Insight into the Possible Formation Mechanism of the Intersex Phenotype of Lanzhou Fat-Tailed Sheep Using Whole-Genome Resequencing

Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animal and Sequencing
2.3. Sequence Quality Checking and Mapping
2.4. Calling and Validation of SNPs and CNVs
2.5. Regional Localization and Depth Statistics for Ovine Homologous Sequences of PIS
2.6. PCR Amplification of PIS Candidate Region Sequences in Sheep
2.7. Sweep Analysis of SNPs in Selected Regions
2.8. Gene Annotation and Functional Enrichment Analysis of Selected Signal Regions
3. Results
3.1. Sequencing, Filtering, and Mapping
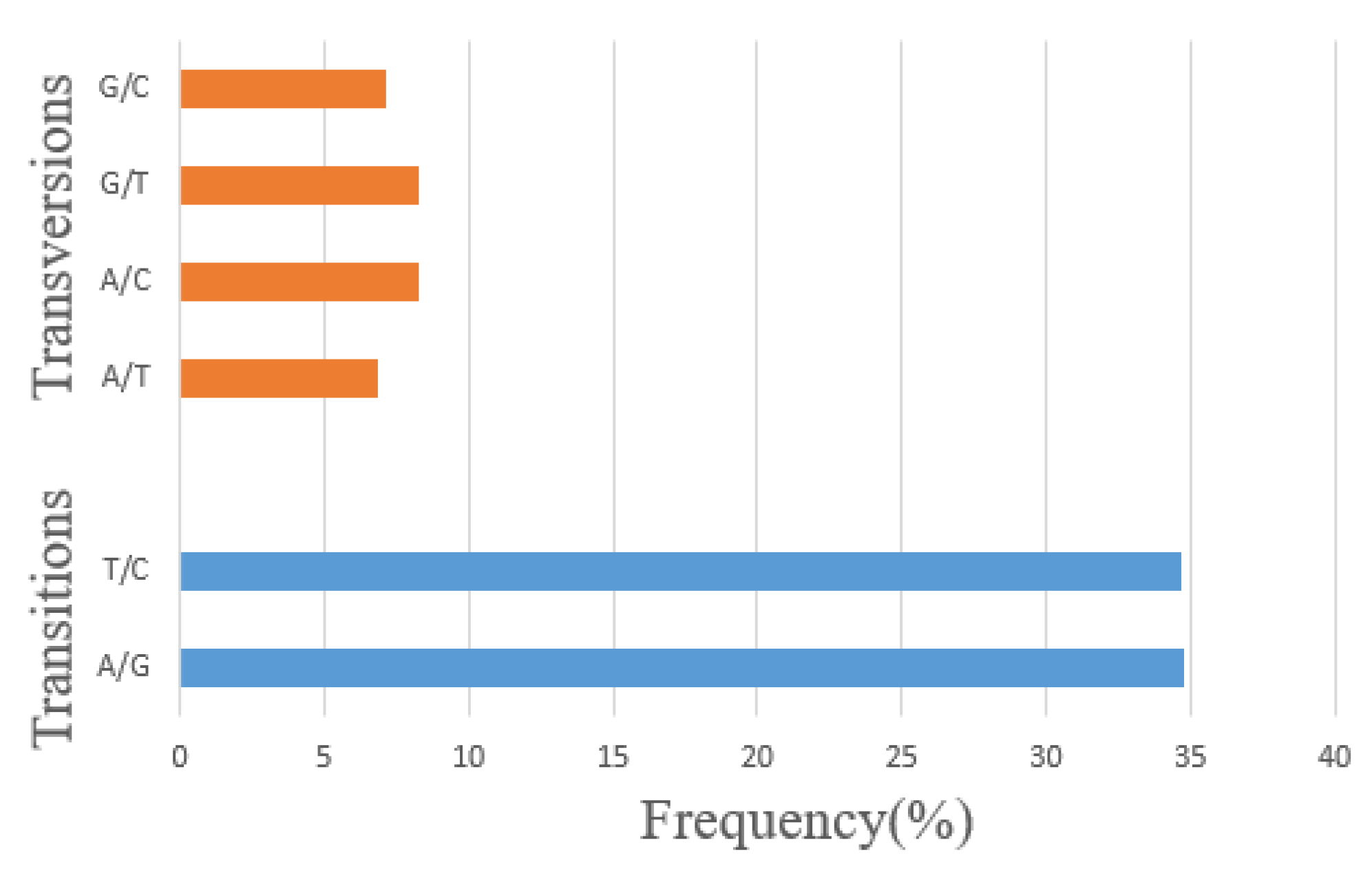
3.2. Single Nucleotide Polymorphisms (SNPs) Calling and Annotation
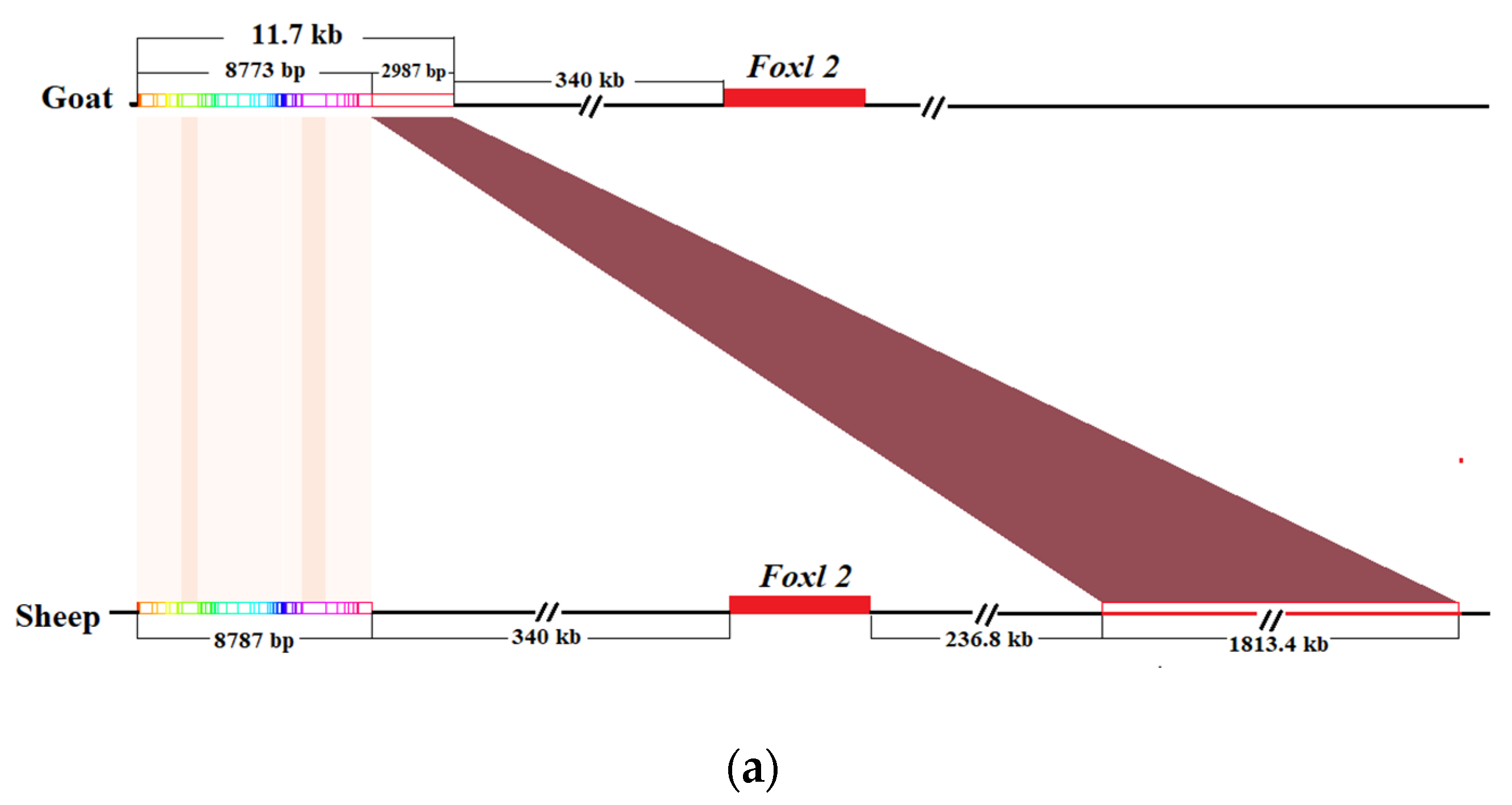
3.3. The Localization Results of Homologous Sequences of Goat PIS Region in Sheep
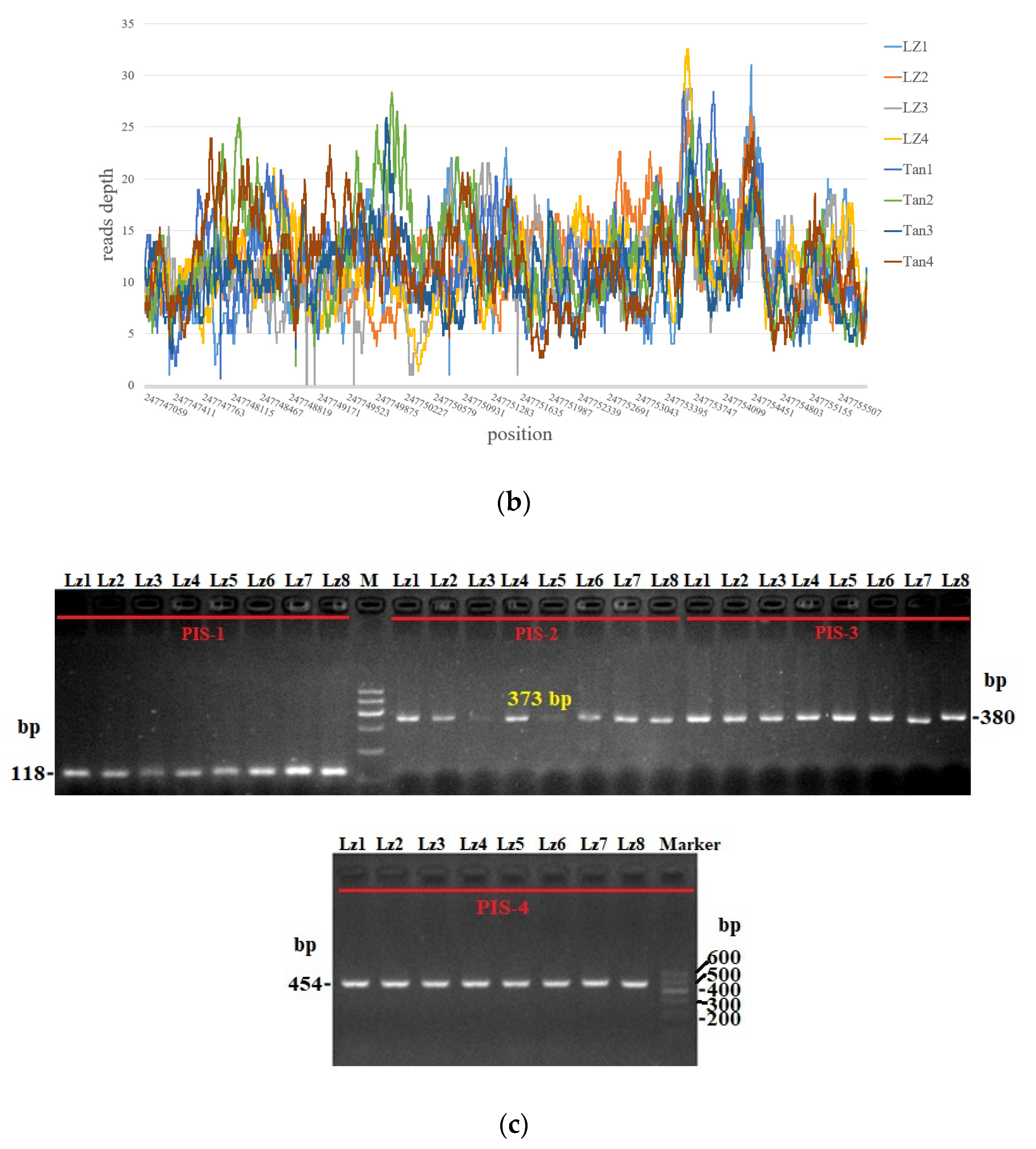
3.4. Statistics of Reads Depth and PCR Amplification of Sheep 8787 bp Candidate Sequences
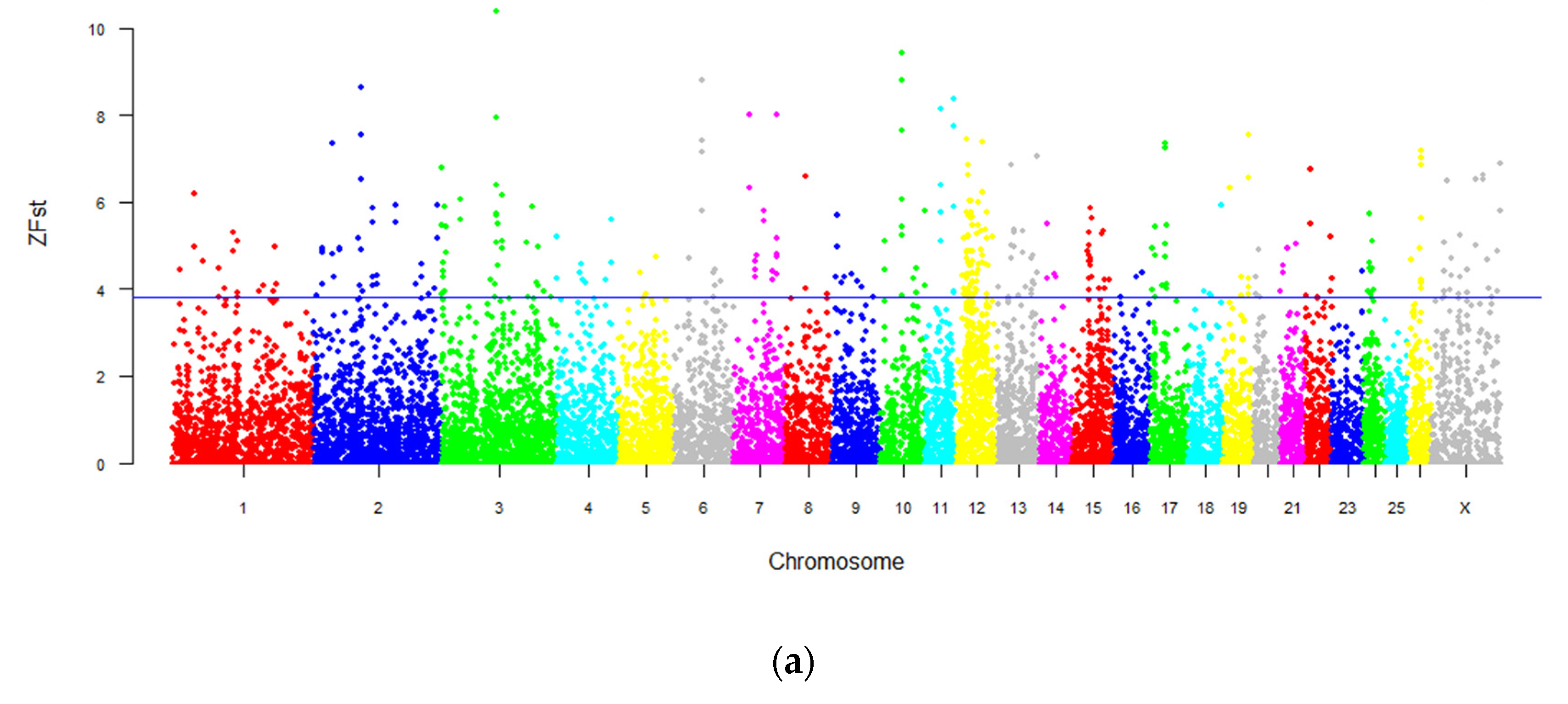
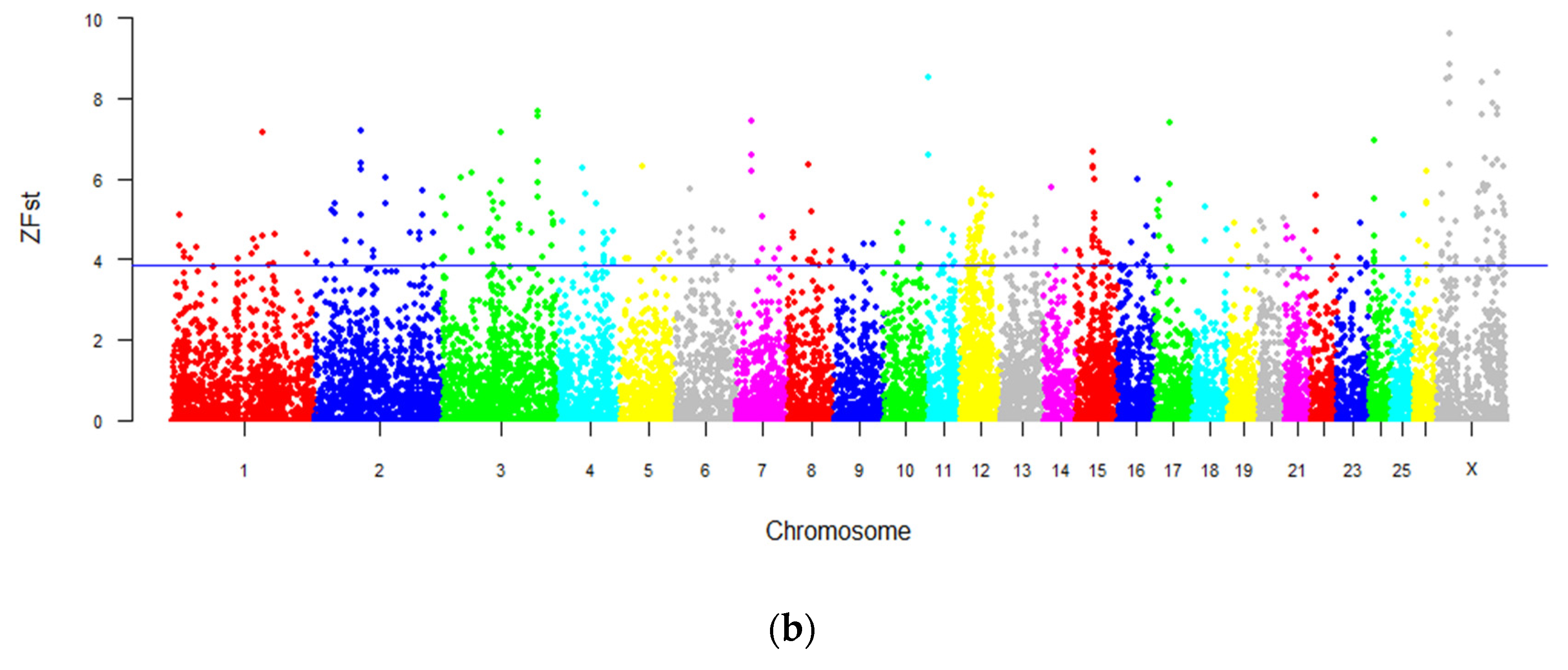
3.5. Genome-Wide Selection Sweeping Analysis in Intersex and Normal Populations
3.6. Genome-Wide Selection Sweeping Analysis in Intersex LFT Sheep and Normal LFT Sheep
