
Phylogenetic Analysis and Codon Usage Bias Reveal the Base of Feline and Canine Chaphamaparvovirus for Cross-Species Transmission

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Samples and Viral Genome Sequence Collection
2.2. Similarity, Genetic Distance, and Phylogenetic Analysis
2.3. Codon Usage Analysis
2.4. Relative Synonymous Codon Usage Analysis
2.5. Codon Usage Pattern Difference Analysis
2.6. Host Adaptability Analysis
2.7. Parity Rule 2, ENC Plot, and Neutrality Analysis
2.8. Comparative Analysis of Mutations, Immune Epitopes, and Structures
2.9. Statistical Analysis
3. Results
3.1. Clinical Samples
3.2. Similarity and Genetic Distance
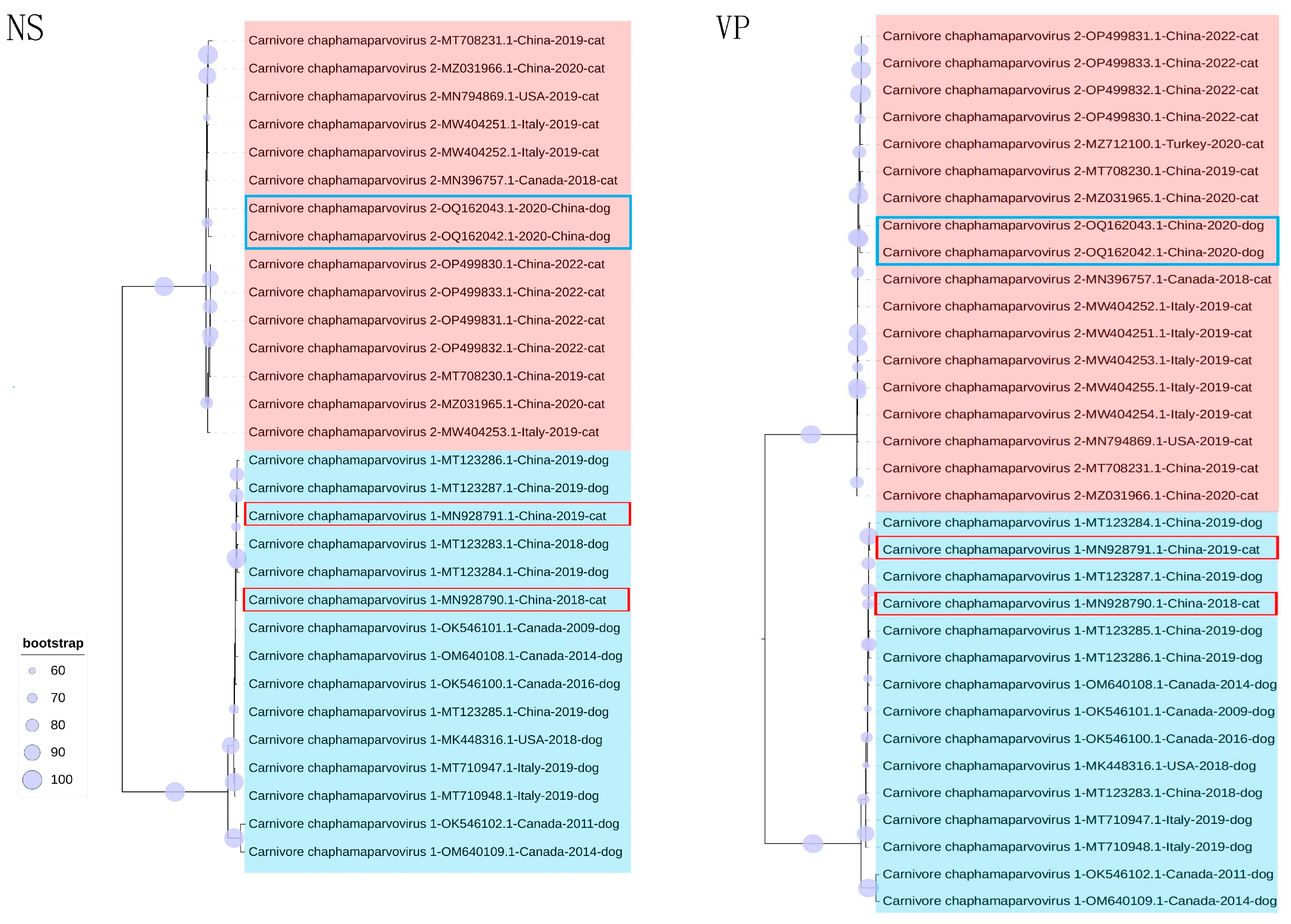
3.3. Phylogenetic Analysis
3.4. Codon Usage Pattern Difference Analysis
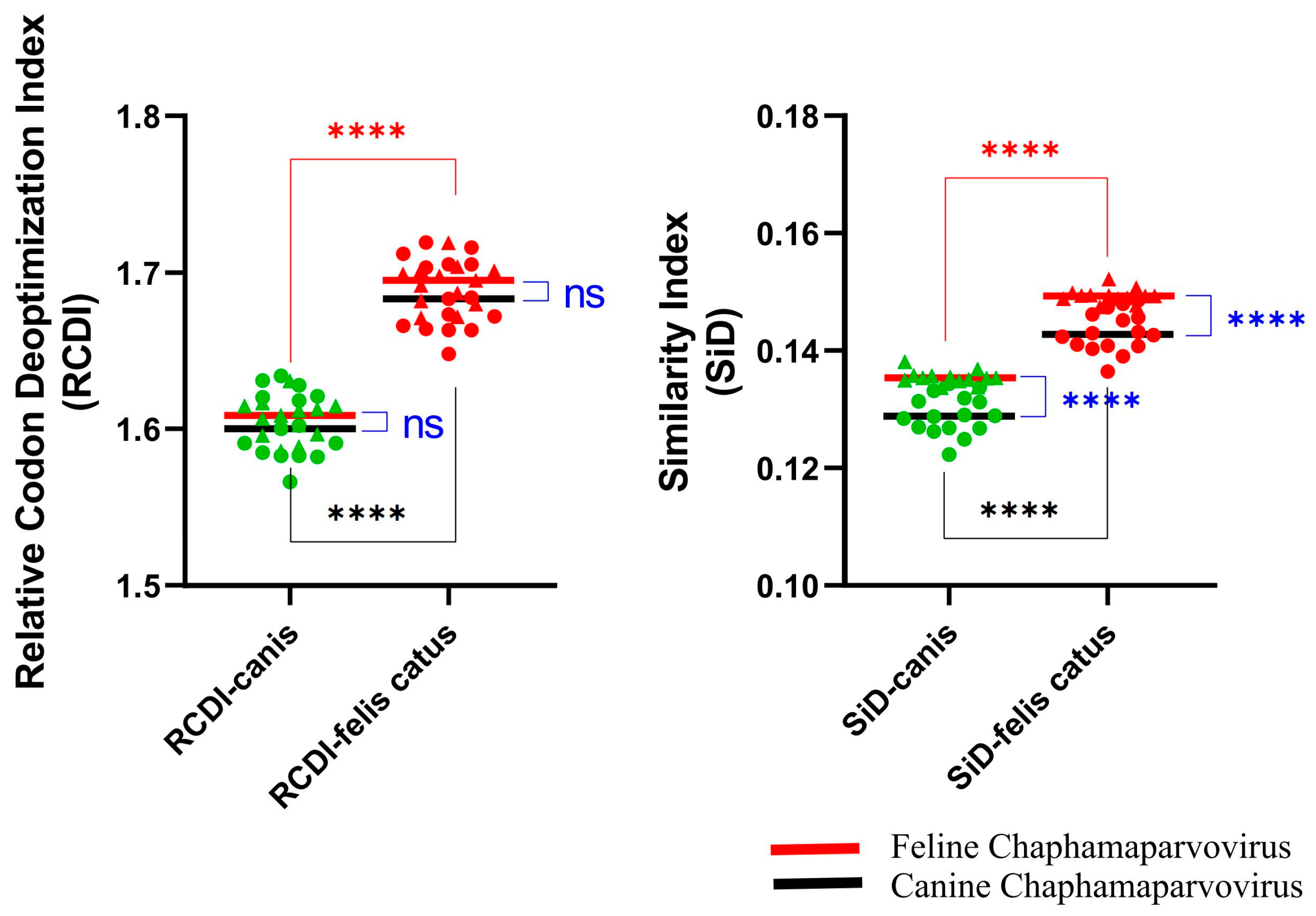
3.5. Host Adaptability Analysis
3.6. CUB and RSCU Analysis
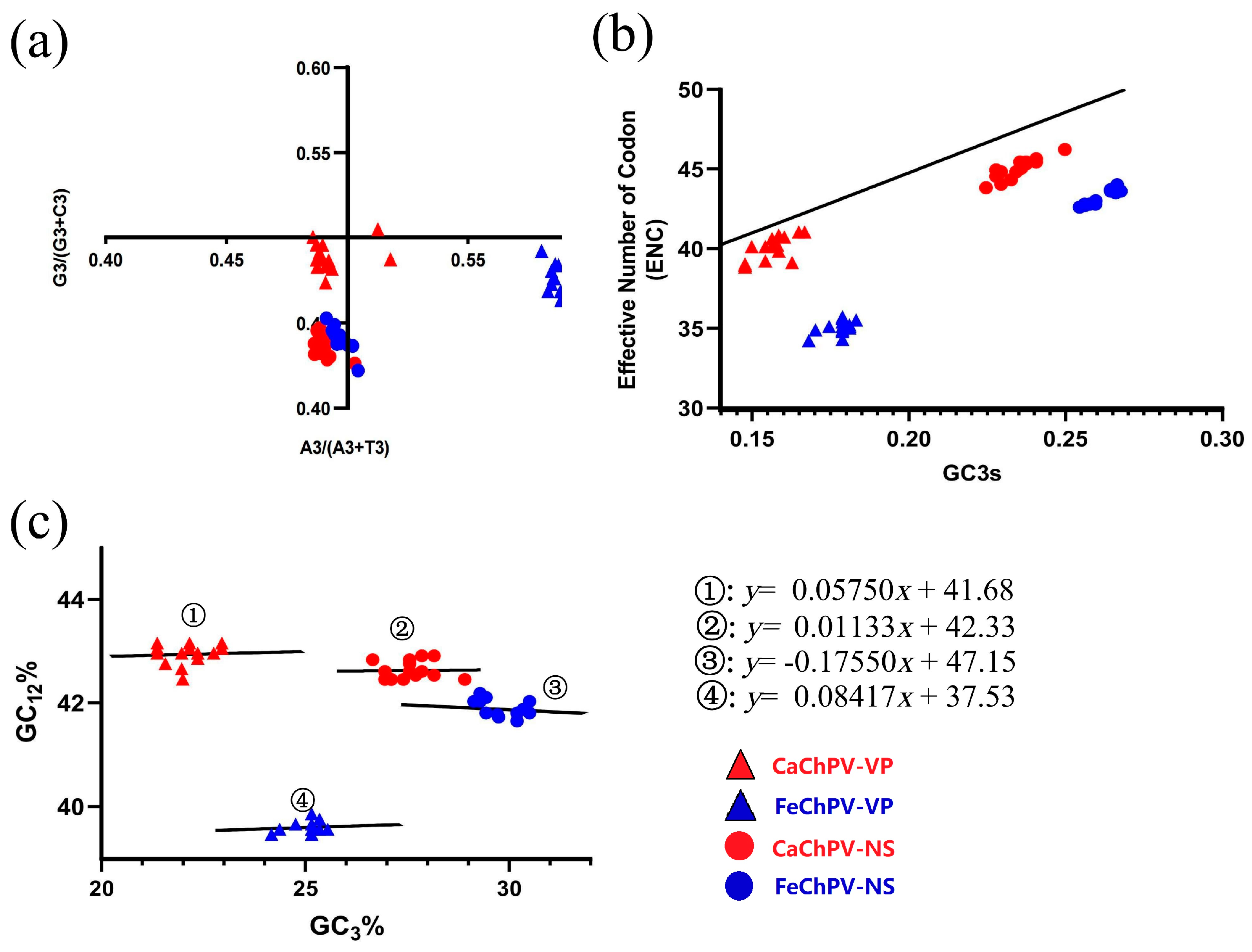
3.7. Parity Rule 2, ENC Plot, and Neutrality Analysis
3.8. Comparative Analysis of Mutations, and Immune Epitopes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Penzes, J.J.; Soderlund-Venermo, M.; Canuti, M.; Eis-Hubinger, A.M.; Hughes, J.; Cotmore, S.F.; Harrach, B. Reorganizing the family Parvoviridae: A revised taxonomy independent of the canonical approach based on host association. Arch. Virol. 2020, 165, 2133–2146. [Google Scholar] [CrossRef]
- Canuti, M.; Verhoeven, J.T.P.; Munro, H.J.; Roul, S.; Ojkic, D.; Robertson, G.J.; Whitney, H.G.; Dufour, S.C.; Lang, A.S. Investigating the Diversity and Host Range of Novel Parvoviruses from North American Ducks Using Epidemiology, Phylogenetics, Genome Structure, and Codon Usage Analysis. Viruses 2021, 13, 193. [Google Scholar] [CrossRef]
- Souza, W.M.; Romeiro, M.F.; Fumagalli, M.J.; Modha, S.; de Araujo, J.; Queiroz, L.H.; Durigon, E.L.; Figueiredo, L.T.M.; Murcia, P.R.; Gifford, R.J. Chapparvoviruses occur in at least three vertebrate classes and have a broad biogeographic distribution. J. Gen. Virol. 2017, 98, 225–229. [Google Scholar] [CrossRef]
- Yang, S.; Liu, Z.; Wang, Y.; Li, W.; Fu, X.; Lin, Y.; Shen, Q.; Wang, X.; Wang, H.; Zhang, W. A novel rodent Chapparvovirus in feces of wild rats. Virol. J. 2016, 13, 133. [Google Scholar] [CrossRef]
