
Detection of the Core Bacteria in Colostrum and Their Association with the Rectal Microbiota and with Milk Composition in Two Dairy Cow Farms


Abstract
Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Study Design and Study Population
2.2. Collection of Colostrum and Feces Samples
2.3. Extraction of Genomic DNA in the Collected Colostrum and Fecal Samples
2.4. The 16S rDNA High-Throughput Sequencing of Microflora in Colostrum and Feces
2.5. Statistical Analyses
3. Results
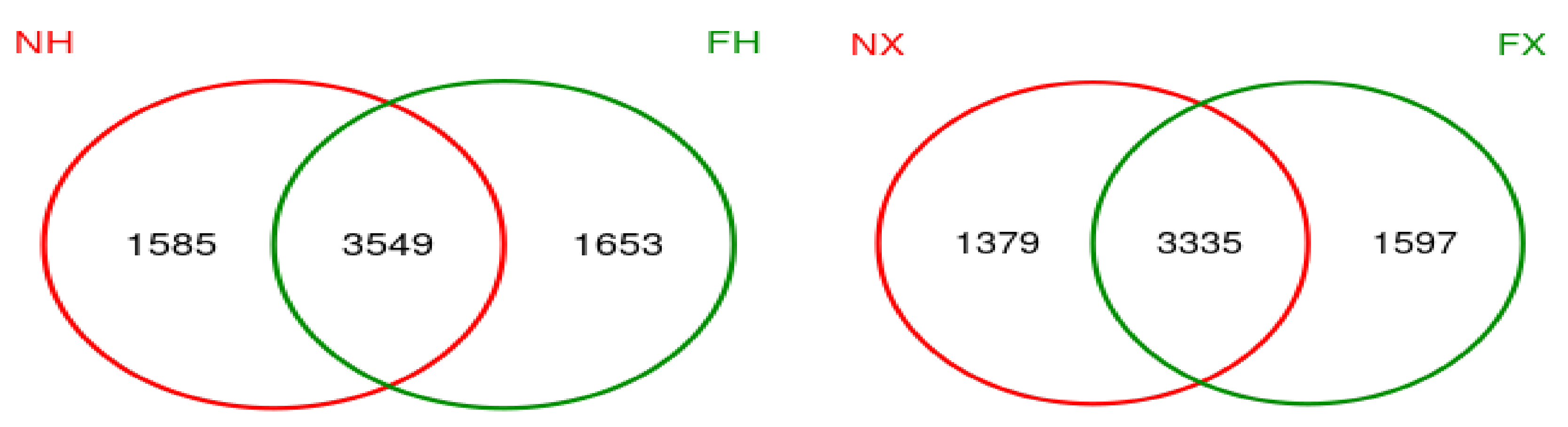
3.1. General DNA Sequencing Observations
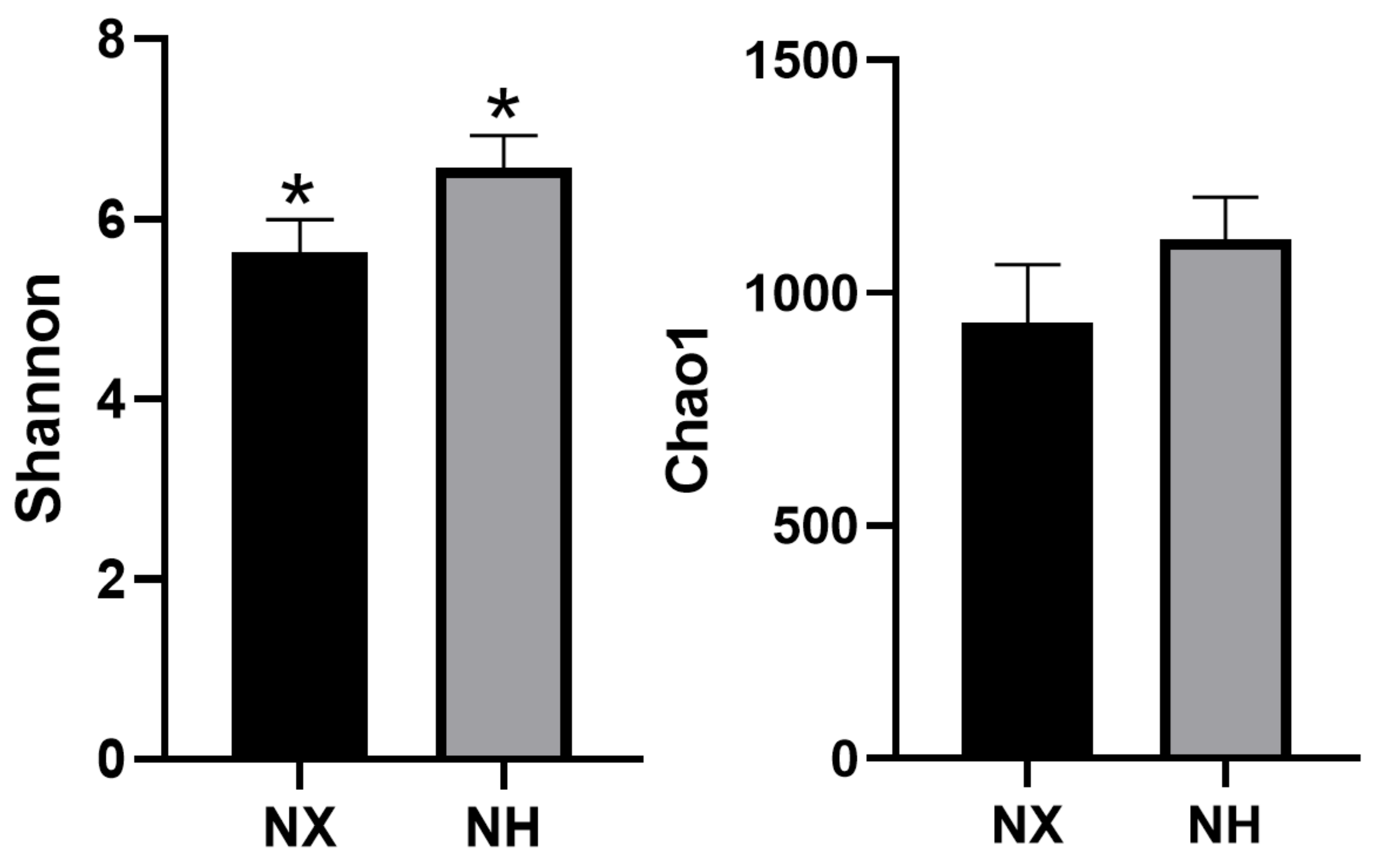
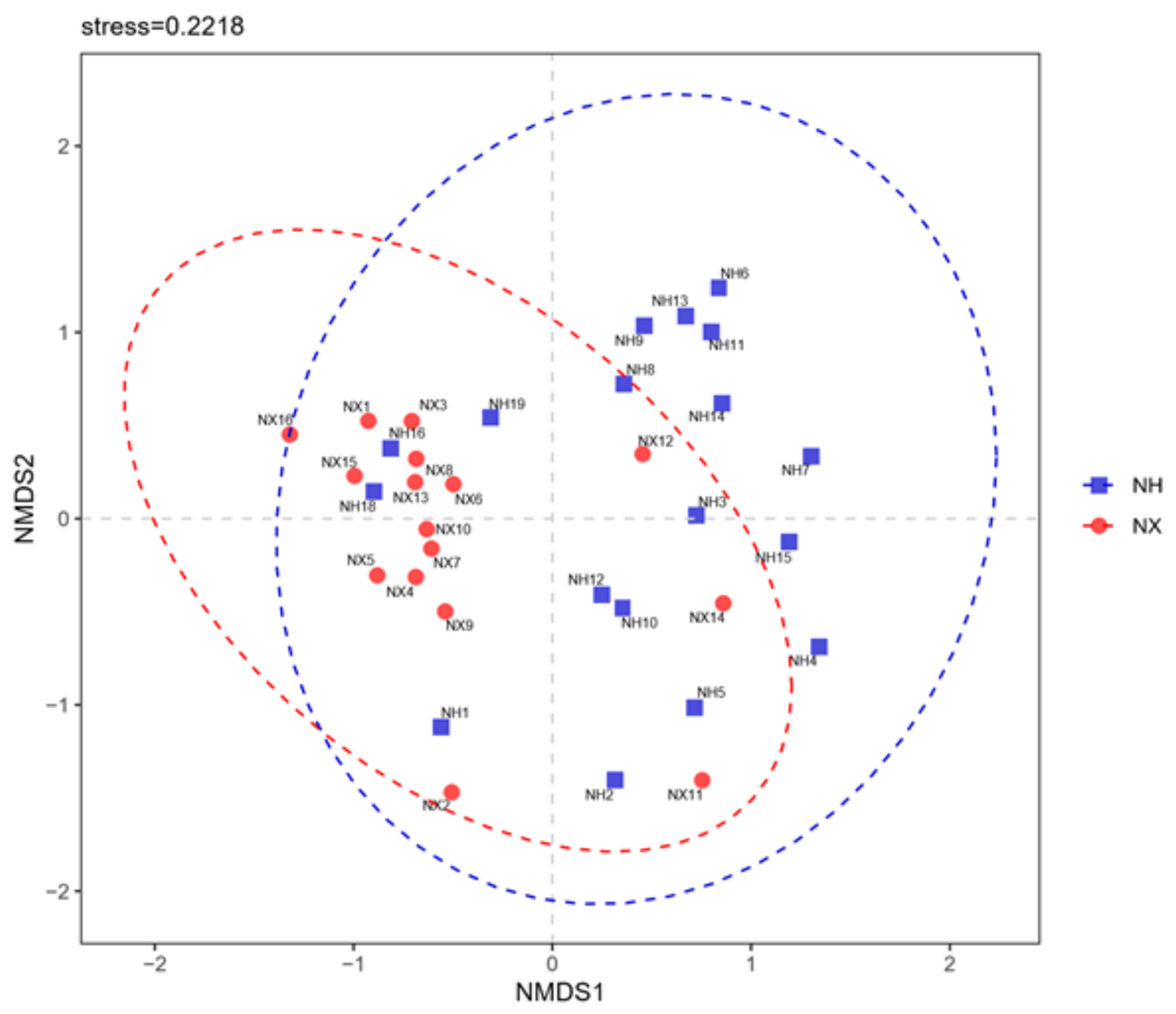
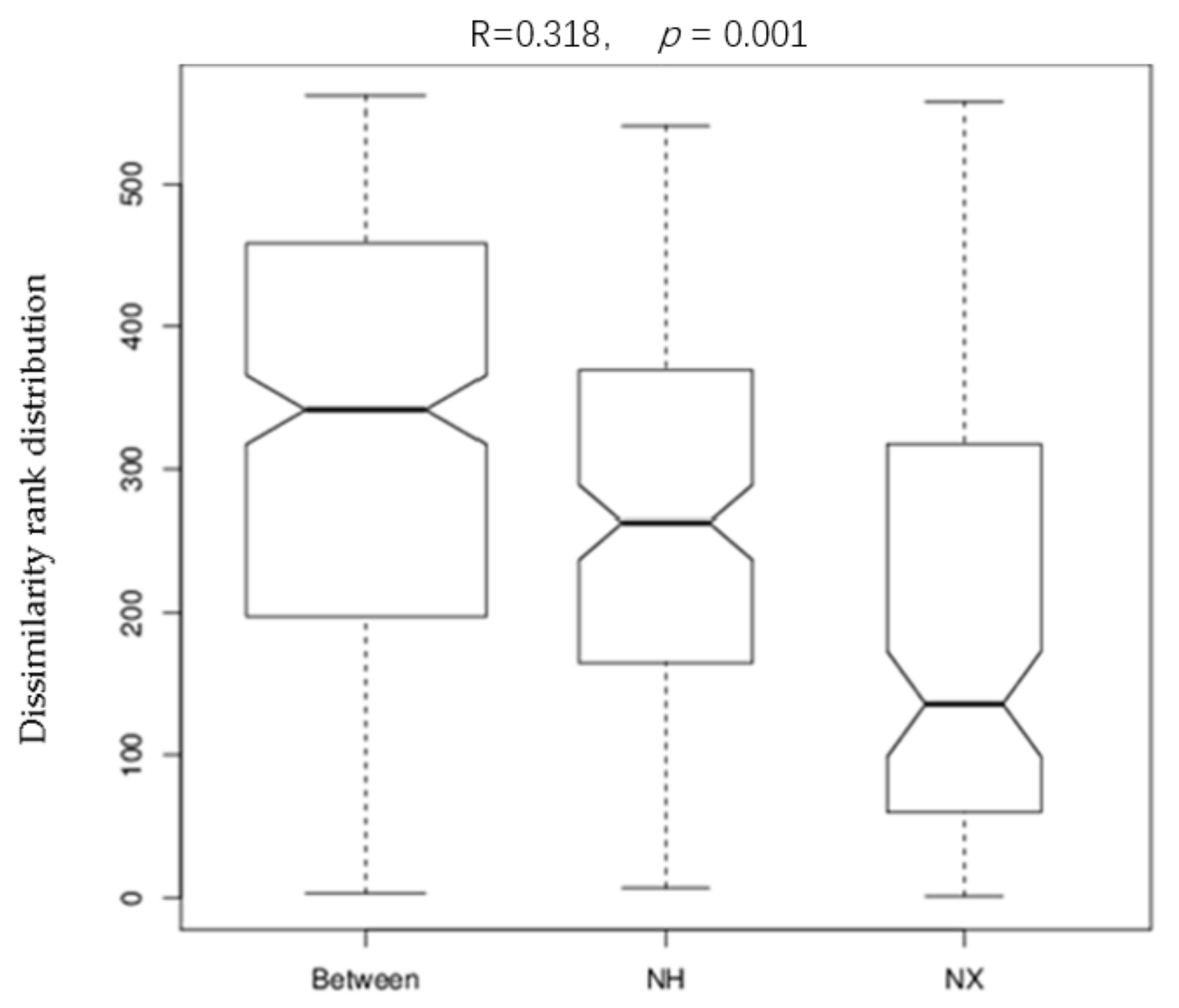
3.2. Alpha and Beta Diversities of Bacterial Composition in Cow Colostrum of the Two Dairy Farms
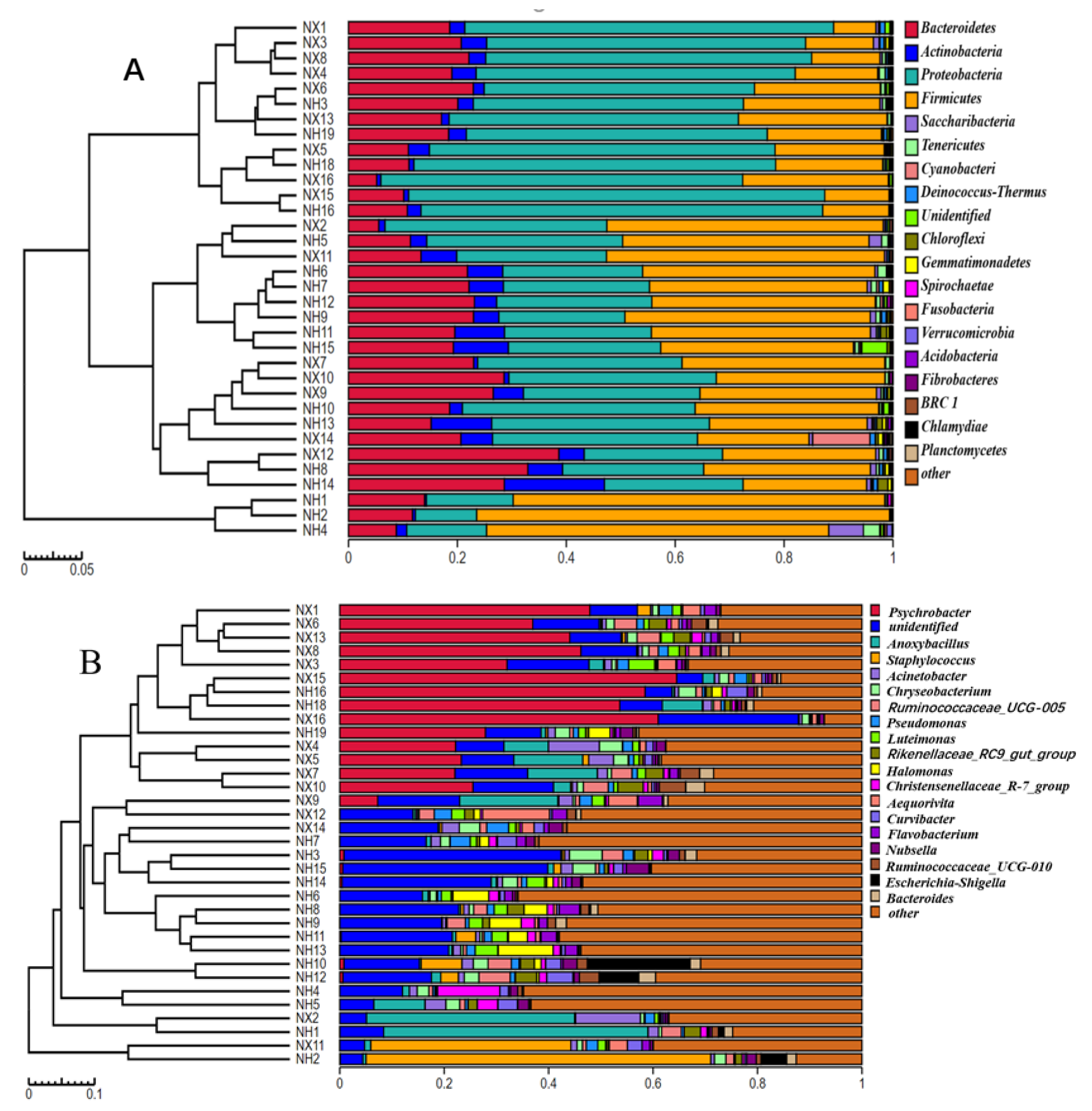
3.3. Taxonomic Analysis of Bacterial Community Structure in Colostrum of the Two Farms
3.4. Analysis of the Core Bacteria in the Collected Colostrum Samples
3.5. Analysis of Commensal Probiotics and Conditional Pathogen in the Colostrum Samples
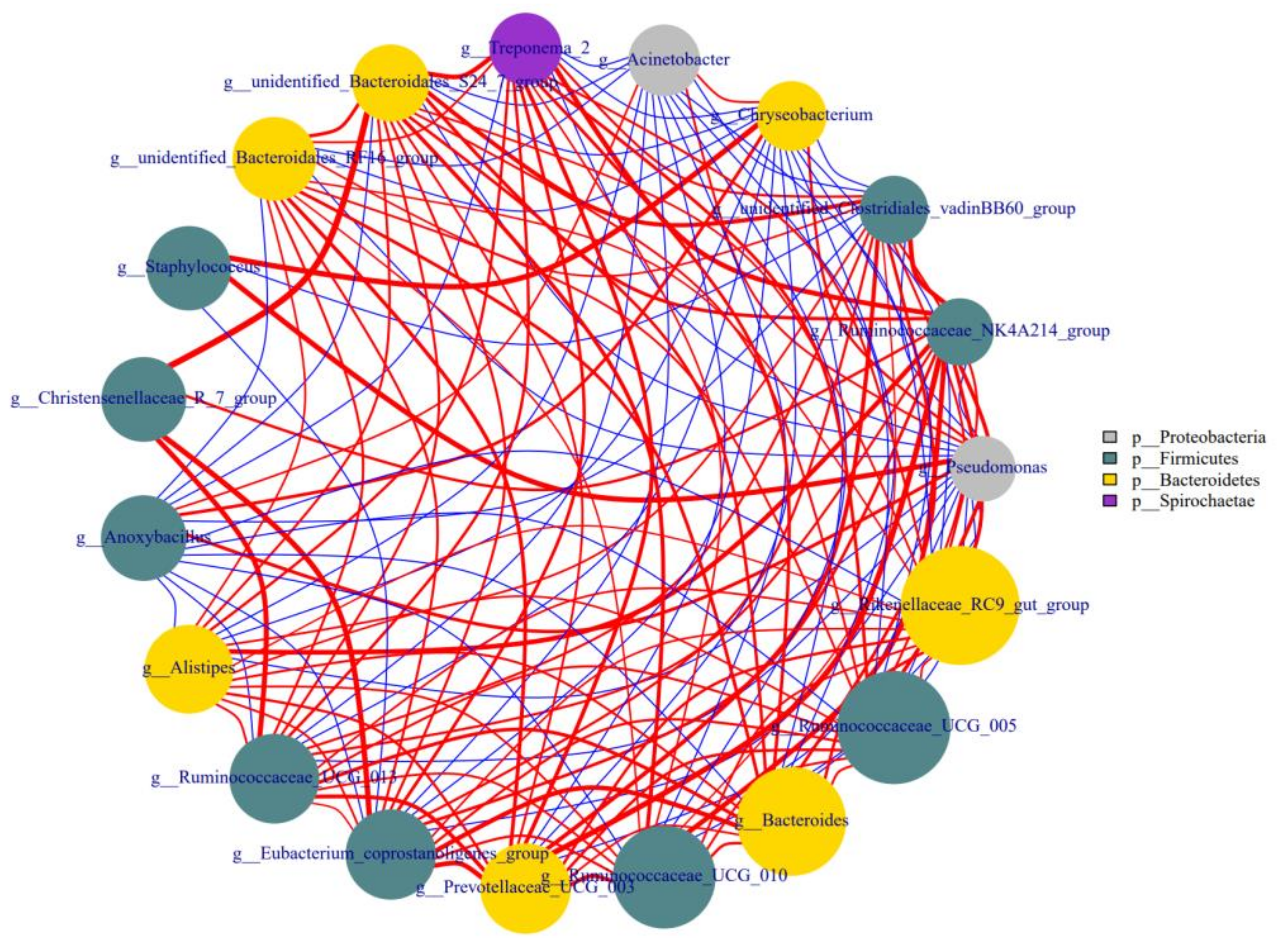
3.6. Correlations among the Different Genera in Colostrum
3.7. Association of the Bacterial Composition in Colostrum and Rectal Feces
3.8. The Correlation between Bacteria and SCC or the Main Composition in Colostrum
4. Discussion
4.1. The Bacterial Compositions in the Colostrum of the Two Dairy Farms Were Different but Relatively Stable at Higher Taxonomic Levels
4.2. Particular Bacteria Existed in the Collected Colostrum and Their Possible Function
