Population Genetic Analysis of Ten Geographically Isolated Tibetan Pig Populations


 and
and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Read Mapping and SNP Calling
2.3. Phylogenic Tree Construction and PCA
2.4. Genetic Diversity and Deleterious Variants
2.5. Selective Sweeps
3. Results and Discussion
3.1. Population Structure Analysis
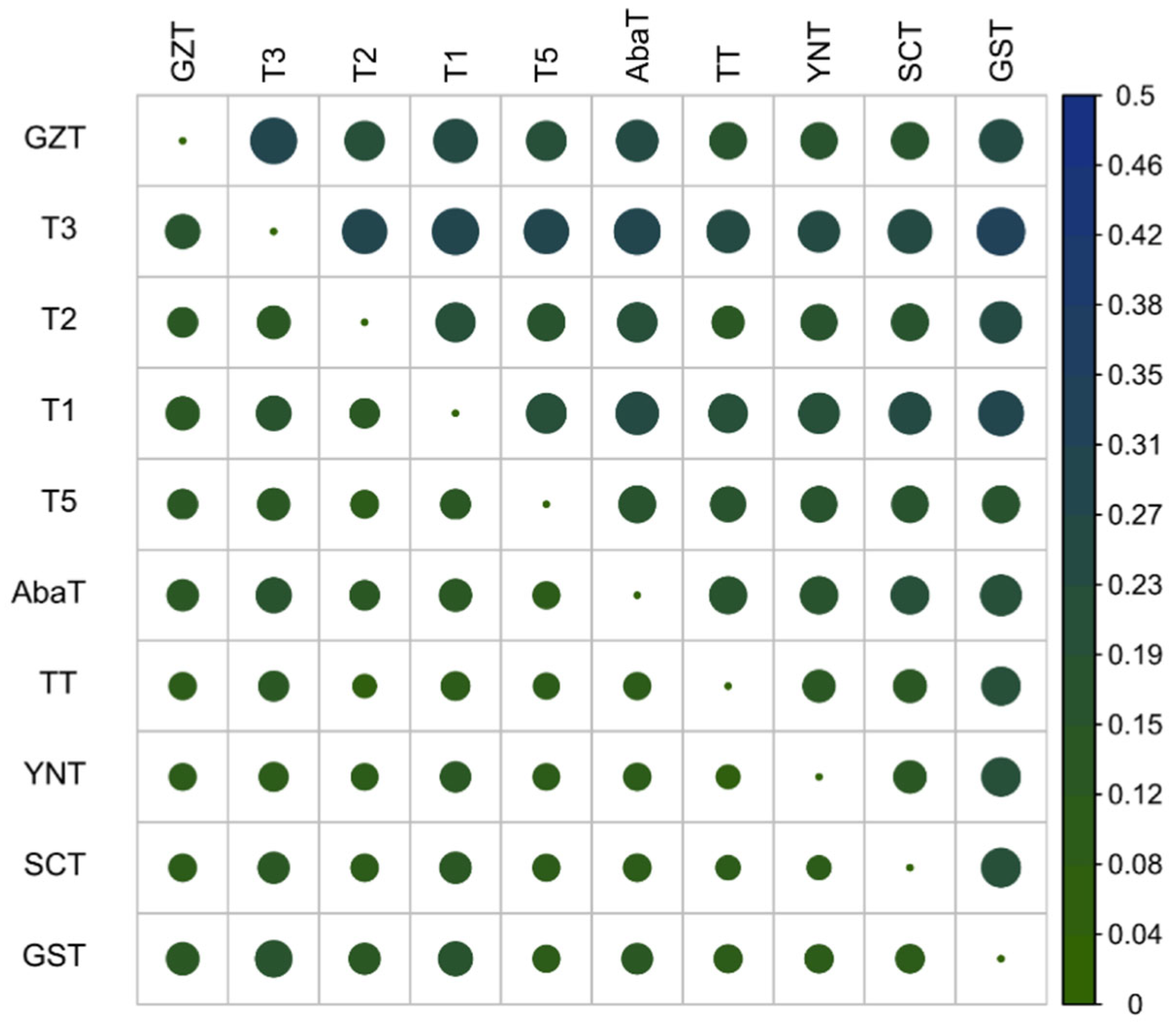
3.2. Genetic Diversity and Differentiation
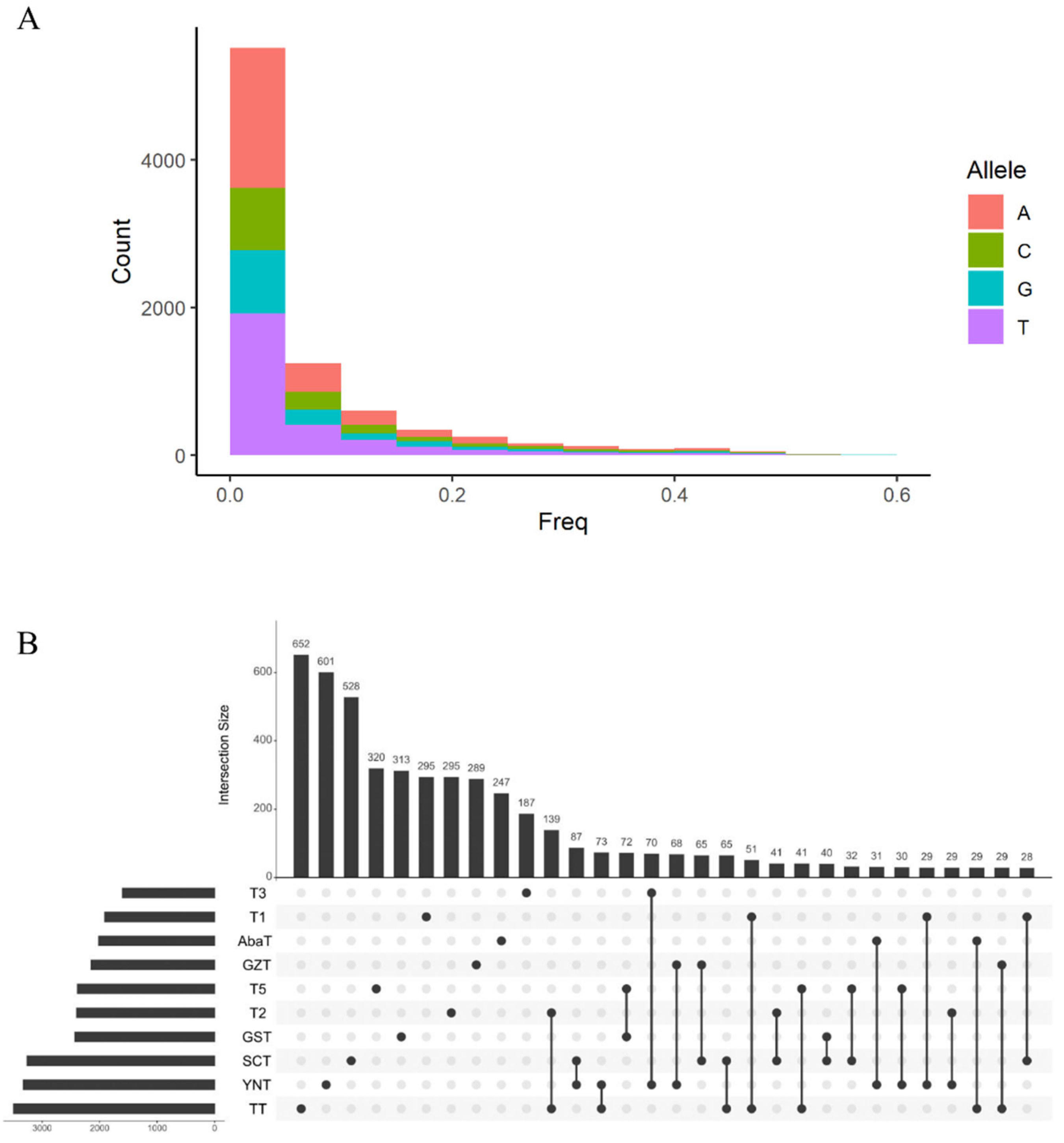
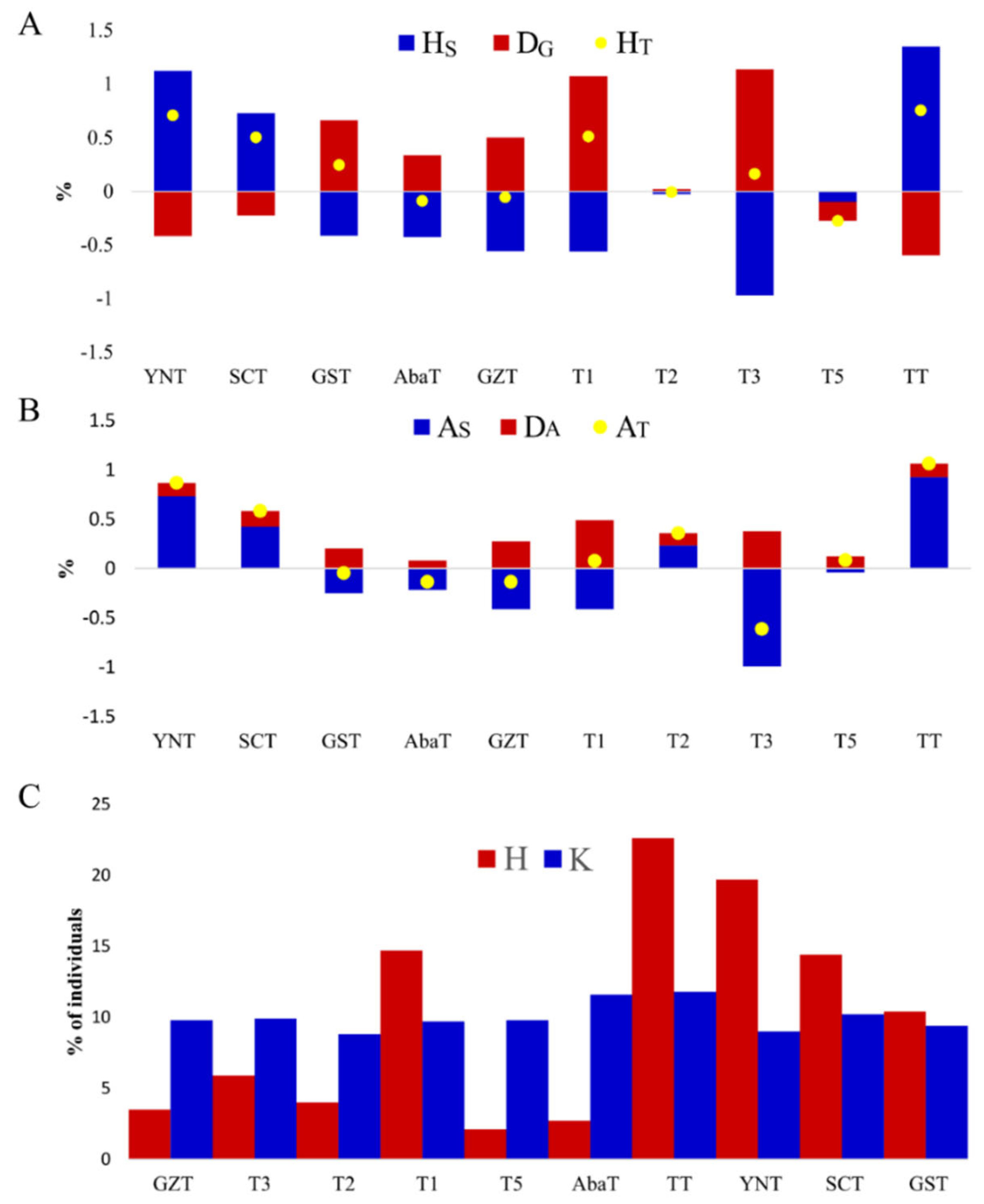
3.3. Deleterious Variants and Their Contribution to Gene and Allele Diversity
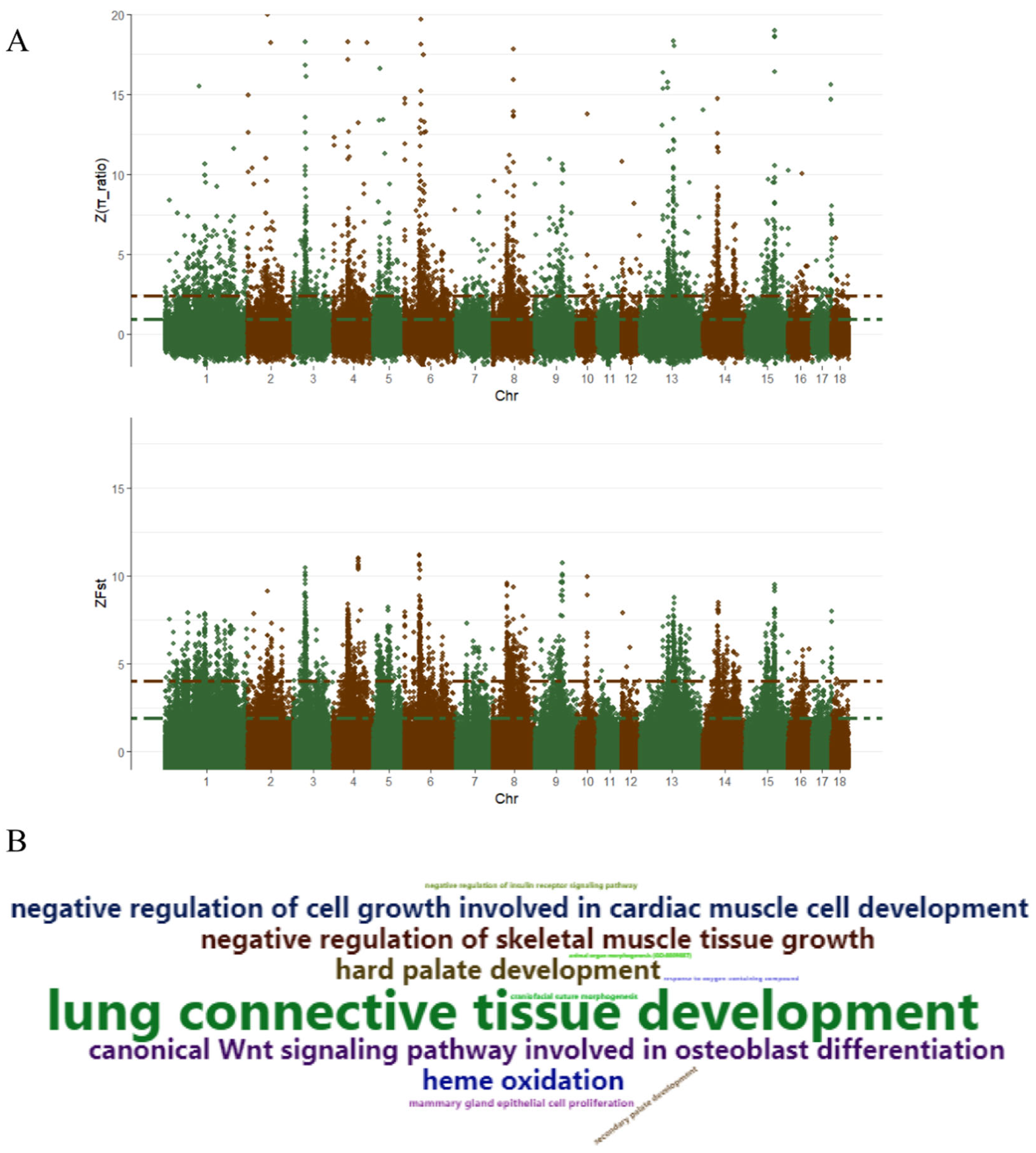
3.4. Selective Sweep
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ma, Y.F.; Han, X.M.; Huang, C.P.; Zhong, L.; Adeola, A.C.; Irwin, D.M.; Xie, H.B.; Zhang, Y.P. Population Genomics Analysis Revealed Origin and High-altitude Adaptation of Tibetan Pigs. Sci. Rep. 2019, 9, 11463. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Qiangba, Y.; Shang, P.; Wang, Z.; Ma, J.; Wang, L.; Zhang, H. A Comprehensive MicroRNA Expression Profile Related to Hypoxia Adaptation in the Tibetan Pig. PLoS ONE 2015, 10, e0143260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chamba, Y.; Shang, P.; Wang, Z.; Ma, J.; Wang, L.; Zhang, H. Comparative transcriptomic and proteomic analyses provide insights into the key genes involved in high-altitude adaptation in the Tibetan pig. Sci. Rep. 2017, 7, 3654. [Google Scholar] [CrossRef] [PubMed]
- Ai, H.; Yang, B.; Li, J.; Xie, X.; Chen, H.; Ren, J. Population history and genomic signatures for high-altitude adaptation in Tibetan pigs. BMC Genom. 2014, 15, 834. [Google Scholar] [CrossRef] [PubMed]
