
Structural and Metabolic Changes in Bone


{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. The Work of Bone Tissue
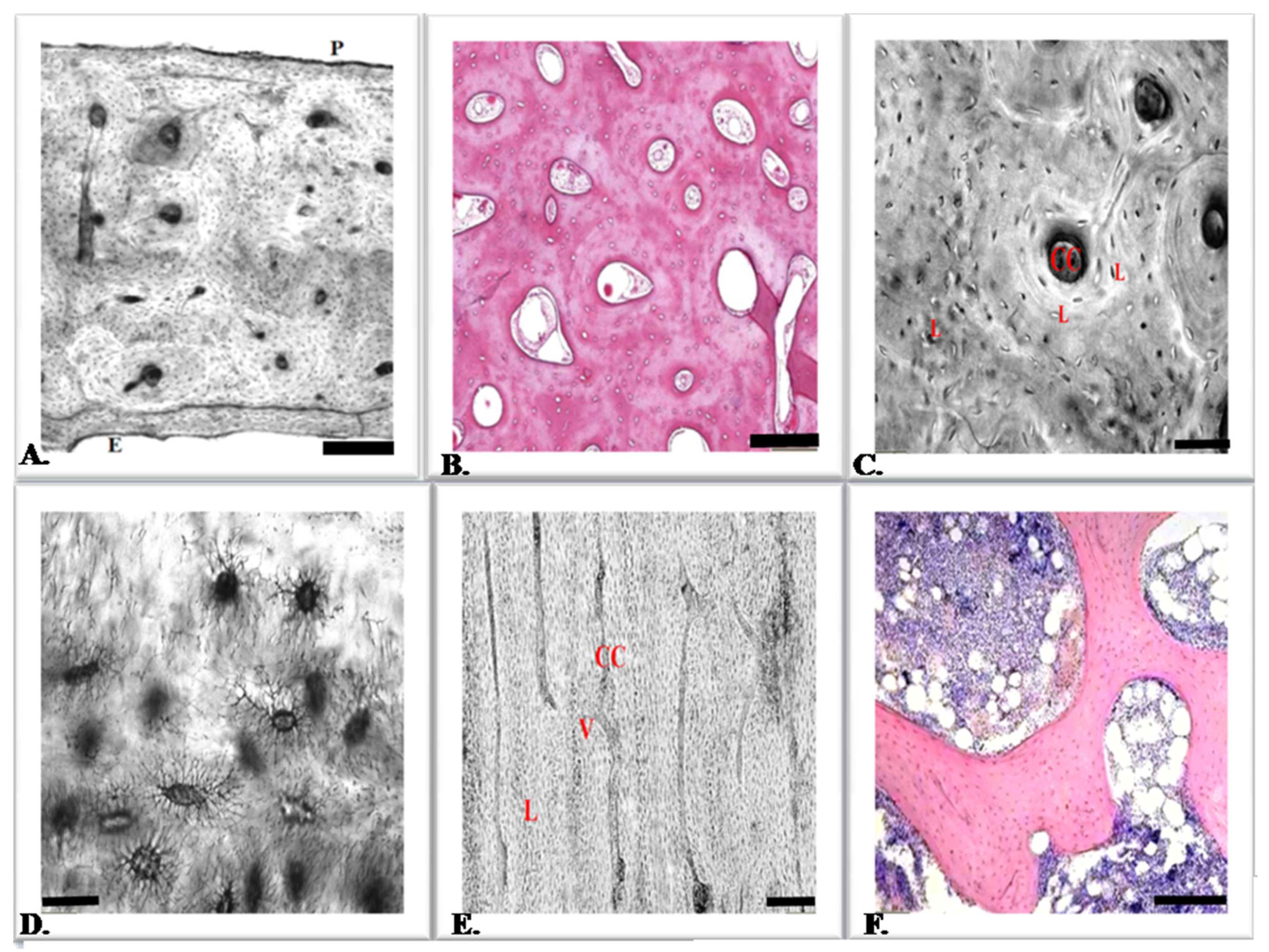
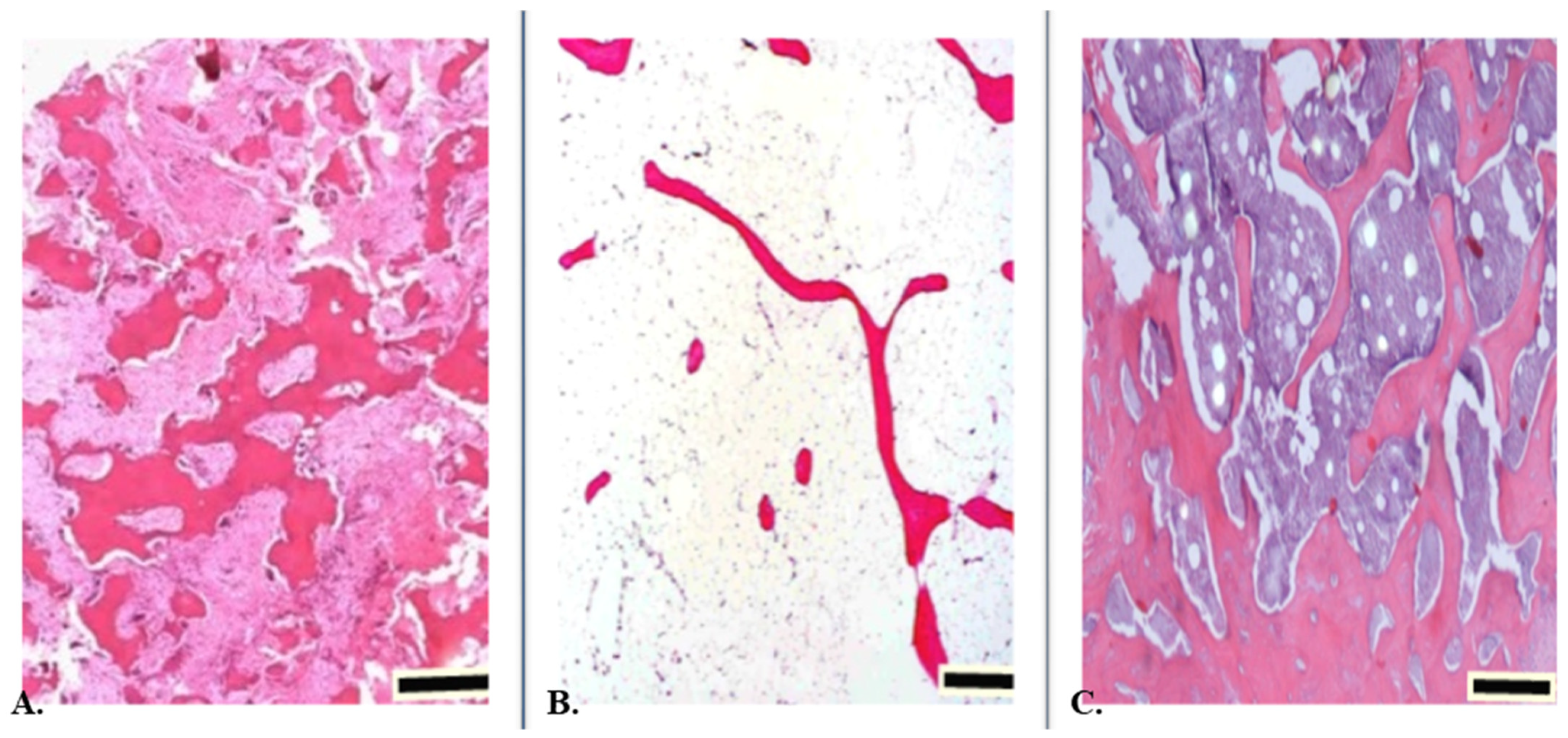
3. Bone Tissue Cells
4. Osteoblasts
5. Osteocytes
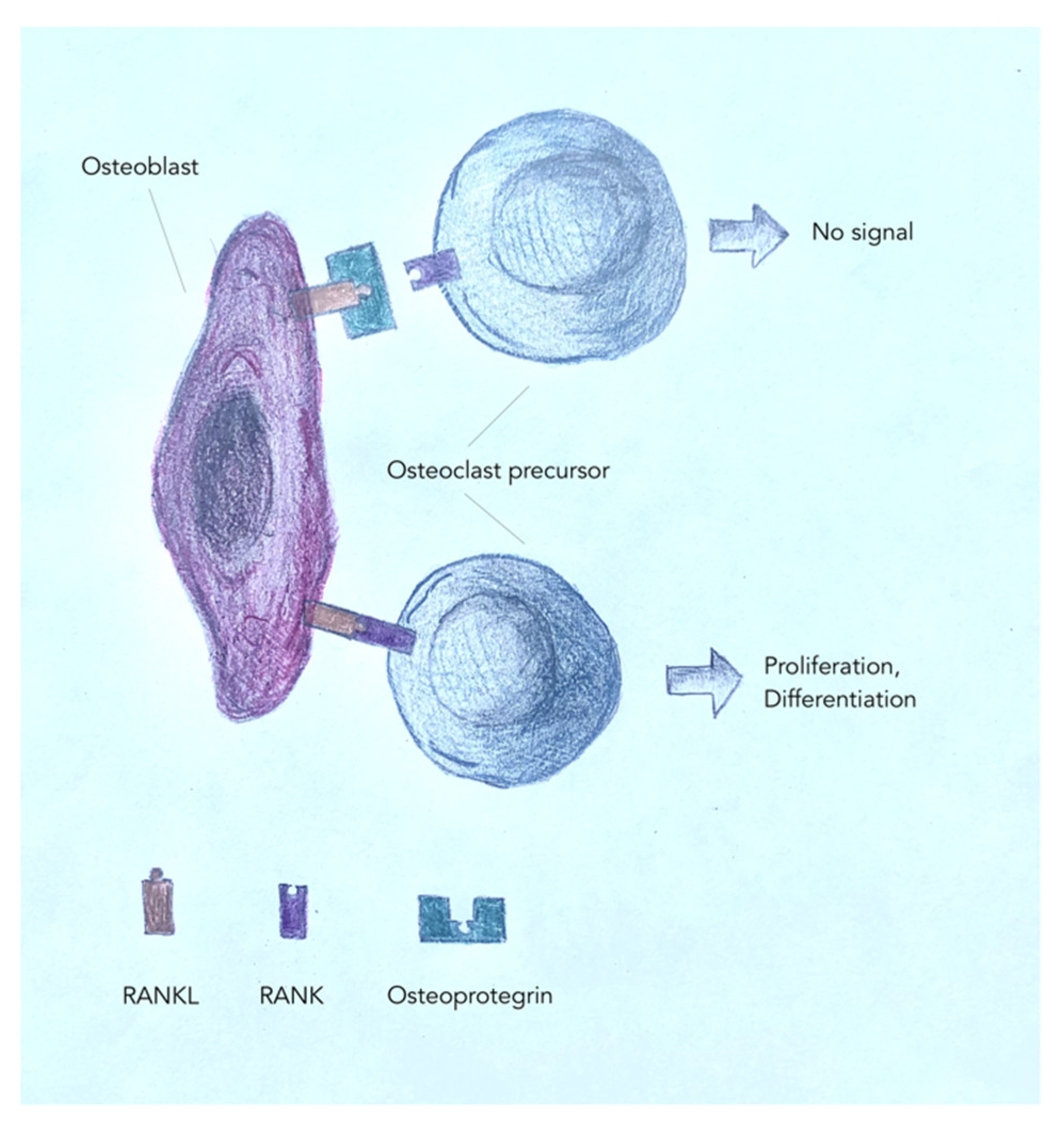
6. Osteoclasts
7. How Do Modeling and Remodeling Occur?
8. Bone Morphogenetic Proteins
9. Calcium-Regulating Hormones
10. Implications of Glucocorticosteroids
11. Bone Metabolism Disorders
12. Abnormal Bone Development
13. Inflammation and Cytokine Activity
14. Osteoporosis
15. Obesity
16. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Raggatt, L.J.; Partridge, N.C. Cellular and Molecular Mechanisms of Bone Remodeling. J. Biol. Chem. 2010, 285, 25103–25108. [Google Scholar] [CrossRef] [PubMed]
- Florencio-Silva, R.; da Silva Sasso, G.R.; Sasso-Cerri, E.; Simões, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. BioMed Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef] [PubMed]
- Mizokami, A.; Kawakubo-Yasukochi, T.; Hirata, M. Osteocalcin and its endocrine functions. Biochem. Pharmacol. 2017, 132, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Avtandilashvili, M.; Tolmachev, S.Y. Modeling the Skeleton Weight of an Adult Caucasian Man. Health Phys. 2019, 117, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Boskey, A.L.; Posner, A.S. Bone structure, composition, and mineralization. Orthop. Clin. N. Am. 1984, 15, 597–612. [Google Scholar] [CrossRef]
- Kozielski, M.; Buchwald, T.; Szybowicz, M.; Błaszczak, Z.; Piotrowski, A.; Ciesielczyk, B. Determination of composition and structure of spongy bone tissue in human head of femur by Raman spectral mapping. J. Mater. Sci. Mater. Med. 2011, 22, 1653–1661. [Google Scholar] [CrossRef] [PubMed]
- De Carmejane, O.; Morris, M.D.; Davis, M.K.; Stixrude, L.; Tecklenburg, M.; Rajachar, R.M.; Kohan, D.H. Bone Chemical Structure Response to Mechanical Stress Studied by High Pressure Raman Spectroscopy. Calcif. Tissue Res. 2005, 76, 207–213. [Google Scholar] [CrossRef]
- Karsenty, G.; Ferron, M. The contribution of bone to whole-organism physiology. Nature 2012, 481, 314–320. [Google Scholar] [CrossRef]
- Rubin, M.R.; Cosman, F.; Lindsay, R.; Bilezikian, J.P. The anabolic effects of parathyroid hormone. Osteoporos. Int. 2002, 13, 267–277. [Google Scholar] [CrossRef]
- Felsenfeld, A.J.; Levine, B.S. Calcitonin, the forgotten hormone: Does it deserve to be forgotten? Clin. Kidney J. 2015, 8, 180–187. [Google Scholar] [CrossRef]
- Han, Y.; You, X.; Xing, W.; Zhang, Z.; Zou, W. Paracrine and endocrine actions of bone-the functions of secretory proteins from osteoblasts, osteocytes, and osteoclasts. Bone Res. 2018, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Shin, Y.; Yen, M.-S.; Sun, S.S. Peak Bone Mass and Patterns of Change in Total Bone Mineral Density and Bone Mineral Contents from Childhood into Young Adulthood. J. Clin. Densitom. 2016, 19, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Lopes, D.; Martins-Cruz, C.; Oliveira, M.B.; Mano, J.F. Bone physiology as inspiration for tissue regenerative therapies. Biomaterials 2018, 185, 240–275. [Google Scholar] [CrossRef] [PubMed]
- Datta, H.K.; Ng, W.F.; Walker, J.A.; Tuck, S.P.; Varanasi, S.S. The cell biology of bone metabolism. J.Clin. Pathol. 2008, 61, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Nakchbandi, I.A. Osteoporosis and fractures in liver disease: Relevance, pathogenesis and therapeutic implications. World J. Gastroenterol. 2014, 20, 9427–9438. [Google Scholar] [CrossRef]