4.3. Relation of Bacteria in Colostrum with Bacteria in Rectal Feces
4.4. Correlation between Bacteria and SCC or Other Compositions in Colostrum
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Biswas, P.; Vecchi, A.; Mantegani, P.; Mantelli, B.; Fortis, C.; Lazzarin, A. Immunomodulatory effects of bovine colostrum in human peripheral blood mononuclear cells. New Microbiol. 2007, 30, 447–454. [Google Scholar] [PubMed]
- Van, H.I.; Goossens, K.; Vandaele, L.; Opsomer, G. Invited review: MicroRNAs in bovine colostrum-Focus on their origin and potential health benefits for the calf. J. Dairy Sci. 2020, 103, 1–15. [Google Scholar] [CrossRef]
- Hernández-Castellano, L.E.; Almeida, A.M.; Castro, N.; Argüello, A. The colostrum proteome, ruminant nutrition and immunity: A review. Curr. Protein Pept. Sci. 2014, 15, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Donnet-Hughes, A.; Perez, P.F.; Doré, J.; Leclerc, M.; Levenez, F.; Benyacoub, J.; Serrant, P.; Segura-Roggero, I.; Schiffrin, E.J. Potential role of the intestinal microbiota of the mother in neonatal immune education. Proc. Nutr. Soc. 2010, 69, 407–415. [Google Scholar] [CrossRef]
- Quigley, L.; O’Sullivan, O.; Stanton, C.; Beresford, T.P.; Ross, R.P.; Fitzgerald, G.F.; Cotter, P.D. The complex microbiota of raw milk. FEMS Microbiol. Rev. 2013, 37, 664–698. [Google Scholar] [CrossRef]
- Pannaraj, P.S.; Li, F.; Cerini, C.; Bender, J.M.; Yang, S.; Rollie, A.; Adisetiyo, H.; Zabih, S.; Lincez, P.J.; Bittinger, K.; et al. Association Between Breast Milk Bacterial Communities and Establishment and Development of the Infant Gut Microbiome. JAMA Pediatrics 2017, 171, 647–654. [Google Scholar] [CrossRef]
