
Faecal Microbiome Transplantation as a Solution to Chronic Enteropathies in Dogs: A Case Study of Beneficial Microbial Evolution
 , , , , ,
, , , , , 
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Case History
2.2. Faecal Microbiota Transplantation
2.3. Control Population
2.4. Microbiota Analysis
2.4.1. Sample Collection and DNA Extraction for Microbiota Analysis
2.4.2. 16S rRNA Gene Amplicon Sequencing
2.4.3. Bioinformatic Analysis
3. Results
3.1. Clinical Outcomes
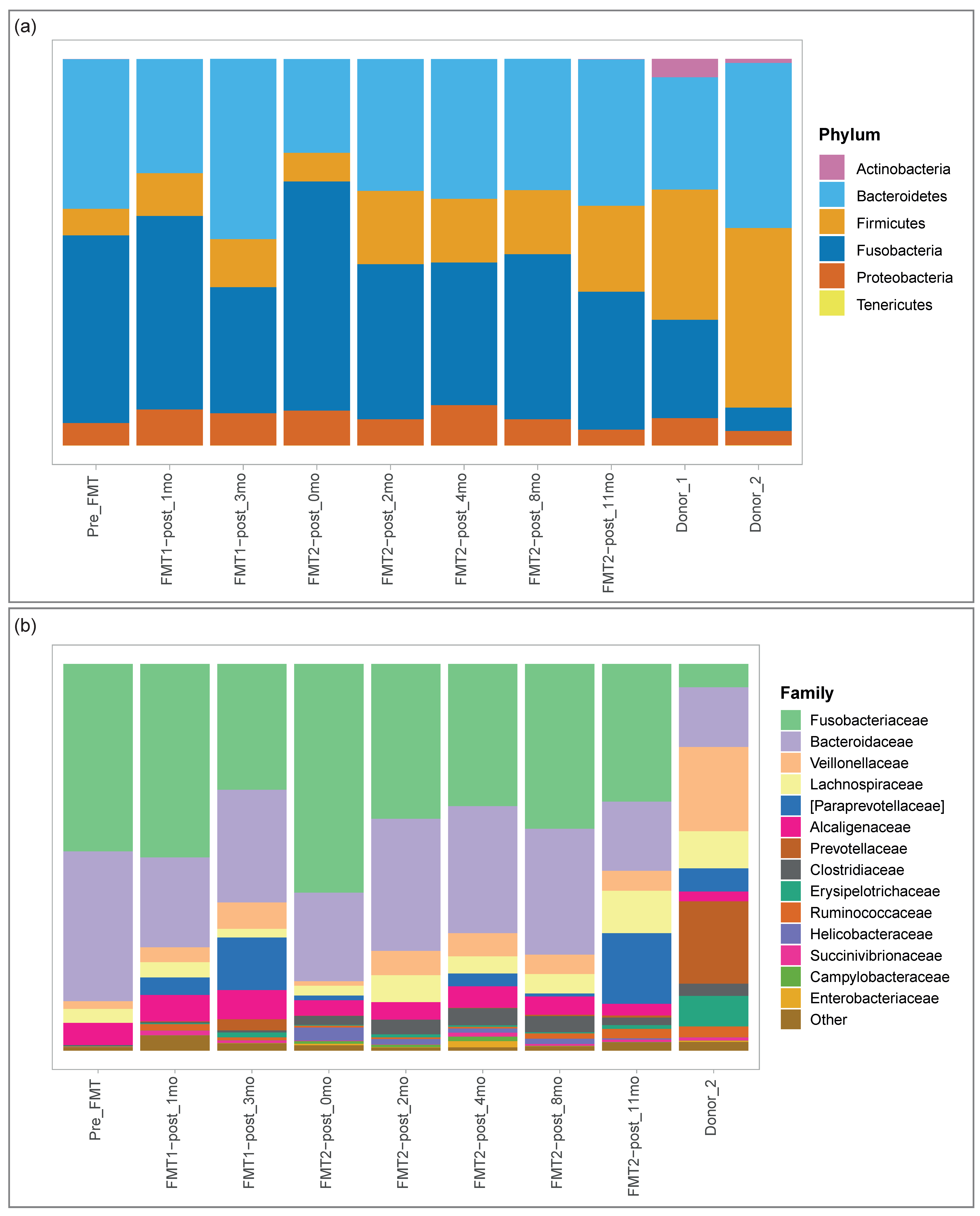
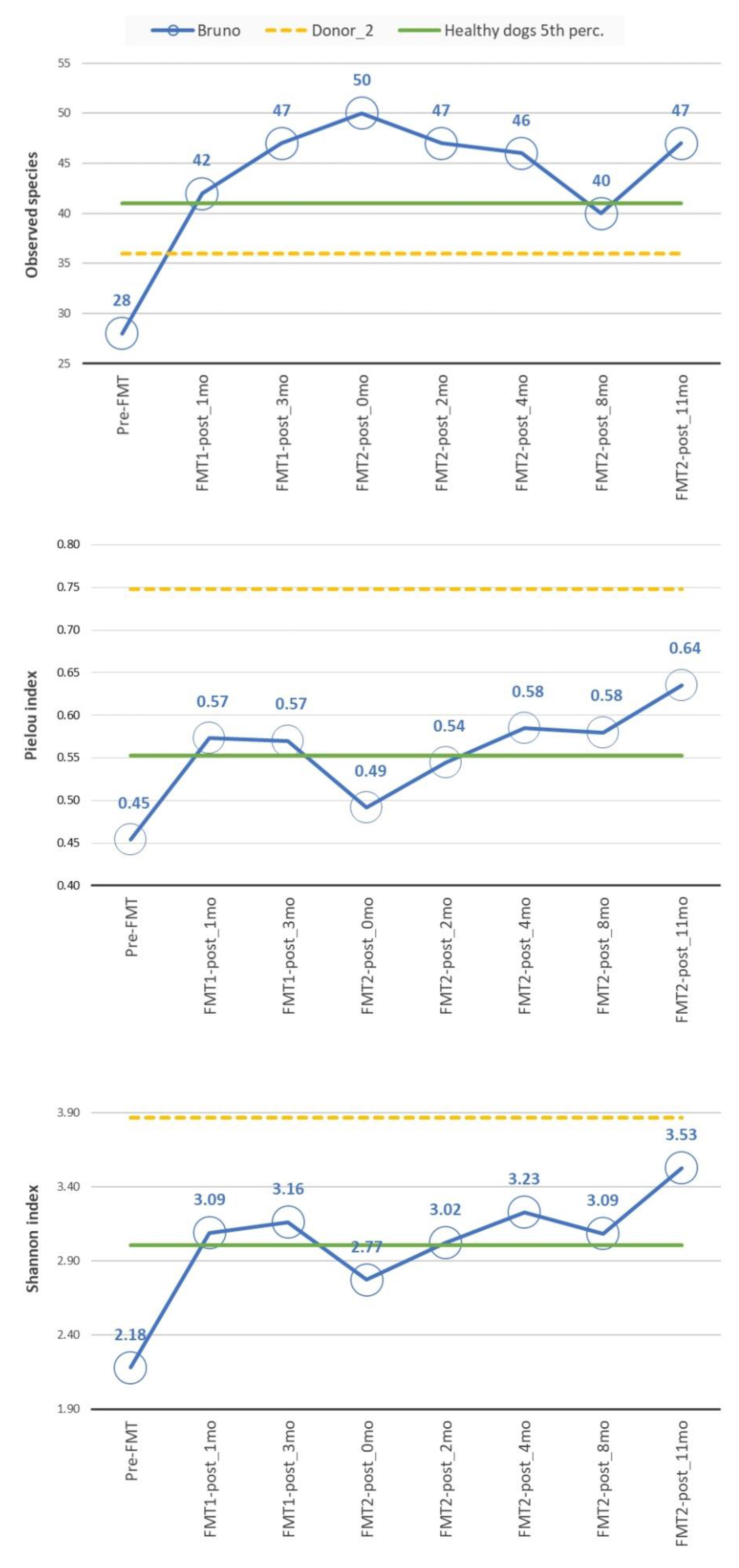
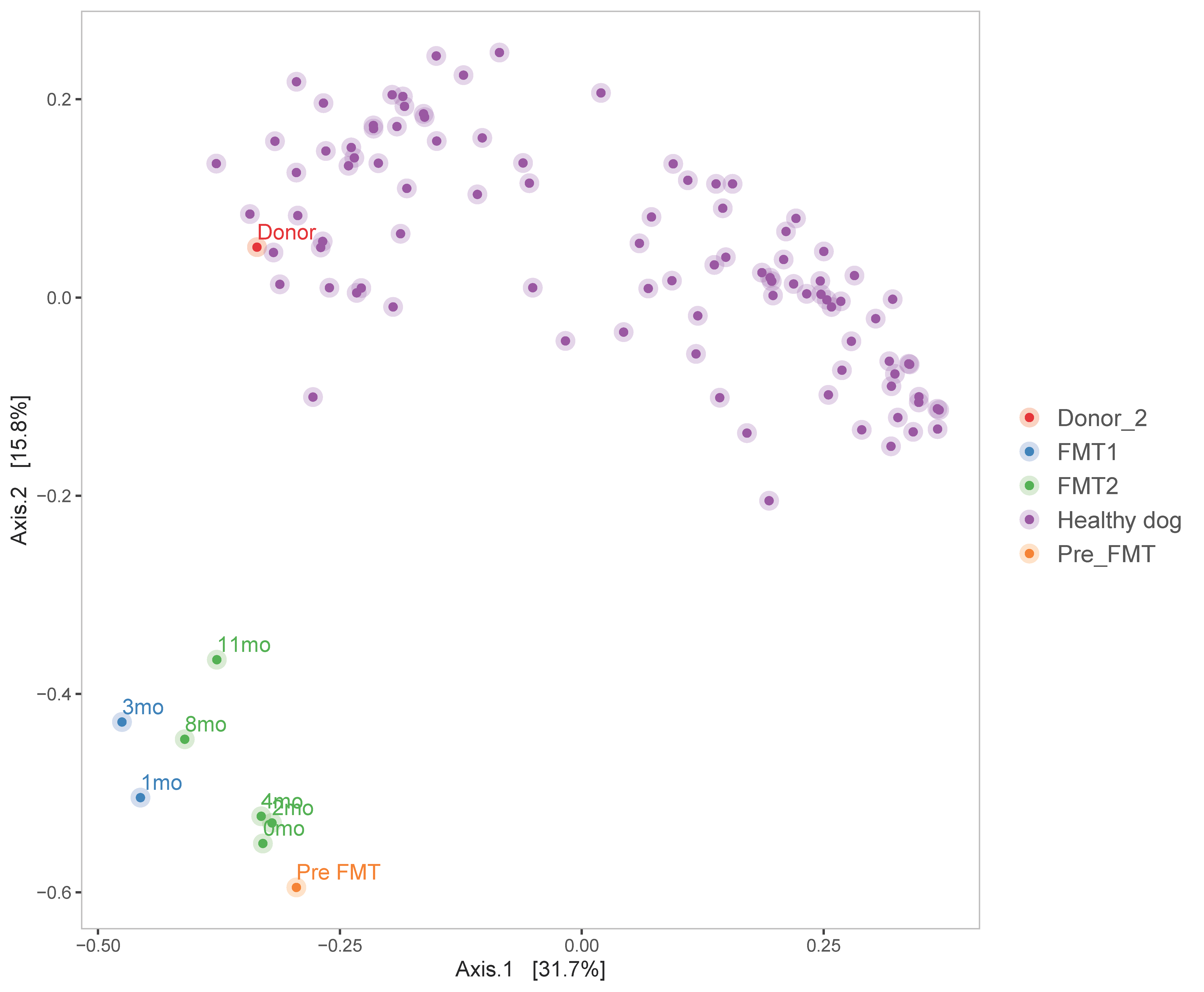
3.2. Microbiome Analysis
3.2.1. Microbial Composition
3.2.2. Diversity Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Dandrieux, J.R.S. Inflammatory bowel disease versus chronic enteropathy in dogs: Are they one and the same? J. Small Anim. Pr. 2016, 57, 589–599. [Google Scholar] [CrossRef]
- Pilla, R.; Suchodolski, J.S. The Role of the Canine Gut Microbiome and Metabolome in Health and Gastrointestinal Disease. Front. Veter. Sci. 2019, 6, 498. [Google Scholar] [CrossRef]
- Suchodolski, J.S.; Markel, M.E.; Garcia-Mazcorro, J.; Unterer, S.; Heilmann, R.M.; Dowd, S.E.; Kachroo, P.; Ivanov, I.; Minamoto, Y.; Dillman, E.M.; et al. The Fecal Microbiome in Dogs with Acute Diarrhea and Idiopathic Inflammatory Bowel Disease. PLoS ONE 2012, 7, e51907. [Google Scholar] [CrossRef]
- Honneffer, J.B.; Minamoto, Y.; Suchodolski, J.S. Microbiota alterations in acute and chronic gastrointestinal inflammation of cats and dogs. World J. Gastroenterol. 2014, 20, 16489–16497. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, U.C.; Nehra, A.; Mathur, A.; Rai, S. A meta-analysis on small intestinal bacterial overgrowth in patients with different subtypes of irritable bowel syndrome. J. Gastroenterol. Hepatol. 