- Hargitai, R.; Boros, A.; Pankovics, P.; Matics, R.; Altan, E.; Delwart, E.; Reuter, G. Detection and genetic characterization of a novel parvovirus (family Parvoviridae) in barn owls (Tyto alba) in Hungary. Arch. Virol. 2021, 166, 231–236. [Google Scholar] [CrossRef]
- Lima, D.A.; Cibulski, S.P.; Tochetto, C.; Varela, A.P.M.; Finkler, F.; Teixeira, T.F.; Loiko, M.R.; Cerva, C.; Junqueira, D.M.; Mayer, F.Q.; et al. The intestinal virome of malabsorption syndrome-affected and unaffected broilers through shotgun metagenomics. Virus Res. 2019, 261, 9–20. [Google Scholar] [CrossRef]
- Liu, X.; Wang, H.; Liu, X.; Li, Y.; Chen, J.; Zhang, J.; Wang, X.; Shen, S.; Wang, H.; Deng, F.; et al. Genomic and transcriptional analyses of novel parvoviruses identified from dead peafowl. Virology 2020, 539, 80–91. [Google Scholar] [CrossRef]
- Vibin, J.; Chamings, A.; Klaassen, M.; Bhatta, T.R.; Alexandersen, S. Metagenomic characterisation of avian parvoviruses and picornaviruses from Australian wild ducks. Sci. Rep. 2020, 10, 12800. [Google Scholar] [CrossRef]
- Xing, X.; Zhou, H.; Tong, L.; Chen, Y.; Sun, Y.; Wang, H.; Zhang, G. First identification of porcine parvovirus 7 in China. Arch. Virol. 2018, 163, 209–213. [Google Scholar] [CrossRef]
- Fahsbender, E.; Altan, E.; Seguin, M.A.; Young, P.; Estrada, M.; Leutenegger, C.; Delwart, E. Chapparvovirus DNA Found in 4% of Dogs with Diarrhea. Viruses 2019, 11, 398. [Google Scholar] [CrossRef]
- Di Profio, F.; Sarchese, V.; Palombieri, A.; Fruci, P.; Massirio, I.; Martella, V.; Fulvio, M.; Di Martino, B. Feline chaphamaparvovirus in cats with enteritis and upper respiratory tract disease. Transbound. Emerg. Dis. 2022, 69, 660–668. [Google Scholar] [CrossRef]
- Chong, R.; Shi, M.; Grueber, C.E.; Holmes, E.C.; Hogg, C.J.; Belov, K.; Barrs, V.R. Fecal Viral Diversity of Captive and Wild Tasmanian Devils Characterized Using Virion-Enriched Metagenomics and Metatranscriptomics. J. Virol. 2019, 93, e00205-19. [Google Scholar] [CrossRef]