- Qadis, A.Q.; Goya, S.; Ikuta, K.; Yatsu, M.; Kimura, A.; Nakanishi, S.; Sato, S. Effects of a bacteria-based probiotic on ruminal pH, volatile fatty acids and bacterial flora of Holstein calves. J. Vet. Med. Sci. 2014, 76, 877–885. [Google Scholar] [CrossRef][Green Version]
- Cantor, M.C.; Stanton, A.L.; Combs, D.K.; Costa, J.H.C. Effect of milk feeding strategy and lactic acid probiotics on growth and behavior of dairy calves fed using an automated feeding system. J. Anim. Sci. 2019, 97, 1052–1065. [Google Scholar] [CrossRef]
- Lima, S.F.; Teixeira, A.G.V.; Lima, F.S.; Ganda, E.K.; Higgins, C.H.; Oikonomou, G.; Bicalho, R.C. The bovine colostrum microbiome and its association with clinical mastitis. J. Dairy Sci. 2017, 100, 3031–3042. [Google Scholar] [CrossRef]
- Biscarini, F.; Cremonesi, P.; Castiglioni, B.; Stella, A.; Bronzo, V.; Locatelli, C.; Moroni, P. A Randomized Controlled Trial of Teat-Sealant and Antibiotic Dry-Cow Treatments for Mastitis Prevention Shows Similar Effect on the Healthy Milk Microbiome. Front. Vet. Sci. 2020, 7, 581. [Google Scholar] [CrossRef]
- McGuire, M.K.; McGuire, M.A. Got bacteria? The astounding, yet not-so-surprising, microbiome of human milk. Curr. Opin. Biotechnol. 2017, 44, 63–68. [Google Scholar] [CrossRef]
- Jost, T.; Lacroix, C.; Braegger, C.P.; Rochat, F.; Chassard, C. Vertical mother-neonate transfer of maternal gut bacteria via breastfeeding. Environ. Microbiol. 2014, 16, 2891–2904. [Google Scholar] [CrossRef]