2020, 35, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Minamoto, Y.; Otoni, C.C.; Steelman, S.M.; Büyükleblebici, O.; Steiner, J.M.; Jergens, A.E.; Suchodolski, J.S. Alteration of the fecal microbiota and serum metabolite profiles in dogs with idiopathic inflammatory bowel disease. Gut Microbes 2015, 6, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Ziese, A.-L.; Suchodolski, J.S. Impact of Changes in Gastrointestinal Microbiota in Canine and Feline Digestive Diseases. Veter. Clin. North. Am. Small Anim. Pr. 2021, 51, 155–169. [Google Scholar] [CrossRef]
- Redfern, A.; Suchodolski, J.; Jergens, A. Role of the gastrointestinal microbiota in small animal health and disease. Veter. Rec. 2017, 181, 370. [Google Scholar] [CrossRef]
- Wang, S.; Martins, R.; Sullivan, M.C.; Friedman, E.S.; Misic, A.M.; El-Fahmawi, A.; de Martinis, E.C.P.; O’Brien, K.; Chen, Y.; Bradley, C.; et al. Diet-induced remission in chronic enteropathy is associated with altered microbial community structure and synthesis of secondary bile acids. Microbiome 2019, 7, 126. [Google Scholar] [CrossRef]
- Makielski, K.; Cullen, J.; O’Connor, A.; Jergens, A.E. Narrative review of therapies for chronic enteropathies in dogs and cats. J. Veter. Intern. Med. 2019, 33, 11–22. [Google Scholar] [CrossRef]
- Khoruts, A.; Sadowsky, M.J. Understanding the mechanisms of faecal microbiota transplantation. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Borody, T.J.; Eslick, G.D.; Clancy, R.L. Fecal microbiota transplantation as a new therapy: From Clostridioides difficile infection to inflammatory bowel disease, irritable bowel syndrome, and colon cancer. Curr. Opin. Pharmacol. 2019, 49, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Mullen, K.R.; Yasuda, K.; Divers, T.J.; Weese, J.S. Equine faecal microbiota transplant: Current knowledge, proposed guidelines and future directions. Equine Veter. Educ. 2018, 30, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Feary, D.J.; Hassel, D.M. Enteritis and Colitis in Horses. Veter. Clin. N. Am. Equine Pr. 2006, 22, 437–479. [Google Scholar] [CrossRef] [PubMed]