- Shang, P.; Li, W.; Liu, G.; Zhang, J.; Li, M.; Wu, L.; Wang, K.; Chamba, Y. Identification of lncRNAs and Genes Responsible for Fatness and Fatty Acid Composition Traits between the Tibetan and Yorkshire Pigs. Int. J. Genom. 2019, 2019, 5070975. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, H.; Mao, H.; Yan, D.; Lu, S.; Lian, L.; Zhao, G.; Yan, Y.; Deng, W.; Shi, X.; et al. The local origin of the Tibetan pig and additional insights into the origin of Asian pigs. PLoS ONE 2011, 6, e28215. [Google Scholar] [CrossRef]
- Li, M.; Tian, S.; Jin, L.; Zhou, G.; Li, Y.; Zhang, Y.; Wang, T.; Yeung, C.K.; Chen, L.; Ma, J.; et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat. Genet. 2013, 45, 1431–1438. [Google Scholar] [CrossRef]
- Ai, H.; Fang, X.; Yang, B.; Huang, Z.; Chen, H.; Mao, L.; Zhang, F.; Zhang, L.; Cui, L.; He, W.; et al. Adaptation and possible ancient interspecies introgression in pigs identified by whole-genome sequencing. Nat. Genet. 2015, 47, 217–225. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Lopez-Cortegano, E.; Perez-Figueroa, A.; Caballero, A. metapop2: Re-implementation of software for the analysis and management of subdivided populations using gene and allelic diversity. Mol. Ecol. Resour. 2019, 19, 1095–1100. [Google Scholar] [CrossRef]
- Bosse, M.; Megens, H.J.; Madsen, O.; Crooijmans, R.P.; Ryder, O.A.; Austerlitz, F.; Groenen, M.A.; de Cara, M.A. Using genome-wide measures of coancestry to maintain diversity and fitness in endangered and domestic pig populations. Genome Res. 2015, 25, 970–981. [Google Scholar] [CrossRef]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef]