- Alex, C.E.; Fahsbender, E.; Altan, E.; Bildfell, R.; Wolff, P.; Jin, L.; Black, W.; Jackson, K.; Woods, L.; Munk, B.; et al. Viruses in unexplained encephalitis cases in American black bears (Ursus americanus). PLoS ONE 2020, 15, e0244056. [Google Scholar] [CrossRef]
- Shi, W.; Shi, M.; Que, T.C.; Cui, X.M.; Ye, R.Z.; Xia, L.Y.; Hou, X.; Zheng, J.J.; Jia, N.; Xie, X.; et al. Trafficked Malayan pangolins contain viral pathogens of humans. Nat. Microbiol. 2022, 7, 1259–1269. [Google Scholar] [CrossRef]
- Fahsbender, E.; Charlys da-Costa, A.; Elise Gill, D.; Augusto de Padua Milagres, F.; Brustulin, R.; Julio Costa Monteiro, F.; Octavio da Silva Rego, M.; Soares D’Athaide Ribeiro, E.; Cerdeira Sabino, E.; Delwart, E. Plasma virome of 781 Brazilians with unexplained symptoms of arbovirus infection include a novel parvovirus and densovirus. PLoS ONE 2020, 15, e0229993. [Google Scholar] [CrossRef]
- Palombieri, A.; Di Profio, F.; Lanave, G.; Capozza, P.; Marsilio, F.; Martella, V.; Di Martino, B. Molecular detection and characterization of Carnivore chaphamaparvovirus 1 in dogs. Vet. Microbiol. 2020, 251, 108878. [Google Scholar] [CrossRef]
- Ji, J.; Hu, W.; Liu, Q.; Zuo, K.; Zhi, G.; Xu, X.; Kan, Y.; Yao, L.; Xie, Q. Genetic Analysis of Cachavirus-Related Parvoviruses Detected in Pet Cats: The First Report From China. Front. Vet. Sci. 2020, 7, 580836. [Google Scholar] [CrossRef]
- Canuti, M.; Mira, F.; Sorensen, R.G.; Rodrigues, B.; Bouchard, E.; Walzthoni, N.; Hopson, M.; Gilroy, C.; Whitney, H.G.; Lang, A.S. Distribution and diversity of dog parvoviruses in wild, free-roaming and domestic canids of Newfoundland and Labrador, Canada. Transbound. Emerg. Dis. 2022, 69, e2694–e2705. [Google Scholar] [CrossRef]
- Li, Y.; Gordon, E.; Idle, A.; Altan, E.; Seguin, M.A.; Estrada, M.; Deng, X.; Delwart, E. Virome of a Feline Outbreak of Diarrhea and Vomiting Includes Bocaviruses and a Novel Chapparvovirus. Viruses 2020, 12, 506. [Google Scholar] [CrossRef]
- Hao, X.; Li, Y.; Chen, B.; Wang, H.; Wang, X.; Xiao, X.; Zhou, P.; Li, S. Detection of FeChPV in a cat shelter outbreak of upper respiratory tract disease in China. Front. Microbiol. 2022, 13, 1064747. [Google Scholar] [CrossRef]