- McKinney, C.A.; Oliveira, B.C.M.; Bedenice, D.; Paradis, M.-R.; Mazan, M.; Sage, S.; Sanchez, A.; Widmer, G. The fecal microbiota of healthy donor horses and geriatric recipients undergoing fecal microbial transplantation for the treatment of diarrhea. PLoS ONE 2020, 15, e0230148. [Google Scholar] [CrossRef] [PubMed]
- Chaitman, J.; Gaschen, F. Fecal Microbiota Transplantation in Dogs. Veter. Clin. N. Am. Small Anim. Pr. 2021, 51, 219–233. [Google Scholar] [CrossRef]
- Burton, E.N.; O’Connor, E.; Ericsson, A.C.; Franklin, C.L. Evaluation of Fecal Microbiota Transfer as Treatment for Postweaning Diarrhea in Research-Colony Puppies. J. Am. Assoc. Lab. Anim. Sci. 2016, 55, 582–587. [Google Scholar]
- Pereira, G.Q.; Gomes, L.A.; Santos, I.S.; Alfieri, A.F.; Weese, J.S.; Costa, M.C. Fecal microbiota transplantation in puppies with canine parvovirus infection. J. Veter. Intern. Med. 2018, 32, 707–711. [Google Scholar] [CrossRef]
- Chaitman, J.; Ziese, A.-L.; Pilla, R.; Minamoto, Y.; Blake, A.B.; Guard, B.C.; Isaiah, A.; Lidbury, J.A.; Steiner, J.M.; Unterer, S.; et al. Fecal Microbial and Metabolic Profiles in Dogs with Acute Diarrhea Receiving Either Fecal Microbiota Transplantation or Oral Metronidazole. Front. Veter. Sci. 2020, 7, 192. [Google Scholar] [CrossRef]
- Bottero, E.; Benvenuti, E.; Ruggiero, P. Trapianto del microbiota fecale (FMT) in 16 cani affetti da IBD idiopatica. Veterinaria 2017, 31, 31–45. [Google Scholar]
- Chaitman, J.; Guard, B.; Sarwar, F.; Lidbury, J.; Steiner, J.; Suchodolski, J. Fecal microbial transplantation decreases the dysbiosis index in dogs presenting with chronic diarrhea. J. Vet. Intern. Med. 2017, 31, 1287. [Google Scholar]