- Schilling, C.H.; Letscher, D.; Palsson, B.O. Theory for the Systemic Definition of Metabolic Pathways and their use in Interpreting Metabolic Function from a Pathway-Oriented Perspective. J. Theor. Biol. 2000, 203, 229–248. [Google Scholar] [CrossRef]
- Fratzl, P.; Gupta, H.S.; Paschalis, E.P.; Roschger, P. Structure and mechanical quality of the collagen–mineral nano-composite in bone. J. Mater. Chem. 2004, 14, 2115–2123. [Google Scholar] [CrossRef]
- Hart, N.H.; Newton, R.U.; Tan, J.; Rantalainen, T.; Chivers, P.; Siafarikas, A.; Nimphius, S. Biological basis of bone strength: Anatomy, physiology and measurement. J. Musculoskelet. Neuronal Interact. 2020, 20, 347–371. [Google Scholar]
- Kontulainen, S.; Sievanen, H.; Kannus, P.; Pasanen, M.; Vuori, I. Effect of long-term impact-loading on mass, size, and estimated strength of humerus and radius of female racquet-sports players: A peripheral quantitative computed tomography study between young and old starters and controls. J. Bone Miner. Res. 2003, 18, 352–359. [Google Scholar] [CrossRef]
- Capulli, M.; Paone, R.; Rucci, N. Osteoblast and osteocyte: Games without frontiers. Arch. Biochem. Biophys. 2014, 561, 3–12. [Google Scholar] [CrossRef]
- Asada, N.; Sato, M.; Katayama, Y. Communication of bone cells with hematopoiesis, immunity and energy metabolism. BoneKEy Rep. 2015, 4, 748. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Molagoda, I.M.N.; Athapaththu, A.M.G.K.; ParkE, K.; Choi, Y.H.; Jeon, Y.J.; Young, K.G. Fermented Oyster (Crassostrea gigas) Extract Cures and Prevents Prednisolone-Induced Bone Resorption by Activating Osteoblast Differentiation. Foods 2022, 11, 678. [Google Scholar] [CrossRef] [PubMed]
- Estrada, K.D.; Retting, K.N.; Chin, A.M.; Lyons, K.M. Smad6 is essential to limit BMP signaling during cartilage development. J. Bone Miner. Res. 2011, 26, 2498–2510. [Google Scholar] [CrossRef]
- Chau, J.; Leong, W.F.; Li, B. Signaling pathways governing osteoblast proliferation, differentiation and function. Histol. Histopathol. 2009, 24, 1593–1606. [Google Scholar] [PubMed]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cell sregulate the haemato-poietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef]
- Vancea, A.; Serban, O.; Fodor, D. Relationship between Osteopontin and Bone Mineral Density. Acta Endocrinol. 2021, 17, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Harada, S.-I.; Rodan, G.A. Control of osteoblast function and regulation of bone mass. Nature 2003, 423, 349–355. [Google Scholar] [CrossRef]
- Chen, G.; Deng, C.; Li, Y.-P. TGF-beta and BMP signaling in osteoblast differentiation and bone formation. Int. J. Biol. Sci. 2012, 8, 272–288. [Google Scholar] [CrossRef]
- Yang, Y.-Q.; Tan, Y.-Y.; Wong, R.; Wenden, A.; Zhang, L.-K.; Rabie, A.B.M. The role of vascular endothelial growth factor in ossi-fication. Int. J. Oral. Sci. 2012, 4, 64–68. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, J.; Cheng, C.M.; Kopchick, J.J.; Bondy, C.A. Evidence supporting dual, IGF-I-independent and IGF-I-dependent, roles for GH in promoting longitudinal bone growth. J. Endocrinol. 2004, 180, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Kiechl, S.; Redlich, K.; Oberhollenzer, F.; Weger, S.; Egger, G.; Mayr, A.; Jocher, J.; Xu, Q.; Pietschmann, P.; et al. Soluble RANKL and risk of nontraumatic fracture. JAMA 2004, 291, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Si, J.; Wang, C.; Zhang, D.; Wang, B.; Hou, W.; Zhou, Y. Osteopontin in Bone Metabolism and Bone Diseases. Med Sci. Monit. 2020, 26, e919159-1–e919159-9. [Google Scholar] [CrossRef]