- Jing, Z.; Ji, P.; Wei, Y.; Hao, F.; Wei, Y. Isolation and identification of a novel canine parvovirus type 2c strain in domestic cats in Dalian, China. Front. Vet. Sci. 2022, 9, 1001604. [Google Scholar] [CrossRef]
- Wu, J.; Gao, X.T.; Hou, S.H.; Guo, X.Y.; Yang, X.S.; Yuan, W.F.; Xin, T.; Zhu, H.F.; Jia, H. Molecular epidemiological and phylogenetic analyses of canine parvovirus in domestic dogs and cats in Beijing, 2010–2013. J. Vet. Med. Sci. 2015, 77, 1305–1310. [Google Scholar] [CrossRef]
- Ji, J.; Cui, H.; Xu, S.Q.; Xu, X.; Liu, Q.; Kan, Y.C.; Xie, Q.M.; Yao, L.G. Molecular Characterization of Feline Chaphamaparvovirus (Carnivore chaphamaparvovirus 2) Firstly Detected in Dogs from China. Transbound. Emerg. Dis. 2023, 2023, 1–8. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, X.; Li, W.; Cui, Y.; Zhang, D.; Xu, F.; Jiang, S.; Zhou, T. Phylogenetic analysis and evolution of feline bocavirus in Anhui Province, eastern China. Comp. Immunol. Microbiol. Infect. Dis. 2021, 77, 101676. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Puigbo, P.; Bravo, I.G.; Garcia-Vallve, S. CAIcal: A combined set of tools to assess codon usage adaptation. Biol. Direct 2008, 3, 38. [Google Scholar] [CrossRef]
- Peng, Q.; Zhang, X.; Li, J.; He, W.; Fan, B.; Ni, Y.; Liu, M.; Li, B. Comprehensive analysis of codon usage patterns of porcine deltacoronavirus and its host adaptability. Transbound. Emerg. Dis. 2022, 69, e2443–e2455. [Google Scholar] [CrossRef]
- Rahman, S.U.; Rehman, H.U.; Rahman, I.U.; Rauf, A.; Alshammari, A.; Alharbi, M.; Haq, N.U.; Suleria, H.A.R.; Raza, S.H.A. Analysis of codon usage bias of lumpy skin disease virus causing livestock infection. Front. Vet. Sci. 2022, 9, 1071097. [Google Scholar] [CrossRef]
- Chen, F.; Yang, J.R. Distinct codon usage bias evolutionary patterns between weakly and strongly virulent respiratory viruses. iScience 2022, 25, 103682. [Google Scholar] [CrossRef]
- Puigbo, P.; Aragones, L.; Garcia-Vallve, S. RCDI/eRCDI: A web-server to estimate codon usage deoptimization. BMC Res. Notes 2010, 3, 87. [Google Scholar] [CrossRef]
- Qin, L.; Ding, S.; Wang, Z.; Jiang, R.; He, Z. Host Plants Shape the Codon Usage Pattern of Turnip Mosaic Virus. Viruses 2022, 14, 2267. [Google Scholar] [CrossRef]