- Lopez-Cortegano, E.; Pouso, R.; Labrador, A.; Perez-Figueroa, A.; Fernandez, J.; Caballero, A. Optimal Management of Genetic Diversity in Subdivided Populations. Front. Genet. 2019, 10, 843. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Li, W.H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 1979, 76, 5269–5273. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef]
- Steffen, W.; Richardson, K.; Rockstrom, J.; Cornell, S.E.; Fetzer, I.; Bennett, E.M.; Biggs, R.; Carpenter, S.R.; de Vries, W.; de Wit, C.A.; et al. Planetary boundaries: Guiding human development on a changing planet. Science 2015, 347, 1259855. [Google Scholar] [CrossRef] [PubMed]
- Frankham, R. Introduction to quantitative genetics (4th edn). Am. J. Hum. Genet. 1996, 12, 280. [Google Scholar] [CrossRef]
- Leberg, P.L. Estimating allelic richness: Effects of sample size and bottlenecks. Mol. Ecol. 2002, 11, 2445–2449. [Google Scholar] [CrossRef]
- Petit, R.J.; Mousadik, A.E.; Pons, O. Identifying Populations for Conservation on the Basis of Genetic Markers. Conserv. Biol. 1998, 12, 844–855. [Google Scholar] [CrossRef]
- Caballero, A.; Toro, M.A. Analysis of genetic diversity for the management of conserved subdivided populations. Conserv. Genet. 2002, 3, 289–299. [Google Scholar] [CrossRef]
- Toro, M.; Pérez-Enciso, M. Optimization of selection response under restricted inbreeding. Genet. Sel. Evol. 1990, 22, 93–107. [Google Scholar] [CrossRef]