3.7. Detection of Genome-Wide Copy Number Variation (CNV) in Intersex and Normal Populations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Marshall Graves, J.A. Weird animal genomes and the evolution of vertebrate sex and sex chromosomes. Annu. Rev. Genet. 2008, 42, 565–586. [Google Scholar] [CrossRef] [PubMed]
- Eaton, O.N. The relation between polled and hermaphroditic characters in dairy goats. Genetics 1945, 30, 51–61. [Google Scholar] [PubMed]
- Pailhoux, E.; Vigier, B.; Chaffaux, S. A 11.7-kb deletion triggers intersexuality and polledness in goats. Nat. Genet. 2001, 29, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Pailhoux, E.; Vigier, B.; Schibler, L. Positional cloning of the PIS mutation in goats and its impact on understanding mammalian sex-differentiation. Genet. Sel. Evol. 2005, 37, S55–S64. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.; Lischer, H.E.L.; Pieńkowska-Schelling, A.; Keller, I.; Häfliger, I.M.; Letko, A.; Schelling, C.; Lühken, G.; Drögemüller, C. New genomic features of the polled intersex syndrome variant in goats unraveled by long-read whole-genome sequencing. Anim. Genet. 2020, 51, 439–448. [Google Scholar] [CrossRef]
- Pannetier, M.; Renault, L.; Jolivet, G. Ovarian-specific expression of a new gene regulated by the goat PIS region and transcribed by a FOXL2 bidirectional promoter. Gnomics 2005, 85, 715–726. [Google Scholar] [CrossRef]