- Alexaki, A.; Kames, J.; Holcomb, D.D.; Athey, J.; Santana-Quintero, L.V.; Lam, P.V.N.; Hamasaki-Katagiri, N.; Osipova, E.; Simonyan, V.; Bar, H.; et al. Codon and Codon-Pair Usage Tables (CoCoPUTs): Facilitating Genetic Variation Analyses and Recombinant Gene Design. J. Mol. Biol. 2019, 431, 2434–2441. [Google Scholar] [CrossRef]
- Puigbo, P.; Bravo, I.G.; Garcia-Vallve, S. E-CAI: A novel server to estimate an expected value of Codon Adaptation Index (eCAI). BMC Bioinform. 2008, 9, 65. [Google Scholar] [CrossRef]
- Yang, K.; Zhang, M.; Liu, Q.; Cao, Y.; Zhang, W.; Liang, Y.; Song, X.; Ji, K.; Shao, Y.; Qi, K.; et al. Epidemiology and Evolution of Emerging Porcine Circovirus-like Viruses in Pigs with Hemorrhagic Dysentery and Diarrhea Symptoms in Central China from 2018 to 2021. Viruses 2021, 13, 2282. [Google Scholar] [CrossRef]
- Sun, J.; Zhao, W.; Wang, R.; Zhang, W.; Li, G.; Lu, M.; Shao, Y.; Yang, Y.; Wang, N.; Gao, Q.; et al. Analysis of the Codon Usage Pattern of HA and NA Genes of H7N9 Influenza A Virus. Int. J. Mol. Sci. 2020, 21, 7129. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]
- Yao, B.; Zhang, L.; Liang, S.; Zhang, C. SVMTriP: A method to predict antigenic epitopes using support vector machine to integrate tri-peptide similarity and propensity. PLoS ONE 2012, 7, e45152. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Baggio, G.; Filippini, F.; Righetto, I. Comparative Surface Electrostatics and Normal Mode Analysis of High and Low Pathogenic H7N7 Avian Influenza Viruses. Viruses 2023, 15, 305. [Google Scholar] [CrossRef]
- Penzes, J.J.; de Souza, W.M.; Agbandje-McKenna, M.; Gifford, R.J. An Ancient Lineage of Highly Divergent Parvoviruses Infects both Vertebrate and Invertebrate Hosts. Viruses 2019, 11, 525. [Google Scholar] [CrossRef]
- Matos, M.; Bilic, I.; Viloux, N.; Palmieri, N.; Albaric, O.; Chatenet, X.; Tvarogova, J.; Dinhopl, N.; Heidl, S.; Liebhart, D.; et al. A novel Chaphamaparvovirus is the etiological agent of hepatitis outbreaks in pheasants (Phasianus colchicus) characterized by high mortality. Transbound. Emerg. Dis. 2022, 69, e2093–e2104. [Google Scholar] [CrossRef]