- Amaral, A.J.; Megens, H.J.; Crooijmans, R.P.; Heuven, H.C.; Groenen, M.A. Linkage disequilibrium decay and haplotype block structure in the pig. Genetics 2008, 179, 569–579. [Google Scholar] [CrossRef]
- Geetha-Loganathan, P.; Nimmagadda, S.; Antoni, L.; Fu, K.; Whiting, C.J.; Francis-West, P.; Richman, J.M. Expression of WNT signalling pathway genes during chicken craniofacial development. Dev. Dyn. 2009, 238, 1150–1165. [Google Scholar] [CrossRef] [PubMed]
- Demeer, B.; Revencu, N.; Helaers, R.; Gbaguidi, C.; Dakpe, S.; Francois, G.; Devauchelle, B.; Bayet, B.; Vikkula, M. Likely Pathogenic Variants in One Third of Non-Syndromic Discontinuous Cleft Lip and Palate Patients. Genes 2019, 10, 833. [Google Scholar] [CrossRef] [PubMed]
- Paiva, K.B.S.; Maas, C.S.; Dos Santos, P.M.; Granjeiro, J.M.; Letra, A. Extracellular Matrix Composition and Remodeling: Current Perspectives on Secondary Palate Formation, Cleft Lip/Palate, and Palatal Reconstruction. Front. Cell Dev. Biol. 2019, 7, 340. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Larson, G.; Ribeiro, H.S.; Li, N.; Andersson, L. Contrasting mode of evolution at a coat color locus in wild and domestic pigs. PLoS Genet. 2009, 5, e1000341. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, W.; Yang, B.; Zhang, Z.; Ai, H.; Ren, J.; Huang, L. Signatures of Selection and Interspecies Introgression in the Genome of Chinese Domestic Pigs. Genome Biol. Evol. 2017, 9, 2592–2603. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lin, C.; Gleiberman, A.; Ohgi, K.A.; Herman, T.; Huang, H.P.; Tsai, M.J.; Rosenfeld, M.G. Tbx19, a tissue-selective regulator of POMC gene expression. Proc. Natl. Acad. Sci. USA 2001, 98, 8674–8679. [Google Scholar] [CrossRef]