- Alberto, F.J.; Boyer, F.; Orozco-terWengel, P.; Streeter, I.; Servin, B.; de Villemereuil, P.; Benjelloun, B.; Librado, P.; Biscarini, F.; Colli, L.; et al. Convergent genomic signatures of domestication in sheep and goats. Nat. Commun. 2018, 9, 813. [Google Scholar] [CrossRef]
- Wong, K.K.; de Leeuw, R.J.; Dosanjh, N.S.; Kimm, L.R.; Cheng, Z.; Horsman, D.E.; MacAulay, C.; Ng, R.T.; Brown, C.J.; Eichler, E.E.; et al. A comprehensive analysis of common copy-number variations in the human genome. Am. J. Hum. Genet. 2007, 80, 91–104. [Google Scholar] [CrossRef]
- Flisikowski, K.; Venhoranta, H.; Nowacka-Woszuk, J. A novel mutation in the maternally imprinted PEG3 domain results in a loss of MIMT1 expression and causes abortions and stillbirths in cattle (Bos Taurus). PLoS ONE 2010, 5, e15116. [Google Scholar] [CrossRef]
- Meyers, S.N.; McDaneld, T.G.; Swist, S.L. A deletion mutation in bovine SLC4A2 is associated with osteopetrosis in Red Angus cattle. BMC Genom. 2010, 11, 337. [Google Scholar] [CrossRef]
- Elferink, M.G.; Vallée, A.; Jungerius, A.P. Partial duplication of the PRLR and SPEF2 genes at the late feathering locus in chicken. BMC Genom. 2008, 9, 391. [Google Scholar] [CrossRef] [PubMed]