- Karthik, V.; Guntur, A.R. Energy Metabolism of Osteocytes. Curr. Osteoporos. Rep. 2021, 19, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Tresguerres, F.G.F.; Torres, J.; López-Quiles, J.; Hernández, G.; Vega, J.A.; Tresguerres, I.F. The osteocyte: A multifunctional cell within the bone. Ann. Anat. Anat. Anz. 2020, 227, 151422. [Google Scholar] [CrossRef] [PubMed]
- Moriishi, T.; Komori, T. Osteocytes: Their Lacunocanalicular Structure and Mechanoresponses. Int. J. Mol. Sci. 2022, 23, 4373. [Google Scholar] [CrossRef]
- Tsourdi, E.; Jahn, K.; Rauner, M.; Busse, B.; Bonewald, L.F. Physiological and pathological osteocytic osteolysis. J. Musculoskelet. Neuronal Interact. 2018, 18, 292–303. [Google Scholar] [PubMed]
- Delgado-Calle, J.M.; Bellido, T. The osteocyte as a signaling cell. Physiol. Rev. 2022, 102, 379–410. [Google Scholar] [CrossRef]
- Franz-Odendaal, T.A.; Hall, B.K.; Witten, P.E. Buried alive: How osteoblasts become osteocytes. Dev. Dyn. 2006, 235, 176–190. [Google Scholar] [CrossRef]
- Chen, H.; Senda, T.; Kubo, K.Y. The osteocyte plays multiple roles in bone remodeling and mineral homeostasis. Med. Mol. Morphol. 2015, 48, 61–68. [Google Scholar] [CrossRef]
- Ubaidus, S.; Li, M.; Sultana, S.; de Freitas, P.H.L.; Oda, K.; Maeda, T.; Takagi, R. Amizuka N.FGF23 is mainly synthesized by osteocytes in the regularly distributed osteocytic lacunar canalicular system established after physiological bone remodeling. J. Electron Microsc. 2009, 58, 381–392. [Google Scholar] [CrossRef]
- Qiao, W.; Yu, S.; Sun, H.; Chen, L.; Wang, R.; Wu, X.; Goltzman, D.; Miao, D. 1,25-Dihydroxyvitamin D insufficiency accelerates age-related bone loss by increasing oxidative stress and cell senescence. Am. J. Transl. Res. 2020, 12, 507–518. [Google Scholar] [PubMed]
- Li, J.; Karim, M.A.; Che, H.; Geng, Q.; Miao, D. Deletion of p16 prevents estrogen deficiency-induced osteoporosis by inhibiting oxidative stress and osteocyte senescence. Am. J. Transl. Res. 2020, 12, 672–683. [Google Scholar] [PubMed]
- Cui, J.; Shibata, Y.; Zhu, T.; Zhou, J.; Zhang, J. Osteocytes in bone aging: Advances, challenges, and future perspectives. Ageing Res. Rev. 2022, 77, 101608. [Google Scholar] [CrossRef]
- Shiozawa, Y. The Roles of Bone Marrow-Resident Cells as a Microenvironment for Bone Metastasis. Adv. Exp. Med. Biol. 2020, 1226, 57–72. [Google Scholar] [PubMed]
- Kollet, O.; Dar, A.; Shivtiel, S.; Kalinkovich, A.; Lapid, K.; Sztainberg, Y.; Tesio, M.; Samstein, R.M.; Goichberg, P.; Spiegel, A.; et al. Faculty Opinions recommendation of Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat. Med. 2006, 12, 657–664. [Google Scholar] [CrossRef]
- Mosley, J.R. Osteoporosis and bone functional adaptation: Mechanobiological regulation of bone architecture in growing and adult bone, a review. J. Rehabil. Res. Dev. 2000, 37, 189–199. [Google Scholar] [PubMed]
- Clarke, B.L.; Khosla, S. Physiology of Bone Loss. Radiol. Clin. N. Am. 2010, 48, 483–495. [Google Scholar] [CrossRef]
- Phan, T.; Xu, J.; Zheng, M. Interaction between osteoblast and osteoclast: Impact in bone disease. Histol. Histopathol. 2004, 19, 1325–1344. [Google Scholar]
- Bouillon, R.; Bischoff-Ferrari, H.; Willett, W. Vitamin D and health: Perspectives from mice and man. J. Bone Miner. Res. 2008, 23, 974–979. [Google Scholar] [CrossRef]
- Jilka, R.L. Biology of the basic multicellular unit and the pathophysiology of osteoporosis. Med. Pediatric Oncol. 2003, 41, 182–185. [Google Scholar] [CrossRef]
- Seeman, E. Invited Review: Pathogenesis of osteoporosis. J. Appl. Physiol. 2003, 95, 2142–2151. [Google Scholar] [CrossRef] [PubMed]