- Niina, A.; Kibe, R.; Suzuki, R.; Yuchi, Y.; Teshima, T.; Matsumoto, H.; Kataoka, Y.; Koyama, H. Improvement in Clinical Symptoms and Fecal Microbiome After Fecal Microbiota Transplantation in a Dog with Inflammatory Bowel Disease. Veter. Med. Res. Rep. 2019, 10, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Sugita, K.; Yanuma, N.; Ohno, H.; Takahashi, K.; Kawano, K.; Morita, H.; Ohmori, K. Oral faecal microbiota transplantation for the treatment of Clostridium difficile-associated diarrhoea in a dog: A case report. BMC Veter. Res. 2019, 15, 1–4. [Google Scholar] [CrossRef]
- Weese, J.; Costa, M.; Webb, J. GI-28 Preliminary clinical and microbiome assessment of stool transplantation in the dog and the cat [abstract]. J. Vet. Intern. Med. 2013, 27, 604–756. [Google Scholar]
- Murphy, T.; Chaitman, J.; Han, E. Use of fecal transplant in eight dogs with refractory Clostridium perfringens associated diarrhea [abstract]. J. Vet. Intern. Med. 2014, 28, 976–1134. [Google Scholar]
- Burchell, R.K.; Pazzi, P.; Biggs, P.J. Others Faecal microbial transplantation in a canine model of haemorrhagic diarrhoea syndrome. J. Vet. Intern. Med. 2019, 33, 1026–1027. [Google Scholar]
- Dwyer, E.L.; Marclay, M.; Suchodolski, J. Others Effect of fecal microbiota transplantation on the fecal microbiome of healthy dogs treated with antibiotics. J. Vet. Intern. Med. 2019, 33, 2467. [Google Scholar]
- Dailey, F.E.; Turse, E.P.; Daglilar, E.; Tahan, V. The dirty aspects of fecal microbiota transplantation: A review of its adverse effects and complications. Curr. Opin. Pharmacol. 2019, 49, 29–33. [Google Scholar] [CrossRef]
- Quera, R.; Espinoza, R.; Estay, C.; Rivera, D. Bacteremia as an adverse event of fecal microbiota transplantation in a patient with Crohn’s disease and recurrent Clostridium difficile infection. J. Crohn’s Coliti 2014, 8, 252–253. [Google Scholar] [CrossRef]
- Baxter, M.; Colville, A. Adverse events in faecal microbiota transplant: A review of the literature. J. Hosp. Infect. 2016, 92, 117–127. [Google Scholar] [CrossRef]