- Baldwin, W.S.; Marko, P.B.; Nelson, D.R. The cytochrome P450 (CYP) gene superfamily in Daphnia pulex. BMC Genom. 2009, 10, 169. [Google Scholar] [CrossRef]
- Saunders, M.A.; Good, J.M.; Lawrence, E.C.; Ferrell, R.E.; Li, W.H.; Nachman, M.W. Human adaptive evolution at Myostatin (GDF8), a regulator of muscle growth. Am. J. Hum. Genet. 2006, 79, 1089–1097. [Google Scholar] [CrossRef]
- Yang, B.; Cui, L.; Perez-Enciso, M.; Traspov, A.; Crooijmans, R.; Zinovieva, N.; Schook, L.B.; Archibald, A.; Gatphayak, K.; Knorr, C.; et al. Genome-wide SNP data unveils the globalization of domesticated pigs. Genet. Sel. Evol. 2017, 49, 71. [Google Scholar] [CrossRef]
- Li, W.T.; Zhang, M.M.; Li, Q.G.; Tang, H.; Zhang, L.F.; Wang, K.J.; Zhu, M.Z.; Lu, Y.F.; Bao, H.G.; Zhang, Y.M.; et al. Whole-genome resequencing reveals candidate mutations for pig prolificacy. Proc. Biol. Sci. 2017, 284. [Google Scholar] [CrossRef]
- Malnic, B.; Godfrey, P.A.; Buck, L.B. The human olfactory receptor gene family. Proc. Natl. Acad. Sci. USA 2004, 101, 2584–2589. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.S.; Yang, H.C.; Otecko, N.O.; Wu, D.D.; Zhang, Y.P. Olfactory genes in Tibetan wild boar. Nat. Genet. 2016, 48, 972–973. [Google Scholar] [CrossRef] [PubMed]
- Bahudhanapati, H.; Tan, J.; Dutta, J.A.; Strock, S.B.; Sembrat, J.; Alvarez, D.; Rojas, M.; Jager, B.; Prasse, A.; Zhang, Y.; et al. MicroRNA-144-3p targets relaxin/insulin-like family peptide receptor 1 (RXFP1) expression in lung fibroblasts from patients with idiopathic pulmonary fibrosis. J. Biol. Chem. 2019, 294, 5008–5022. [Google Scholar] [CrossRef] [PubMed]