- Canalis, E.; Delany, A.M. Mechanisms of Glucocorticoid Action in Bone. Ann. N. Y. Acad. Sci. 2002, 966, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Hauge, E.M.; Qvesel, D.; Eriksen, E.F.; Mosekilde, L.; Melsen, F. Cancellous bone remodeling occurs in specialized compartments lined by cells expressing osteoblastic markers. J. Bone Miner. Res. 2001, 16, 1575–1582. [Google Scholar] [CrossRef] [PubMed]
- Everts, V.; Delaisse, J.M.; Korper, W.; Jansen, D.C.; Tigchelaar-Gutter, W.; Saftig, P.; Beertsen, W. The bone lining cell: Its role in cleaning Howship’s lacunae and initiating bone formation. J. Bone Miner. Res. 2002, 17, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Parfitt, A.M. The bone remodeling compartment: A circulatory function for bone lining cells. J. Bone Miner. Res. 2001, 16, 1583–1585. [Google Scholar] [CrossRef] [PubMed]
- Tencerova, M.; Ferencakova, M.; Kassem, M. Bone marrow adipose tissue: Role in bone remodeling and energy metabolism. Best Pract. Res. Clin. Endocrinol. Metab. 2021, 35, 101545. [Google Scholar] [CrossRef]
- Manolagas, S.C. Birth and Death of Bone Cells: Basic Regulatory Mechanisms and Implications for the Pathogenesis and Treatment of Osteoporosis. Endocr. Rev. 2000, 21, 115–137. [Google Scholar] [CrossRef]
- Tobeiha, M.; Moghadasian, M.H.; Amin, N.; Jafarnejad, S. RANKL/RANK/OPG Pathway: A Mechanism Involved in Exercise-Induced Bone Remodeling. BioMed Res. Int. 2020, 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bleuming, S.A.; He, X.C.; Kodach, L.L.; Hardwick, J.C.; Koopman, F.A.; Kate, F.J.T.; van Deventer, S.J.; Hommes, D.W.; Peppelenbosch, M.P.; Offerhaus, G.J.; et al. Bone Morphogenetic Protein Signaling Suppresses Tumorigenesis at Gastric Epithelial Transition Zones in Mice. Cancer Res. 2007, 67, 8149–8155. [Google Scholar] [CrossRef]
- Lin, G.L.; Hankenson, K.D. Integration of BMP, Wnt, and notch signaling pathways in osteoblast differentiation. J. Cell. Biochem. 2011, 112, 3491–3501. [Google Scholar] [CrossRef] [PubMed]
- Beederman, M.; Lamplot, J.D.; Nan, G.; Wang, J.; Liu, X.; Yin, L.; Li, R.; Shui, W.; Zhang, H.; Kim, S.H.; et al. BMP signaling in mesenchymal stem cell differentiation and bone formation. Biomed. Sci. Eng. 2013, 6, 32–52. [Google Scholar] [CrossRef] [PubMed]
- Khosla, S. Minireview: The OPG/RANKL/RANK system. Endocrinology 2001, 142, 5050–5055. [Google Scholar] [CrossRef] [PubMed]
- Mallorie, A.; Shine, B. Normal bone physiology, remodelling and its hormonal regulation. Surgery 2022, 40, 163–168. [Google Scholar] [CrossRef]
- Lee, N.K.; Sowa, H.; Hinoi, E.; Ferron, M.; Ahn, J.D.; Confavreux, C.; Dacquin, R.; Mee, P.J.; McKee, M.D.; Jung, D.Y.; et al. Endocrine regulation of energy metabolism by the skeleton. Cell 2007, 130, 456–469. [Google Scholar] [CrossRef]
- Orwoll, E.S. Treatment of osteoporosis in men. Calcif Tissue Int. 2004, 75, 114–119. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stewart, A.F. Hyperparathyroidism, humoral hypercalcemia of malignancy, and the anabolic actions of parathyroid hormone and parathyroid hormone-related protein on the skeleton. J. Bone Miner. Res. 2002, 17, 758–762. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, P.; Mosekilde, L. Fractures in patients with hyperthyroidism and hypothyroidism: A nationwide follow-up study in 16,249 patients. Thyroid 2002, 12, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Norman, A.W.; Okamura, W.H.; Bishop, J.E.; Henry, H.L. Update on biological actions of 1 alpha,25(OH)(2)-vitamin D(3) (rapid effects) and 24R,25(OH)(2)-vitamin D(3). Mol. Cell Endocrinol. 2002, 197, 1–13. [Google Scholar] [CrossRef]