- Rodríguez, J.M. The origin of human milk bacteria: Is there a bacterial entero-mammary pathway during late pregnancy and lactation? Adv. Nutr. 2014, 5, 779–784. [Google Scholar] [CrossRef]
- Addis, M.F.; Tanca, A.; Uzzau, S.; Oikonomou, G.; Bicalho, R.C.; Moroni, P. The bovine milk microbiota: Insights and perspectives from -omics studies. Mol. BioSyst. 2016, 12, 2359–2372. [Google Scholar] [CrossRef]
- Derakhshani, H.; Plaizier, J.C.; De Buck, J.; Barkema, H.W.; Khafipour, E. Association of bovine major histocompatibility complex (BoLA) gene polymorphism with colostrum and milk microbiota of dairy cows during the first week of lactation. Microbiome 2018, 6, 203–221. [Google Scholar] [CrossRef]
- Liu, L.; Xu, X.; Cao, Y.; Cai, C.; Cui, H.; Yao, J. Nitrate decreases methane production also by increasing methane oxidation through stimulating NC10 population in ruminal culture. AMB Express 2017, 7, 76. [Google Scholar] [CrossRef]
- Liu, J.H.; Zhang, M.L.; Zhang, R.Y.; Zhu, W.Y.; Mao, S.Y. Comparative studies of the composition of bacterial microbiota associated with the ruminal content, ruminal epithelium and in the faeces of lactating dairy cows. Microb. Biotechnol. 2016, 9, 257–268. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Li, Q.Q.; Kang, J.M.; Ma, Z.; Li, X.P.; Liu, L.; Hu, X.Z. Microbial succession and metabolite changes during traditional serofluid dish fermentation. LWT-Food Sci. Technol. 2017, 84, 771–779. [Google Scholar] [CrossRef]