- Bu, G.; Huang, G.; Fu, H. Characterization of the novel duplicated PRLR gene at the late-feathering K locus in Lohmann chickens. J. Mol. Endocrinol. 2013, 51, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Norris, B.J.; Whan, V.A. A gene duplication affecting expression of the ovine ASIP gene is responsible for white and black sheep. Genome Res. 2008, 18, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.H.; Sun, K.; Bian, Y.N.; Zhao, Q.; Wang, Z.; Ji, C.N.; Li, C. Developmental validation of an X-Insertion/Deletion polymorphism panel and application in HAN population of China. Sci. Rep. 2015, 5, 18336. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, J.; Niu, Y.; Li, Y.; Zhou, S.; Li, C.; Ma, B.; Kou, Q.; Petersen, B.; Sonstegard, T.; et al. Low incidence of SNVs and indels in trio genomes of Cas9-mediated multiplex edited sheep. BMC Genom. 2018, 19, 397. [Google Scholar] [CrossRef]
- Zhou, S.; Cai, B.; He, C.; Wang, Y.; Ding, Q.; Liu, J.; Liu, Y.; Ding, Y.; Zhao, X.; Li, G.; et al. Programmable base editing of the sheep genome revealed no genome-wide off-target mutations. Front. Genet. 2019, 10, 215. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Jiang, Y.; Xie, M.; Chen, W.; Talbot, R.; Maddox, J.F.; Faraut, T.; Wu, C.; Muzny, D.M.; Li, Y.; Zhang, W.; et al. The sheep genome illuminates biology of the rumen and lipid metabolism. Science 2014, 344, 1168–1173. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zheng, Z.; Cai, Y.; Chen, T. CNVcaller: Highly efficient and widely applicable software for detecting copy number variations in large populations. Gigascience 2017, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Jiang, J.; Yang, S.; Hou, Y.; Liu, G.E.; Zhang, S.; Zhang, Q.; Sun, D. CNV discovery for milk composition traits in dairy cattle using whole genome resequencing. BMC Genom. 2017, 18, 265. [Google Scholar] [CrossRef] [PubMed]
- Redon, R.; Ishikawa, S.; Fitch, K.R.; Feuk, L.; Perry, G.H.; Andrews, T.D.; Fiegler, H.; Shapero, M.H.; Carson, A.R.; Chen, W.; et al. Global variation in copy number in the human genome. Nature 2006, 444, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Sudmant, P.H.; Mallick, S.; Nelson, B.J.; Hormozdiari, F.; Krumm, N.; Huddleston, J.; Coe, B.P.; Baker, C.; Nordenfelt, S.; Bamshad, M.; et al. Global diversity, population stratification, and selection of human copy-number variation. Science 2015, 349, aab3761. [Google Scholar] [CrossRef]
- Kent, W. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef]