- Abe, E.; Marians, R.C.; Yu, W.; Wu, X.-B.; Ando, T.; Li, Y.; Iqbal, J.; Eldeiry, L.; Rajendren, G.; Blair, H.C.; et al. TSH Is a Negative Regulator of Skeletal Remodeling. Cell 2003, 115, 151–162. [Google Scholar] [CrossRef]
- Falahati-Nini, A.; Riggs, B.L.; Atkinson, E.J.; O’Fallon, W.M.; Eastell, R.; Khosla, S. Relative contributions of testosterone and estrogen in regulating bone resorption and formation in normal elderly men. J. Clin. Investig. 2000, 106, 1553–1560. [Google Scholar] [CrossRef]
- Ahlborg, H.G.; Johnell, O.; Turner, C.H.; Rannevik, G.; Karlsson, M.K. Bone loss and bone size after menopause. N. Engl. J. Med. 2003, 349, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Yakar, S.; Rosen, C.J. From mouse to man: Redefining the role of insulin-like growth factor-I in the acquisition of bone mass. Exp. Biol. Med. 2003, 228, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Kanis, J.A.; Johansson, H.; Oden, A.; Johnell, O.; De Laet, C.; Melton, L.J., III; Tenenhouse, A.; Reeve, J.; Silman, A.J.; Pols, H.A.; et al. A meta-analysis of prior corticosteroid use and fracture risk. J. Bone Miner. Res. 2004, 19, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Kraut, D.; Gerstenfeld, L.C.; Graves, D.T. Diabetes interferes with the bone formation by affecting the expression of transcription factors that regulate osteoblast differentiation. Endocrinology 2003, 144, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Miyakoshi, N.; Tsuchida, T.; Kasukawa, Y.; Sato, K.; Itoi, E. Effects of combined treatment of insulin and human par-athyroid hormone (1–34) on cancellous bone mass and structure in streptozotocin-induced diabetic rats. Bone 2003, 33, 108–114. [Google Scholar] [CrossRef]
- Elefteriou, F.; Takeda, S.; Ebihara, K.; Magre, J.; Patano, N.; Kim, C.A.; Ogawa, Y.; Liu, X.; Ware, S.M.; Craigen, W.J.; et al. Serum leptin level is a regulator of bone mass. Proc. Natl. Acad. Sci. USA 2004, 101, 3258–3263. [Google Scholar] [CrossRef] [PubMed]
- Cornish, J.; Callon, K.E.; Bava, U.; Lin, C.; Naot, D.; Hill, B.L.; Grey, A.B.; Broom, N.; Myers, D.E.; Nicholson, G.C.; et al. Leptin directly regulates bone cell function in vitro and reduces bone fragility in vivo. J. Endocrinol. 2002, 175, 405–415. [Google Scholar] [CrossRef]
- Stein, E.; Shane, E. Secondary osteoporosis. Endocrinol. Metab. Clin. N. Am. 2003, 32, 115–134. [Google Scholar] [CrossRef]
- Reid, I.R.; A Baldock, P.; Cornish, J. Effects of Leptin on the Skeleton. Endocr. Rev. 2018, 39, 938–959. [Google Scholar] [CrossRef]
- Schiellerup, S.P.; Skov-Jeppesen, K.; Windeløv, J.A.; Svane, M.S.; Holst, J.J.; Hartmann, B.; Rosenkilde, M.M. Gut Hormones and Their Effect on Bone Metabolism. Potential Drug Therapies in Future Osteoporosis Treatment. Front. Endocrinol. 2019, 10, 75. [Google Scholar] [CrossRef]
- Martin, C.S.; Cooper, M.S.; Hardy, R.S. Endogenous Glucocorticoid Metabolism in Bone: Friend or Foe. Front. Endocrinol. 2021, 12, 733611. [Google Scholar] [CrossRef] [PubMed]
- Lane, N.E.; Yao, W.; Balooch, M.; Nalla, R.K.; Balooch, G.; Habelitz, S.; Kinney, J.H.; Bonewald, L.F. Glucocorticoid-Treated Mice Have Localized Changes in Trabecular Bone Material Properties and Osteocyte Lacunar Size That Are Not Observed in Placebo-Treated or Estrogen-Deficient Mice. J. Bone Miner. Res. 2005, 21, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, R.S.; Chen, J.-R.; Powers, C.C.; Stewart, S.A.; Landes, R.D.; Bellido, T.; Jilka, R.L.; Parfitt, A.M.; Manolagas, S.C. Promotion of osteoclast survival and antagonism of bisphosphonate-induced osteoclast apoptosis by glucocorticoids. J. Clin. Investig. 2002, 109, 1041–1048. [Google Scholar] [CrossRef]