- Hang, B.P.T.; Wredle, E.; Dicksved, J. Analysis of the developing gut microbiota in young dairy calves-impact of colostrum microbiota and gut disturbances. Trop. Anim. Health Prod. 2020, 53, 50. [Google Scholar] [CrossRef]
- Lyons, K.E.; Ryan, C.A.; Dempsey, E.M.; Ross, R.P.; Stanton, C. Breast Milk, a Source of Beneficial Microbes and Associated Benefits for Infant Health. Nutrients 2020, 12, 1039. [Google Scholar] [CrossRef]
- Krewinkel, M.; Gosch, M.; Rentschler, E.; Fischer, L. Epilactose production by 2 cellobiose 2-epimerases in natural milk. J. Dairy Sci. 2014, 97, 155–161. [Google Scholar] [CrossRef]
- Watanabe, J.; Nishimukai, M.; Taguchi, H.; Senoura, T.; Hamada, S.; Matsui, H.; Yamamoto, T.; Wasaki, J.; Hara, H.; Ito, S. Prebiotic properties of epilactose. J. Dairy Sci. 2008, 91, 4518–4526. [Google Scholar] [CrossRef]
- Nilsson, Å. Role of Sphingolipids in Infant Gut Health and Immunity. J. Pediatrics 2016, 173, S53–S59. [Google Scholar] [CrossRef]
- Ranc, A.; Dubourg, G.; Fournier, P.E.; Raoult, D.; Fenollar, F. Delftia tsuruhatensis, an Emergent Opportunistic Healthcare-Associated Pathogen. Emerg. Infect. Dis. 2018, 24, 594–596. [Google Scholar] [CrossRef]
- An, S.; Berg, G. Stenotrophomonas maltophilia. Trends Microbiol. 2018, 26, 637–638. [Google Scholar] [CrossRef]
- Stacy, A.; Andrade-Oliveira, V.; McCulloch, J.A.; Hild, B.; Oh, J.H.; Perez-Chaparro, P.J.; Sim, C.K.; Lim, A.I.; Link, V.M.; Enamorado, M.; et al. Infection trains the host for microbiota-enhanced resistance to pathogens. Cell 2021, 184, 615–627.e17. [Google Scholar] [CrossRef]