- Edmondson, E.F.; Hsieh, W.T.; Kramer, J.A.; Breed, M.W.; Roelke-Parker, M.E.; Stephens-Devalle, J.; Pate, N.M.; Bassel, L.L.; Hollingshead, M.G.; Karim, B.O.; et al. Naturally Acquired Mouse Kidney Parvovirus Infection Produces a Persistent Interstitial Nephritis in Immunocompetent Laboratory Mice. Vet. Pathol. 2020, 57, 915–925. [Google Scholar] [CrossRef]
- Han, Z.; Xiao, J.; Song, Y.; Zhao, X.; Sun, Q.; Lu, H.; Zhang, K.; Li, J.; Li, J.; Si, F.; et al. Highly diverse ribonucleic acid viruses in the viromes of eukaryotic host species in Yunnan province, China. Front. Microbiol. 2022, 13, 1019444. [Google Scholar] [CrossRef]
- Zhao, C.; Zhang, X.; Si, X.; Ye, L.; Lawrence, K.; Lu, Y.; Du, C.; Xu, H.; Yang, Q.; Xia, Q.; et al. Hedgehogs as Amplifying Hosts of Severe Fever with Thrombocytopenia Syndrome Virus, China. Emerg. Infect. Dis. 2022, 28, 2491–2499. [Google Scholar] [CrossRef]
- Kannekens-Jager, M.M.; de Rooij, M.M.T.; de Groot, Y.; Biesbroeck, E.; de Jong, M.K.; Pijnacker, T.; Smit, L.A.M.; Schuurman, N.; Broekhuizen-Stins, M.J.; Zhao, S.; et al. SARS-CoV-2 infection in dogs and cats is associated with contact to COVID-19-positive household members. Transbound. Emerg. Dis. 2022, 69, 4034–4040. [Google Scholar] [CrossRef]
- Seang, S.; Burrel, S.; Todesco, E.; Leducq, V.; Monsel, G.; Le Pluart, D.; Cordevant, C.; Pourcher, V.; Palich, R. Evidence of human-to-dog transmission of monkeypox virus. Lancet 2022, 400, 658–659. [Google Scholar] [CrossRef]
- Chen, F.; Wu, P.; Deng, S.; Zhang, H.; Hou, Y.; Hu, Z.; Zhang, J.; Chen, X.; Yang, J.R. Dissimilation of synonymous codon usage bias in virus-host coevolution due to translational selection. Nat. Ecol. Evol. 2020, 4, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wang, W.; Chan, J.F.; Wang, G.; Huang, Y.; Yi, Y.; Zhu, Z.; Peng, R.; Hu, X.; Wu, Y.; et al. Identification of a Novel Ichthyic Parvovirus in Marine Species in Hainan Island, China. Front. Microbiol. 2019, 10, 2815. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, J.; Zhou, Y.; Yue, H.; Zhou, N.; Tang, C. Circulation of heterogeneous Carnivore protoparvovirus 1 in diarrheal cats and prevalence of an A91S feline panleukopenia virus variant in China. Transbound. Emerg. Dis. 2022, 69, e2913–e2925. [Google Scholar] [CrossRef]