- Conaway, H.H.; Henning, P.; Lie, A.; Tuckermann, J.; Lerner, U.H. Activation of dimeric glucocorticoid receptors in osteoclast progenitors potentiates RANKL induced mature osteoclast bone resorbing activity. Bone 2016, 93, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Chevalley, T.; Strong, D.D.; Mohan, S.; Baylink, D.J.; Linkhart, T.A. Evidence for a role for insulin-like growth factor binding proteins in glucocorticoid inhibition of normal human osteoblast-like cell proliferation. Eur. J. Endocrinol. 1996, 134, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.S.; Raza, K.; Cooper, M.S. Glucocorticoid metabolism in rheumatoid arthritis. Ann. N. Y. Acad. Sci. 2014, 1318, 18–26. [Google Scholar] [CrossRef]
- Di Dalmazi, G.; Pagotto, U.; Pasquali, R.; Vicennati, V. Glucocorticoids and Type 2 Diabetes: From Physiology to Pathology. J. Nutr. Metab. 2012, 2012, 525093. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Kitazawa, R.; Yamaguchi, A.; Kitazawa, S. Dexamethasone promotes osteoclastogenesis by inhibiting osteoprotegerin through multiple levels. J. Cell. Biochem. 2008, 103, 335–345. [Google Scholar] [CrossRef]
- Yao, W.; Cheng, Z.; Busse, C.; Pham, A.; Nakamura, M.C.; Lane, N.E. Glucocorticoid excess in mice results in early activation of osteoclastogenesis and adipogenesis and prolonged suppression of osteogenesis: A longitudinal study of gene expression in bone tissue from glucocorticoid-treated mice. Arthritis Rheum 2008, 58, 1674–1686. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Thompson, C.B. Cellular Metabolism and Disease: What Do Metabolic Outliers Teach Us? Cell 2012, 148, 1132–1144. [Google Scholar] [CrossRef]
- Long, F. Energy Metabolism and Bone. Bone 2018, 115, 1. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, A.; Schneider, V.; Shackelford, L.; West, S.; Oganov, V.; Bakulin, A.; Voronin, L. Bone mineral and lean tissue loss after long duration space flight. J. Musculoskelet. Neuronal Interact. 2000, 1, 157–160. [Google Scholar] [PubMed]
- Shaker, J.L. Paget’s disease of bone: A review of epidemiology, pathophysiology and management. Ther. Adv. Musculoskelet. Dis. 2009, 1, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Mackie, E.J.; Ahmed, Y.A.; Tatarczuch, L.; Chen, K.-S.; Mirams, M. Endochondral ossification: How cartilage is converted into bone in the developing skeleton. Int. J. Biochem. Cell Biol. 2008, 40, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Rey, J.P.; Ellies, D.L. Wnt modulators in the biotech pipeline. Dev. Dyn. 2010, 239, 102–114. [Google Scholar] [CrossRef]
- Takei, Y.; Minamizaki, T.; Yoshiko, Y. Functional Diversity of Fibroblast Growth Factors in Bone Formation. Int. J. Endocrinol. 2015, 2015, 1–12. [Google Scholar] [CrossRef]
- Bodine, P.V. Wnt signaling in bone development. In Bone and Development; Springer: Berlin/Heidelberg, Germany, 2010; pp. 137–152. [Google Scholar]
- Kobayashi, Y.; Uehara, S.; Koide, M.; Takahashi, N. The regulation of osteoclast differentiation by Wnt signals. BoneKey Rep. 2015, 4, 713. [Google Scholar] [CrossRef]
- Salhotra, A.; Shah, H.N.; Levi, B.; Longaker, M.T. Mechanisms of bone development and rep air. Nat. Rev. Mol. Cell Biol. 2020, 21, 696–711. [Google Scholar] [CrossRef]
- Boivin, G.; Meunier, P.J. Changes in Bone Remodeling Rate Influence the Degree of Mineralization of Bone. Connect. Tissue Res. 2002, 43, 535–537. [Google Scholar] [CrossRef]
- Terashima, A.; Okamoto, K.; Nakashima, T.; Akira, S.; Ikuta, K.; Takayanagi, H. Sepsis-Induced Osteoblast Ablation Causes Immunodeficiency. Immunity 2016, 44, 1434–1443. [Google Scholar] [CrossRef]