- Macpherson, A.J.; de Agüero, M.G.; Ganal-Vonarburg, S.C. How nutrition and the maternal microbiota shape the neonatal immune system. Nat. Rev. Immunol. 2017, 17, 508–517. [Google Scholar] [CrossRef]
- Mishra, A.; Lai, G.C.; Yao, L.J.; Aung, T.T.; Shental, N.; Rotter-Maskowitz, A.; Shepherdson, E.; Singh, G.S.N.; Pai, R.; Shanti, A.; et al. Microbial exposure during early human development primes fetal immune cells. Cell 2021, 184, 3394–3409.e20. [Google Scholar] [CrossRef]
- Macpherson, A.J.; Uhr, T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science 2004, 303, 1662–1665. [Google Scholar] [CrossRef]
- Zhao, L.; Ouyang, H.; Zhang, N.; Wang, C.; Ji, B.; Zhou, F. Effects of Huangjiu, Baijiu and Red Wine Combined With High-Fat Diet on Glucose and Lipid Metabolism: Aggravate or Alleviate? Alcohol Alcohol. 2021, 56, 334–347. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.K.; Koo, H.C.; Kim, S.H.; Hwang, S.Y.; Jung, W.K.; Kim, J.M.; Shin, S.; Kim, R.T.; Park, Y.H. The analysis of milk components and pathogenic bacteria isolated from bovine raw milk in Korea. J. Dairy Sci. 2007, 90, 5405–5414. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, U.K.; Kaltwasser, H. Urease from Staphylococcus saprophyticus: Purification, characterization and comparison to Staphylococcus xylosus urease. Arch. Microbiol. 1994, 161, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Bhinderwala, F.; Lehman, M.K.; Thomas, V.C.; Chaudhari, S.S.; Yamada, K.J.; Foster, K.W.; Powers, R.; Kielian, T.; Fey, P.D. Urease is an essential component of the acid response network of Staphylococcus aureus and is required for a persistent murine kidney infection. PLoS Pathog. 