- Franzo, G.; Tucciarone, C.M.; Cecchinato, M.; Drigo, M. Canine parvovirus type 2 (CPV-2) and Feline panleukopenia virus (FPV) codon bias analysis reveals a progressive adaptation to the new niche after the host jump. Mol. Phylogenet Evol. 2017, 114, 82–92. [Google Scholar] [CrossRef]
- Day, T.; Kennedy, D.A.; Read, A.F.; Gandon, S. Pathogen evolution during vaccination campaigns. PLoS Biol. 2022, 20, e3001804. [Google Scholar] [CrossRef]
- Brand, M.; Kesmir, C. Evolution of SARS-CoV-2-specific CD4(+) T cell epitopes. Immunogenetics 2023, 75, 283–293. [Google Scholar] [CrossRef]
- Chen, Y.; Xue, Y.; Yang, J. Gilteritinib: Repurposing of AXL-targeting kinase inhibitors against COVID-19. J. Med. Virol. 2023, 95, e28592. [Google Scholar] [CrossRef]
- Mandya Naganayak, M.; Kuralayanapalya Puttahonnappa, S.; Indrabalan, U.B.; Paramanandham, K.; Jacob, S.S.; Subramaniam, S.; Patil, S.S.; Seethakempanahalli Kempanna, K.; Goroshi, S. An extensive analysis of Codon usage pattern, Evolutionary rate, and Phylogeographic reconstruction in Foot and mouth disease (FMD) serotypes (A, Asia 1, and O) of six major climatic zones of India: A comparative study. Acta Trop. 2022, 236, 106674. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Viruses | Dogs (Canis lupus Familiaris) (Mean ± SD) | Cats (Felis catus) (Mean ± SD) | |
|---|---|---|---|
| CAI | FeChPV | 0.7624 ± 0.0008 | 0.7089 ± 0.0009 |
| CaChPV | 0.7485 ± 0.0020 | 0.6950 ± 0.0020 | |
| eCAI | FeChPV | 0.7694 ± 0.0017 | 0.7157 ± 0.0016 |
| CaChPV | 0.7657 ± 0.0029 | 0.7113 ± 0.0030 | |
| Normalised CAI (CAI/eCAI) | FeChPV | 0.9909 ± 0.0024 | 0.9906 ± 0.0033 |
| CaChPV | 0.9775 ± 0.0038 | 0.9771 ± 0.0042 |
| Virus | Gene | Range of ENC | The Average of ENC (X ± S) |
|---|---|---|---|
| FeChPV | NS | 42.6–44.0 | 43.123 ± 0.461 |
| VP | 34.2–35.7 | 35.081 ± 0.417 | |
| CaChPV | NS | 43.9–46.3 | 45.047 ± 0.613 |
| VP | 39.1–41.2 | 40.16 ± 0.761 |
| Strains | Mutation Sites |
|---|---|
| CaChPV-MN928790.1 | I56T, Y68C, F131S, D402N, R449K |