- Li, J.; Zhang, S.; Erdenee, S.; Sun, X.; Dang, R.; Huang, Y.; Lei, C.; Chen, H.; Xu, H.; Cai, Y.; et al. Nucleotide variants in prion-related protein (testis-specific) gene (PRNT) and effects on Chinese and Mongolian sheep phenotypes. Prion 2018, 12, 185–196. [Google Scholar] [CrossRef]
- Lai, F.N.; Zhai, H.L.; Cheng, M.; Ma, J.Y.; Cheng, S.F.; Ge, W.; Zhang, G.L.; Wang, J.J.; Zhang, R.Q.; Wang, X.; et al. Whole-genome scanning for the litter size trait associated genes and SNPs under selection in dairy goat (Capra hircus). Sci. Rep. 2016, 6, 38096. [Google Scholar] [CrossRef]
- Guo, J.H.; Tao, H.X.; Li, P.F.; Li, L.; Zhong, T.; Wang, L.J.; Ma, J.; Chen, X.; Song, T.; Zhang, H. Whole-genome sequencing reveals selection signatures associated with important traits in six goat breeds. Sci. Rep. 2018, 8, 10405. [Google Scholar] [CrossRef]
- Zhou, Y.; Connor, E.E.; Wiggans, G.R.; Lu, Y.; Tempelman, R.J.; Schroeder, S.G.; Chen, H.; Liu, G.E. Genome-wide copy number variant analysis reveals variants associated with 10 diverse production traits in Holstein cattle. BMC Genom. 2018, 19, 314. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- E, G.X.; Jin, M.L.; Zhao, Y.J.; Li, X.L.; Li, L.H.; Yang, B.G.; Duan, X.H.; Hunag, Y.F. Genome-wide analysis of Chongqing native intersexual goats using next-generation sequencing. 3 Biotech 2019, 9, 99. [Google Scholar] [CrossRef] [PubMed]