2019, 15, e1007538. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of the OTUs | Genus of the Core OTUs Belonged to | NX | NH | SEM | p-Value |
|---|---|---|---|---|---|
| 36 | Acinetobacter | 0.32 | 0.28 | 0.14 | 0.787 |
| 40 | Acinetobacter | 1.32 | 0.62 | 0.70 | 0.323 |
| 58 | Curvibacter | 0.82 | 1.51 | 0.43 | 0.103 |
| 60 | Anoxybacillus | 6.54 | 4.42 | 3.88 | 0.587 |
| 1156 | Chryseobacterium | 1.46 | 1.76 | 0.38 | 0.441 |
| 1161 | Nubsella | 0.78 | 1.38 | 0.32 | 0.062 |
| 2182 | Unidentified genus | 0.18 | 0.50 | 0.17 | 0.065 |
| 2280 | Ruminococcaceae_UCG-005 | 1.18 | 1.27 | 0.43 | 0.837 |
| 3169 | Sphingomonas | 0.37 | 0.58 | 0.18 | 0.256 |
| 3187 | Bradyrhizobium | 0.61 | 0.70 | 0.27 | 0.742 |
| Core Genus | NX | NH | SEM | p-Value |
|---|---|---|---|---|
| Unidentified genus | 12.3 | 17.38 | 2.99 | 0.100 |
| Anoxybacillus | 6.55 | 4.42 | 3.88 | 0.587 |
| Staphylococcus | 2.78 | 4.71 | 0.45 | 0.669 |
| Acinetobacter | 2.65 | 1.41 | 0.86 | 0.192 |
| Pseudomonas | 1.84 a | 1.10 b | 0.33 | 0.032 |
| Ruminococcaceae_UCG-005 | 1.69 | 1.88 | 0.60 | 0.762 |
| Luteimonas | 1.62 | 1.19 | 0.45 | 0.342 |
| Chryseobacterium | 1.59 | 2.13 | 0.48 | 0.269 |
| Flavobacterium | 1.32 | 1.04 | 0.39 | 0.462 |
| Rikenellaceae_RC9_gut_group | 1.30 | 1.41 | 0.47 | 0.829 |
| Pedobacter | 0.92 | 0.62 | 0.36 | 0.415 |
| Bacteroides | 0.89 | 1.02 | 0.35 | 0.701 |
| Curvibacter | 0.82 | 1.51 | 0.43 | 0.103 |