- De Filipp, Z.; Bloom, P.P.; Soto, M.T.; Mansour, M.K.; Sater, M.R.; Huntley, M.H.; Turbett, S.; Chung, R.T.; Chen, Y.-B.; Hohmann, E.L. Drug-Resistant E. coli Bacteremia Transmitted by Fecal Microbiota Transplant. N. Engl. J. Med. 2019, 381, 2043–2050. [Google Scholar] [CrossRef]
- Cammarota, G.; Ianiro, G.; Tilg, H.; Rajilić-Stojanović, M.; Kump, P.; Satokari, R.; Sokol, H.; Arkkila, P.; Pintus, C.; Hart, A.; et al. European consensus conference on faecal microbiota transplantation in clinical practice. Gut 2017, 66, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Scarsella, E.; Stefanon, B.; Cintio, M.; Licastro, D.; Sgorlon, S.; Monego, S.D.; Sandri, M. Learning machine approach reveals microbial signatures of diet and sex in dog. PLoS ONE 2020, 15, e0237874. [Google Scholar] [CrossRef]
- Takahashi, S.; Tomita, J.; Nishioka, K.; Hisada, T.; Nishijima, M. Development of a Prokaryotic Universal Primer for Simultaneous Analysis of Bacteria and Archaea Using Next-Generation Sequencing. PLoS ONE 2014, 9, e105592. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- De Santis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Reeve, J.; Zhang, L.; Huang, S.; Wang, X.; Chen, J. GMPR: A robust normalization method for zero-inflated count data with application to microbiome sequencing data. PeerJ 2018, 6, e4600. [Google Scholar] [CrossRef]
- Risely, A. Applying the core microbiome to understand host–microbe systems. J. Anim. Ecol. 2020, 89, 1549–1558. [Google Scholar] [CrossRef]
- Suchodolski, J.S.; Dowd, S.E.; Wilke, V.; Steiner, J.M.; Jergens, A.E. 16S rRNA Gene Pyrosequencing Reveals Bacterial Dysbiosis in the Duodenum of Dogs with Idiopathic Inflammatory Bowel Disease. PLoS ONE 2012, 7, e39333. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef]
- Minamoto, Y.; Minamoto, T.; Isaiah, A.; Sattasathuchana, P.; Buono, A.; Rangachari, V.R.; McNeely, I.H.; Lidbury, J.; Steiner, J.M.; Suchodolski, J.S. Fecal short-chain fatty acid concentrations and dysbiosis in dogs with chronic enteropathy. J. Veter. Intern. Med. 2019, 33, 1608–1618. [Google Scholar] [CrossRef]