- Moalem, S.; Babul-Hirji, R.; Stavropolous, D.J.; Wherrett, D.; Bägli, D.J.; Thomas, P.; Chitayat, D. XX male sex reversal with genital abnormalities associated with a de novo SOX3 gene duplication. Am. J. Med Genet. 2012, 158, 1759–1764. [Google Scholar] [CrossRef]
- Kropatsch, R.; Dekomien, G.; Akkad, D.A.; Gerding, W.M.; Petrasch-Parwez, E.; Young, N.D.; Altmüller, J.; Nürnberg, P.; Gasser, R.B.; Epplen, J.T. SOX9 duplication linked to intersex in deer. PLoS ONE 2013, 8, e73734. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Zheng, D.; Zhang, L.; Li, C.; Zhang, X.; Wang, F.; Guan, Q.; Fang, L.; Zhao, J.; Xu, C. Clinical and molecular characterization of 5α-reductase type 2 deficiency due to mutations (p.Q6X, p. R246Q) in SRD5A2 gene. Endocr. J. 2018, 65, 645–655. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CNV Regions | Related Genes | Vst-Values | Sample Genotype | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chromosome | Start Position | End Position | LZ1 | LZ2 | LZ3 | LZ4 | Tan1 | Tan2 | Tan3 | Tan4 | ||
| 1 | 50776001 | 50778000 | LHX8 | 0.5713 | AB | AB | AA | AA | AA | AA | AA | AA |