| Nubsella | 0.78 | 1.38 | 0.32 | 0.062 |
| Bacillus | 0.70 | 0.69 | 0.37 | 0.970 |
| Eubacterium_coprostanoligenes_group | 0.68 | 0.86 | 0.27 | 0.513 |
| Bradyrhizobium | 0.61 | 0.70 | 0.27 | 0.742 |
| Christensenellaceae_R-7_group | 0.50 b | 2.01 a | 0.48 | 0.031 |
| Sphingomonas | 0.40 | 0.66 | 0.20 | 0.208 |
| Brevundimonas | 0.36 | 0.42 | 0.17 | 0.727 |
| Paracoccus | 0.20 b | 0.48 a | 0.10 | 0.010 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, B.; Tang, G.; Guo, W.; Lei, J.; Yao, J.; Xu, X. Detection of the Core Bacteria in Colostrum and Their Association with the Rectal Microbiota and with Milk Composition in Two Dairy Cow Farms. Animals 2021, 11, 3363. https://doi.org/10.3390/ani11123363
Chen B, Tang G, Guo W, Lei J, Yao J, Xu X. Detection of the Core Bacteria in Colostrum and Their Association with the Rectal Microbiota and with Milk Composition in Two Dairy Cow Farms. Animals. 2021; 11(12):3363. https://doi.org/10.3390/ani11123363
Chicago/Turabian StyleChen, Bin, Guangfu Tang, Weiqing Guo, Jie Lei, Junhu Yao, and Xiurong Xu. 2021. "Detection of the Core Bacteria in Colostrum and Their Association with the Rectal Microbiota and with Milk Composition in Two Dairy Cow Farms" Animals 11, no. 12: 3363. https://doi.org/10.3390/ani11123363
APA StyleChen, B., Tang, G., Guo, W., Lei, J., Yao, J., & Xu, X. (2021). Detection of the Core Bacteria in Colostrum and Their Association with the Rectal Microbiota and with Milk Composition in Two Dairy Cow Farms. Animals, 11(12), 3363. https://doi.org/10.3390/ani11123363