- Hofbauer, L.C.; Khosla, S.; Dunstan, C.R.; Lacey, D.L.; Boyle, W.J.; Riggs, B.L. The roles of osteoprotegerin and osteopro tegerin ligand in the paracrine regulation of bone resorption. J Bone Miner Res. 2000, 15, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Luntzer, M. Interaction between bone and immune cells: Implications for postmenopausal osteoporosis. Semin. Cell Dev. Biol. 2021, 123, 14–21. [Google Scholar] [CrossRef]
- Epsley, S.; Tadros, S.; Farid, A.; Kargilis, D.; Mehta, S.; Rajapakse, C.S. The Effect of Inflammation on Bone. Front. Physiol. 2021, 11, 511799. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, M.; Arora, S.; Li, J.; Rahmani, R.; Sun, L.; Steinlauf, A.F.; Mechanick, J.I.; Zaidi, M. Bone, Inflammation, and Inflammatory Bowel Disease. Curr. Osteoporos. Rep. 2001, 9, 251–257. [Google Scholar] [CrossRef]
- Lehouck, A.; Boonen, S.; Decramer, M.; Janssens, W. COPD, bone metabolism, and osteoporosis. Chest. 2011, 139, 648–657. [Google Scholar] [CrossRef]
- Kanis, J.A.; McCloskey, E.V.; Johansson, H.; Cooper, C.; Rizzoli, R.; Reginster, J.-Y.; Scientific Advisory Board of the European Society for Clinical and Economic Aspects of Osteoporosis and Osteoarthritis (ESCEO) and the Committee of Scientific Advisors of the International Osteoporosis Foundation (IOF). European guidance for the diagnosis and management of osteoporosis in postmenopausal women. Osteoporos. Int. 2012, 24, 23–57. [Google Scholar] [CrossRef]
- Saxena, Y.; Routh, S.; Mukhopadhaya, A. Immunoporosis: Role of Innate Immune Cells in Osteoporosis. Front. Immunol. 2021, 12, 687037. [Google Scholar] [CrossRef]
- Fang, H.; Zhang, H.; Wang, Z.; Zhou, Z.; Li, Y.; Lu, L. Systemic immune-inflammationindex acts as a novel diagnostic biomarker for postmenopausal osteoporosis an could predict the risk of osteoporotic fracture. J. Clin. Lab. Anal. 2020, 34, 23016. [Google Scholar] [CrossRef]
- Srivastava, R.K.; Dar, H.Y.; Mishra, P.K. Immunoporosis: Immunology of Osteoporosis—Role of T Cells. Front. Immunol. 2018, 9, 657. [Google Scholar] [CrossRef]
- Suzuki, K. Chronic Inflammation as an Immunological Abnormality and Effectiveness of Exercise. Biomolecules 2019, 9, 223. [Google Scholar] [CrossRef] [PubMed]
- Raisz, L.G.; Rodan, G.A. Pathogenesis of osteoporosis. Endocrinol. Metab. Clin. N. Am. 2003, 32, 15–24. [Google Scholar] [CrossRef]
- Gao, Y.; Patil, S.; Jia, J. The Development of Molecular Biology of Osteoporosis. Int. J. Mol. Sci. 2021, 22, 8182. [Google Scholar] [CrossRef] [PubMed]
- Downey, P.A.; I Siegel, M. Bone Biology and the Clinical Implications for Osteoporosis. Phys. Ther. 2006, 86, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Mo, C.; Ke, J.; Zhao, D.; Zhang, B. Role of the renin–angiotensin–aldosterone system in bone metabolism. J. Bone Miner. Metab. 2020, 38, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Skerry, T.M. The response of bone to mechanical loading and disuse: Fundamental principles and influences on osteoblast/osteocyte homeostasis. Arch Biochem Biophys. 2008, 473, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Seeman, E. Periosteal Bone Formation—A Neglected Determinant of Bone Strength. N. Engl. J. Med. 2003, 349, 320–323. [Google Scholar] [CrossRef]
- Riggs, B.L.; Khosla, S.; Melton, L.J., 3rd. Sex steroids and the construction and conservation of the adult skeleton. Endocr. Rev. 2002, 23, 279–302. [Google Scholar] [CrossRef] [PubMed]
- Sheu, A.; Diamond, T. Bone mineral density: Testing for osteoporosis. Aust. Prescr. 2016, 39, 35–39. [Google Scholar] [CrossRef]
- Marshall, D.; Johnell, O.; Wedel, H. Meta-analysis of how well measures of bone mineral density predict occurrence of oste-oporotic fractures. BMJ 1996, 312, 1254–1259. [Google Scholar] [CrossRef]