- Vital, M.; Gao, J.; Rizzo, M.; Harrison, T.; Tiedje, J.M. Diet is a major factor governing the fecal butyrate-producing community structure across Mammalia, Aves and Reptilia. ISME J. 2015, 9, 832–843. [Google Scholar] [CrossRef] [PubMed]
- Leipig-Rudolph, M.; Busch, K.; Prescott, J.F.; Gohari, I.M.; Leutenegger, C.M.; Hermanns, W.; Wolf, G.; Hartmann, K.; Verspohl, J.; Unterer, S. Intestinal lesions in dogs with acute hemorrhagic diarrhea syndrome associated with netF-positive Clostridium perfringens type A. J. Veter. Diagn. Investig. 2018, 30, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Herstad, K.M.V.; Gajardo, K.; Bakke, A.M.; Moe, L.; Ludvigsen, J.; Rudi, K.; Rud, I.; Sekelja, M.; Skancke, E. A diet change from dry food to beef induces reversible changes on the faecal microbiota in healthy, adult client-owned dogs. BMC Veter. Res. 2017, 13, 147. [Google Scholar] [CrossRef]
- Guard, B.C.; Honneffer, J.B.; Jergens, A.E.; Jonika, M.M.; Toresson, L.; Lawrence, Y.A.; Webb, C.B.; Hill, S.; Lidbury, J.A.; Steiner, J.M.; et al. Longitudinal assessment of microbial dysbiosis, fecal unconjugated bile acid concentrations, and disease activity in dogs with steroid-responsive chronic inflammatory enteropathy. J. Veter. Intern. Med. 2019, 33, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Giaretta, P.R.; Rech, R.R.; Guard, B.C.; Blake, A.B.; Blick, A.K.; Steiner, J.M.; Lidbury, J.A.; Cook, A.K.; Hanifeh, M.; Spillmann, T.; et al. Comparison of intestinal expression of the apical sodium-dependent bile acid transporter between dogs with and without chronic inflammatory enteropathy. J. Veter. Intern. Med. 2018, 32, 1918–1926. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Baeza, Y.; Hyde, E.R.; Suchodolski, J.S.; Knight, R. Dog and human inflammatory bowel disease rely on overlapping yet distinct dysbiosis networks. Nat. Microbiol. 2016, 1, 16177. [Google Scholar] [CrossRef] [PubMed]