| CaChPV-MN928791.1 | Y22H, Q365R, R449K, H484P, |
| FeChPV-OQ162042.1 | M19T, G112R, F151S, L174P, S197T, S206A, S325R, E352G, S445N, |
| FeChPV-OQ162043.1 | G112R, L174P, S197T, S325R, |
| Strains | Location | Epitope | Score |
|---|---|---|---|
| CaChPV-MN928790.1 | 388–407 | WGPWTWKDIYGIGSNTRMYS | 1.000 |
| 475–494 | PEMIEMQELHHTDDEEIEVI | 0.980 | |
| 123–142 | WKDSSMKDSSIAYKEGMFKS | 0.908 | |
| 24–43 | NNTLATIVAAETGGNAINTG | 0.797 | |
| 363–382 | TTQGCFQVTLHLACKKRRSR | 0.758 | |
| CaChPV-MN928791.1 | 479–498 | EMQELPHTDDEEIEIITADE | 1.000 |
| 388–407 | WGPWTWKDIYGIGSDTRMYS | 0.858 | |
| 24–43 | NNTLATIVAAETGGNAINTG | 0.724 | |
| Other CaChPV strains | 475–494 | PEMIEMQELHHTDDEEIEVI | / * |
| 388–407 | WGPWTWKDIYGIGSDTRMYS | / | |
| 24–43 | NNTLATIVAAETGGNAINTG | / | |
| 123–142 | WKDSSMKDSSIAYKEGMFKS | / | |
| 363–382 | TTQGCFQVTLHLACKKRRSR | / | |
| FeChPV-OQ162042.1 | 144–163 | VTNPLKDSSIAYKEGMFKQG | 1.000 |
| FeChPV-OQ162043.1 | 145–164 | TNPLKDFSIAYKEGMFKQGT | 1.000 |
| Other FeChPV strains | 145–164 | TNPLKDFSIAYKEGMFKQGT | 1.000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, X.; Zhang, Y.; Pan, Y.; Yang, K.; Tong, X.; Wang, Y. Phylogenetic Analysis and Codon Usage Bias Reveal the Base of Feline and Canine Chaphamaparvovirus for Cross-Species Transmission. Animals 2023, 13, 2617. https://doi.org/10.3390/ani13162617
Guo X, Zhang Y, Pan Y, Yang K, Tong X, Wang Y. Phylogenetic Analysis and Codon Usage Bias Reveal the Base of Feline and Canine Chaphamaparvovirus for Cross-Species Transmission. Animals. 2023; 13(16):2617. https://doi.org/10.3390/ani13162617
Chicago/Turabian StyleGuo, Xu, Yingying Zhang, Yang Pan, Kankan Yang, Xinxin Tong, and Yong Wang. 2023. "Phylogenetic Analysis and Codon Usage Bias Reveal the Base of Feline and Canine Chaphamaparvovirus for Cross-Species Transmission" Animals 13, no. 16: 2617. https://doi.org/10.3390/ani13162617
APA StyleGuo, X., Zhang, Y., Pan, Y., Yang, K., Tong, X., & Wang, Y. (2023). Phylogenetic Analysis and Codon Usage Bias Reveal the Base of Feline and Canine Chaphamaparvovirus for Cross-Species Transmission. Animals, 13(16), 2617. https://doi.org/10.3390/ani13162617