| 4 | 58119201 | 58121600 | IMMP2L | 0.7097 | AB | AB | AA | AA | AA | AA | AA | AA |
| 9 | 71660801 | 71662800 | ZFPM2 | 0.8611 | AB | AB | AA | AA | AA | AA | AA | AA |
| 16 | 778801 | 780800 | SLIT3 | 0.4357 | Ad | Ad | AA | AA | AA | AA | AA | AA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Xu, H.; Liu, X.; Xu, H.; Cai, Y.; Lan, X. Insight into the Possible Formation Mechanism of the Intersex Phenotype of Lanzhou Fat-Tailed Sheep Using Whole-Genome Resequencing. Animals 2020, 10, 944. https://doi.org/10.3390/ani10060944
Li J, Xu H, Liu X, Xu H, Cai Y, Lan X. Insight into the Possible Formation Mechanism of the Intersex Phenotype of Lanzhou Fat-Tailed Sheep Using Whole-Genome Resequencing. Animals. 2020; 10(6):944. https://doi.org/10.3390/ani10060944
Chicago/Turabian StyleLi, Jie, Han Xu, Xinfeng Liu, Hongwei Xu, Yong Cai, and Xianyong Lan. 2020. "Insight into the Possible Formation Mechanism of the Intersex Phenotype of Lanzhou Fat-Tailed Sheep Using Whole-Genome Resequencing" Animals 10, no. 6: 944. https://doi.org/10.3390/ani10060944
APA StyleLi, J., Xu, H., Liu, X., Xu, H., Cai, Y., & Lan, X. (2020). Insight into the Possible Formation Mechanism of the Intersex Phenotype of Lanzhou Fat-Tailed Sheep Using Whole-Genome Resequencing. Animals, 10(6), 944. https://doi.org/10.3390/ani10060944