energetics at rest and during locomotion. Environ. Physiol. Amphib. 1992, 314–377. [Google Scholar]
- Martins, S.G.; Zilhão, R.; Thorsteinsdóttir, S.; Carlos, A.R. Linking oxidative stress and DNA damage to changes in the expression of extracellular matrix components. Front. Genet. 2021, 12, 673002. [Google Scholar]
- Hua, X.; Cowman, P.; Warren, D.; Bromham, L. Longevity is linked to mitochondrial mutation rates in rockfish: A test using Poisson regression. Mol. Biol. Evol. 2015, 32, 2633–2645. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C.J. The genetics of ageing. Nature 2010, 464, 504–512. [Google Scholar] [PubMed]
- Dang, W.; Steffen, K.K.; Perry, R.; Dorsey, J.A.; Johnson, F.B.; Shilatifard, A.; Kaeberlein, M.; Kennedy, B.K.; Berger, S.L. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 2009, 459, 802–807. [Google Scholar] [CrossRef]
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S.-i. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013, 18, 416–430. [Google Scholar] [CrossRef]
- Yoh, S.M.; Lucas, J.S.; Jones, K.A. The Iws1: Spt6: CTD complex controls cotranscriptional mRNA biosynthesis and HYPB/Setd2-mediated histone H3K36 methylation. Genes Dev. 2008, 22, 3422–3434. [Google Scholar] [CrossRef] [PubMed]
- Cazzalini, O.; Sommatis, S.; Tillhon, M.; Dutto, I.; Bachi, A.; Rapp, A.; Nardo, T.; Scovassi, A.I.; Necchi, D.; Cardoso, M.C. CBP and p300 acetylate PCNA to link its degradation with nucleotide excision repair synthesis. Nucleic Acids Res. 2014, 42, 8433–8448. [Google Scholar] [CrossRef] [PubMed]
- Tanno, M.; Ogihara, M.; Taguchi, T. Age-related changes in proliferating cell nuclear antigen levels. Mech. Ageing Dev. 1996, 92, 53–66. [Google Scholar] [CrossRef]
- Keane, M.; Semeiks, J.; Webb, A.E.; Li, Y.I.; Quesada, V.; Craig, T.; Madsen, L.B.; van Dam, S.; Brawand, D.; Marques, P.I. Insights into the evolution of longevity from the bowhead whale genome. Cell Rep. 2015, 10, 112–122. [Google Scholar] [CrossRef]
- Wang, S.-C.; Nakajima, Y.; Yu, Y.-L.; Xia, W.; Chen, C.-T.; Yang, C.-C.; McIntush, E.W.; Li, L.-Y.; Hawke, D.H.; Kobayashi, R. Tyrosine phosphorylation controls PCNA function through protein stability. Nat. Cell Biol. 2006, 8, 1359–1368. [Google Scholar] [CrossRef]
- Yu, Y.; Cai, J.-P.; Tu, B.; Wu, L.; Zhao, Y.; Liu, X.; Li, L.; McNutt, M.A.; Feng, J.; He, Q. Proliferating cell nuclear antigen is protected from degradation by forming a complex with MutT Homolog2. J. Biol. Chem. 2009, 284, 19310–19320. [Google Scholar] [CrossRef]
- Baple, E.L.; Chambers, H.; Cross, H.E.; Fawcett, H.; Nakazawa, Y.; Chioza, B.A.; Harlalka, G.V.; Mansour, S.; Sreekantan-Nair, A.; Patton, M.A. Hypomorphic PCNA mutation underlies a human DNA repair disorder. J. Clin. Investig. 2014, 124, 3137–3146. [Google Scholar] [CrossRef] [PubMed]
- Riera, C.E.; Huising, M.O.; Follett, P.; Leblanc, M.; Halloran, J.; Van Andel, R.; de Magalhaes Filho, C.D.; Merkwirth, C.; Dillin, A. TRPV1 pain receptors regulate longevity and metabolism by neuropeptide signaling. Cell 2014, 157, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Sillén, A.; Anton-Lamprecht, I.; Braun-Quentin, C.; Kraus, C.S.; Sayli, B.S.; Ayuso, C.; Jagell, S.; Küster, W.; Wadelius, C. Spectrum of mutations and sequence variants in the FALDH gene in patients with Sjögren-Larsson syndrome. Hum. Mutat. 1998, 12, 377. [Google Scholar] [CrossRef]
- White, J.K.; Gerdin, A.-K.; Karp, N.A.; Ryder, E.; Buljan, M.; Bussell, J.N.; Salisbury, J.; Clare, S.; Ingham, N.J.; Podrini, C. Genome-wide generation and systematic phenotyping of knockout mice reveals new roles for many genes. Cell 2013, 154, 452–464. [Google Scholar] [CrossRef]
- Xue, Y.; Shen, S.Q.; Jui, J.; Rupp, A.C.; Byrne, L.C.; Hattar, S.; Flannery, J.G.; Corbo, J.C.; Kefalov, V.J. CRALBP supports the mammalian retinal visual cycle and cone vision. J. Clin. Investig. 2015, 125, 727. [Google Scholar] [CrossRef]
- He, X.; Lobsiger, J.; Stocker, A. Bothnia dystrophy is caused by domino-like rearrangements in cellular retinaldehyde-binding protein mutant R234W. Proc. Natl. Acad. Sci. USA 2009, 106, 18545–18550. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, B. Evolutionary Insights into the Relationship of Frogs, Salamanders, and Caecilians and Their Adaptive Traits, with an Emphasis on Salamander Regeneration and Longevity. Animals 2023, 13, 3449. https://doi.org/10.3390/ani13223449
Lu B. Evolutionary Insights into the Relationship of Frogs, Salamanders, and Caecilians and Their Adaptive Traits, with an Emphasis on Salamander Regeneration and Longevity. Animals. 2023; 13(22):3449. https://doi.org/10.3390/ani13223449
Chicago/Turabian StyleLu, Bin. 2023. "Evolutionary Insights into the Relationship of Frogs, Salamanders, and Caecilians and Their Adaptive Traits, with an Emphasis on Salamander Regeneration and Longevity" Animals 13, no. 22: 3449. https://doi.org/10.3390/ani13223449
APA StyleLu, B. (2023). Evolutionary Insights into the Relationship of Frogs, Salamanders, and Caecilians and Their Adaptive Traits, with an Emphasis on Salamander Regeneration and Longevity. Animals, 13(22), 3449. https://doi.org/10.3390/ani13223449