- Jain, R.K.; Vokes, T. Dual-energy X-ray Absorptiometry. J. Clin. Densitom. 2017, 20, 291–303. [Google Scholar] [CrossRef]
- Binkley, N.C.; Schmeer, P.; Wasnich, R.D.; Lenchik, L. What are the criteria by which a densitometric diagnosis of osteoporosis can be made in males and non-Whites. J. Clin. Densitom. 2002, 5, S19–S27. [Google Scholar] [CrossRef]
- Kanis, J.A. Osteoporosis III: Diagnosis of osteoporosis and assessment of fracture risk. Lancet 2002, 359, 1929–1936. [Google Scholar] [CrossRef]
- Rössner, S. Obesity: The disease of the twenty-first century. Int. J. Obes. Relat. Metab. Disord. 2002, 26, S2–S4. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose Tissue Remodeling: Its Role in Energy Metabolism and Metabolic Disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Gesta, S.; Tseng, Y.-H.; Kahn, C.R. Developmental Origin of Fat: Tracking Obesity to Its Source. Cell 2007, 131, 242–256. [Google Scholar] [CrossRef] [PubMed]
- Richard, D.; Picard, F. Brown fat biology and thermogenesis. Front. Biosci.-Landmark 2011, 16, 1233–1260. [Google Scholar] [CrossRef] [PubMed]
- Carobbio, S.; Pellegrinelli, V.; Vidal-Puig, A. Adipose Tissue Function and Expandability as Determinants of Lipotoxicity and the Metabolic Syndrome. Adv. Exp. Med. Biol. 2017, 960, 161–196. [Google Scholar] [CrossRef] [PubMed]
- Conde, J.; Scotece, M.; Gomez, R.; Lopez, V.; Gomez-Reino, J.J.; Gualillo, O. Adipokines and osteoarthritis: Novel mol-ecules involved in the pathogenesis and progression of disease. Arthritis 2011, 2011, 203901. [Google Scholar] [CrossRef] [PubMed]
- China, S.P.; Sanyal, S.; Chattopadhyay, N. Adiponectin signaling and its role in bone metabolism. Cytokine 2018, 112, 116–131. [Google Scholar] [CrossRef]
- Frühbeck, G.; Gomez-Ambrosi, J.; Muruzabal, F.J.; Burrell, M.A. The adipocyte: A model for integration of endocrine and metabolic signaling in energy metabolism regulation. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E827–E847. [Google Scholar] [CrossRef]
- Tencerova, M.; Okla, M.; Kassem, M. Insulin Signaling in Bone Marrow Adipocytes. Curr. Osteoporos. Rep. 2019, 17, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, I.; Larson, B.; Vuoristo, J.; Cui, J.; Prockop, D. Adipogenic differentiation of human adult stem cells from bone marrow stroma (MSCs). J. Bone Miner. Res. 2004, 19, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Bosello, O.; Donataccio, M.P.; Cuzzolaro, M. Obesity or obesities? Controversies on the association between body mass index and premature mortality. Eat. Weight Disord. Stud. Anorexia, Bulim. Obes. 2016, 21, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Pirro, M.; Fabbriciani, G.; Leli, C.; Callarelli, L.; Manfredelli, M.R.; Fioroni, C.; Mannarino, M.R.; Scarponi, A.M.; Mannarino, E. High weight or body mass index increase the risk of vertebral fractures in postmenopausal osteoporotic women. J. Bone Miner. Metab. 2010, 28, 88–93. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wawrzyniak, A.; Balawender, K. Structural and Metabolic Changes in Bone. Animals 2022, 12, 1946. https://doi.org/10.3390/ani12151946
Wawrzyniak A, Balawender K. Structural and Metabolic Changes in Bone. Animals. 2022; 12(15):1946. https://doi.org/10.3390/ani12151946
Chicago/Turabian StyleWawrzyniak, Agata, and Krzysztof Balawender. 2022. "Structural and Metabolic Changes in Bone" Animals 12, no. 15: 1946. https://doi.org/10.3390/ani12151946
APA StyleWawrzyniak, A., & Balawender, K. (2022). Structural and Metabolic Changes in Bone. Animals, 12(15), 1946. https://doi.org/10.3390/ani12151946