- Willcox, J.M.; Summerlee, A.J. Relaxin protects astrocytes from hypoxia in vitro. PLoS ONE 2014, 9, e90864. [Google Scholar] [CrossRef]
- Wu, D.D.; Ding, X.D.; Wang, S.; Wojcik, J.M.; Zhang, Y.; Tokarska, M.; Li, Y.; Wang, M.S.; Faruque, O.; Nielsen, R.; et al. Pervasive introgression facilitated domestication and adaptation in the Bos species complex. Nat. Ecol. Evol. 2018, 2, 1139–1145. [Google Scholar] [CrossRef]
- Abe, K.; Narumi, S.; Suwanai, A.S.; Adachi, M.; Muroya, K.; Asakura, Y.; Nagasaki, K.; Abe, T.; Hasegawa, T. Association between monoallelic TSHR mutations and congenital hypothyroidism: A statistical approach. Eur. J. Endocrinol. 2018, 178, 137–144. [Google Scholar] [CrossRef]
- Tatour, Y.; Sanchez-Navarro, I.; Chervinsky, E.; Hakonarson, H.; Gawi, H.; Tahsin-Swafiri, S.; Leibu, R.; Lopez-Molina, M.I.; Fernandez-Sanz, G.; Ayuso, C.; et al. Mutations in SCAPER cause autosomal recessive retinitis pigmentosa with intellectual disability. J. Med. Genet. 2017, 54, 698–704. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | n | Origin | HO | HE | AR | F | fii |
|---|---|---|---|---|---|---|---|
| YNT | 6 | Qinghai-Tibet Plateau, Yunnan province | 0.3728 | 0.3714 | 1.5902 | 0.0083 | 0.7456 |
| SCT | 6 | Qinghai-Tibet Plateau, Sichuan province | 0.3594 | 0.3760 | 1.5700 | 0.0514 | 0.7539 |
| GST | 4 | Qinghai-Tibet Plateau, Gansu province | 0.4385 | 0.4151 | 1.5256 | 0.0189 | 0.7828 |