- Buffie, C.G.; Bucci, V.; Stein, R.R.; McKenney, P.T.; Ling, L.; Gobourne, A.; No, D.; Liu, H.; Kinnebrew, M.A.; Viale, A.; et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 2015, 517, 205–208. [Google Scholar] [CrossRef]
- Moon, C.D.; Young, W.; MacLean, P.H.; Cookson, A.L.; Bermingham, E.N. Metagenomic insights into the roles ofProteobacteriain the gastrointestinal microbiomes of healthy dogs and cats. Microbiologyopen 2018, 7, e00677. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berlanda, M.; Innocente, G.; Simionati, B.; Di Camillo, B.; Facchin, S.; Giron, M.C.; Savarino, E.; Sebastiani, F.; Fiorio, F.; Patuzzi, I. Faecal Microbiome Transplantation as a Solution to Chronic Enteropathies in Dogs: A Case Study of Beneficial Microbial Evolution. Animals 2021, 11, 1433. https://doi.org/10.3390/ani11051433
Berlanda M, Innocente G, Simionati B, Di Camillo B, Facchin S, Giron MC, Savarino E, Sebastiani F, Fiorio F, Patuzzi I. Faecal Microbiome Transplantation as a Solution to Chronic Enteropathies in Dogs: A Case Study of Beneficial Microbial Evolution. Animals. 2021; 11(5):1433. https://doi.org/10.3390/ani11051433
Chicago/Turabian StyleBerlanda, Michele, Giada Innocente, Barbara Simionati, Barbara Di Camillo, Sonia Facchin, Maria Cecilia Giron, Edoardo Savarino, Federico Sebastiani, Francesca Fiorio, and Ilaria Patuzzi. 2021. "Faecal Microbiome Transplantation as a Solution to Chronic Enteropathies in Dogs: A Case Study of Beneficial Microbial Evolution" Animals 11, no. 5: 1433. https://doi.org/10.3390/ani11051433
APA StyleBerlanda, M., Innocente, G., Simionati, B., Di Camillo, B., Facchin, S., Giron, M. C., Savarino, E., Sebastiani, F., Fiorio, F., & Patuzzi, I. (2021). Faecal Microbiome Transplantation as a Solution to Chronic Enteropathies in Dogs: A Case Study of Beneficial Microbial Evolution. Animals, 11(5), 1433. https://doi.org/10.3390/ani11051433