| AbaT | 4 | Qinghai-Tibet Plateau, Tibetan Qiang Autonomous Prefecture of Ngawa | 0.3262 | 0.4178 | 1.5279 | 0.2194 | 0.7832 |
| GZT | 5 | Qinghai-Tibet Plateau, Tibetan Autonomous Prefecture of Garzê | 0.2671 | 0.3951 | 1.5150 | 0.3240 | 0.7836 |
| TT | 6 | Qinghai-Tibet Plateau, Tibet | 0.3324 | 0.3670 | 1.6028 | 0.0987 | 0.7409 |
| T1 | 5 | Qinghai-Tibet Plateau | 0.2872 | 0.3956 | 1.5151 | 0.2742 | 0.7835 |
| T2 | 5 | Qinghai-Tibet Plateau | 0.2569 | 0.3874 | 1.5574 | 0.3399 | 0.7698 |
| T3 | 3 | Qinghai-Tibet Plateau | 0.4036 | 0.4682 | 1.4769 | 0.1384 | 0.8128 |
| T5 | 6 | Qinghai-Tibet Plateau | 0.2379 | 0.3681 | 1.5394 | 0.3537 | 0.7712 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shang, P.; Li, W.; Tan, Z.; Zhang, J.; Dong, S.; Wang, K.; Chamba, Y. Population Genetic Analysis of Ten Geographically Isolated Tibetan Pig Populations. Animals 2020, 10, 1297. https://doi.org/10.3390/ani10081297
Shang P, Li W, Tan Z, Zhang J, Dong S, Wang K, Chamba Y. Population Genetic Analysis of Ten Geographically Isolated Tibetan Pig Populations. Animals. 2020; 10(8):1297. https://doi.org/10.3390/ani10081297
Chicago/Turabian StyleShang, Peng, Wenting Li, Zhankun Tan, Jian Zhang, Shixiong Dong, Kejun Wang, and Yangzom Chamba. 2020. "Population Genetic Analysis of Ten Geographically Isolated Tibetan Pig Populations" Animals 10, no. 8: 1297. https://doi.org/10.3390/ani10081297
APA StyleShang, P., Li, W., Tan, Z., Zhang, J., Dong, S., Wang, K., & Chamba, Y. (2020). Population Genetic Analysis of Ten Geographically Isolated Tibetan Pig Populations. Animals, 10(8), 1297. https://doi.org/10.3390/ani10081297