1. Introduction
The diversity of animal genetic resources plays a crucial role in meeting human needs and is essential for maintaining the efficiency of production systems, particularly as they are threatened by climate change [
1]. Maintaining local breeds is thus vital for facing an uncertain future, but many of these breeds have been abandoned or are currently threatened by more competitive cosmopolitan breeds [
2].
In local breeds, often with small population size, the increase of inbreeding is one of the major concerns, because of the associated loss of genetic diversity and the deleterious impact of inbreeding depression [
3]. The Ramo Grande cattle breed (
Figure 1) was developed in the Azores islands starting in the 15th century, resulting from a founder population brought by the Portuguese, which may have received additional genetic influences over the centuries. The Azores are a group of volcanic islands located in the middle of the Atlantic, and Ramo Grande cattle currently have a registered census of about 1300 cows and 80 bulls, dispersed by seven of the nine islands that make-up the archipelago. There is reduced gene-flow between islands, except for the use of artificial insemination with semen from a common group of bulls, but some inflow of genes from mainstream breeds may have occurred in recent past [
4]. Most likely, this breed was selected over the centuries for adaptation to the unique harsh environment of the Azores, potentially harboring specific genetic variants that are worth conserving and possibly of high value for the future. Currently, the breed is used for meat production and for draught in agricultural activities, but it also plays a significant role in cultural traditions, where Ramo Grande ox pairs often participate in local corteges.
A program aimed at the genetic management of Ramo Grande cattle has been in place for nearly 20 years, essentially based on the collection of phenotypic and pedigree data, with the goal of selecting for some production traits while maintaining inbreeding under control [
5]. Still, the estimated effective population size (
Ne) based on pedigree information was 28 [
5] which is nearly one-half of the recommendation of the FAO for the maintenance of genetic diversity [
6].
The use of medium-density single nucleotide polymorphism (SNP) arrays allows an alternative but accurate estimation of inbreeding (
F), even in the absence of pedigree information, and provides insight into the levels of linkage disequilibrium (LD) and the corresponding Ne [
7]. Currently, the estimation of F is often based on the analysis of runs of homozygosity (
FROH) [
8], which are contiguous lengths of homozygous genotypes that are present in an individual due to the transmission of identical haplotypes from parents to offspring [
9].
FROH provides a good indication of individual autozygosity at the genome-wide level and allows the distinction between ancient and recent inbreeding, as recombination tends to disrupt long chromosome segments [
10] and long ROH (~10 Mb) thus arise from recent inbreeding. On the other hand, the analysis of LD allows the estimation of
Ne, which is a useful way to evaluate the loss of genetic diversity over time [
11].
In addition to conserving genetic diversity of local breeds, the improvement of their productive performance while maintaining adaptability to local environmental conditions is desired, with the goal of making them more competitive and thus self-sustaining. Therefore, identifying polymorphisms in genes that may influence important traits is also crucial for the current management/improvement program of this breed.
Genomic tools developed in recent years may provide insight and a thorough understanding of the threats to genetic diversity of local breeds [
12], and reveal genetic variability for traits associated with production ability, including adaptation traits that may be of importance for sustainable agriculture in a changing climate [
13]. In this study, we used the 50K v3 Illumina Beadchip to assess the threats to genetic diversity in Ramo Grande, by estimating the occurrence of ROH, as well as the levels of LD and the corresponding Ne in past generations. Moreover, in order to identify polymorphisms significantly associated with relevant traits, we used a genome-wide association analysis, and investigated the functional impact of the identified associations by examining gene-interaction networks, allowing a better understanding of the genetic influences and candidate genes affecting the traits of interest [
14].
4. Discussion
In this study we have performed the first genome-wide characterization of the Ramo Grande cattle breed, including assessment of inbreeding and identification of genetic markers associated with production traits. This local breed from the Azores archipelago is characterized by a small population census, spread by various islands, and very well adapted to local conditions unique to the volcanic territory where it has evolved. Also unique to this breed is the cultural role that it plays in local folklore, specifically by the participation of ox pairs in corteges organized according to customary traditions.
The results of genotyping using the Illumina 50K SNP chip showed that only about 68% of the assayed SNPs are polymorphic for this breed, which is in accordance with other studies with local cattle breeds that were not included in the SNP chip discovery panel [
14,
17], and indicate that ascertainment bias may be a concern when SNP chips are applied to minor breeds.
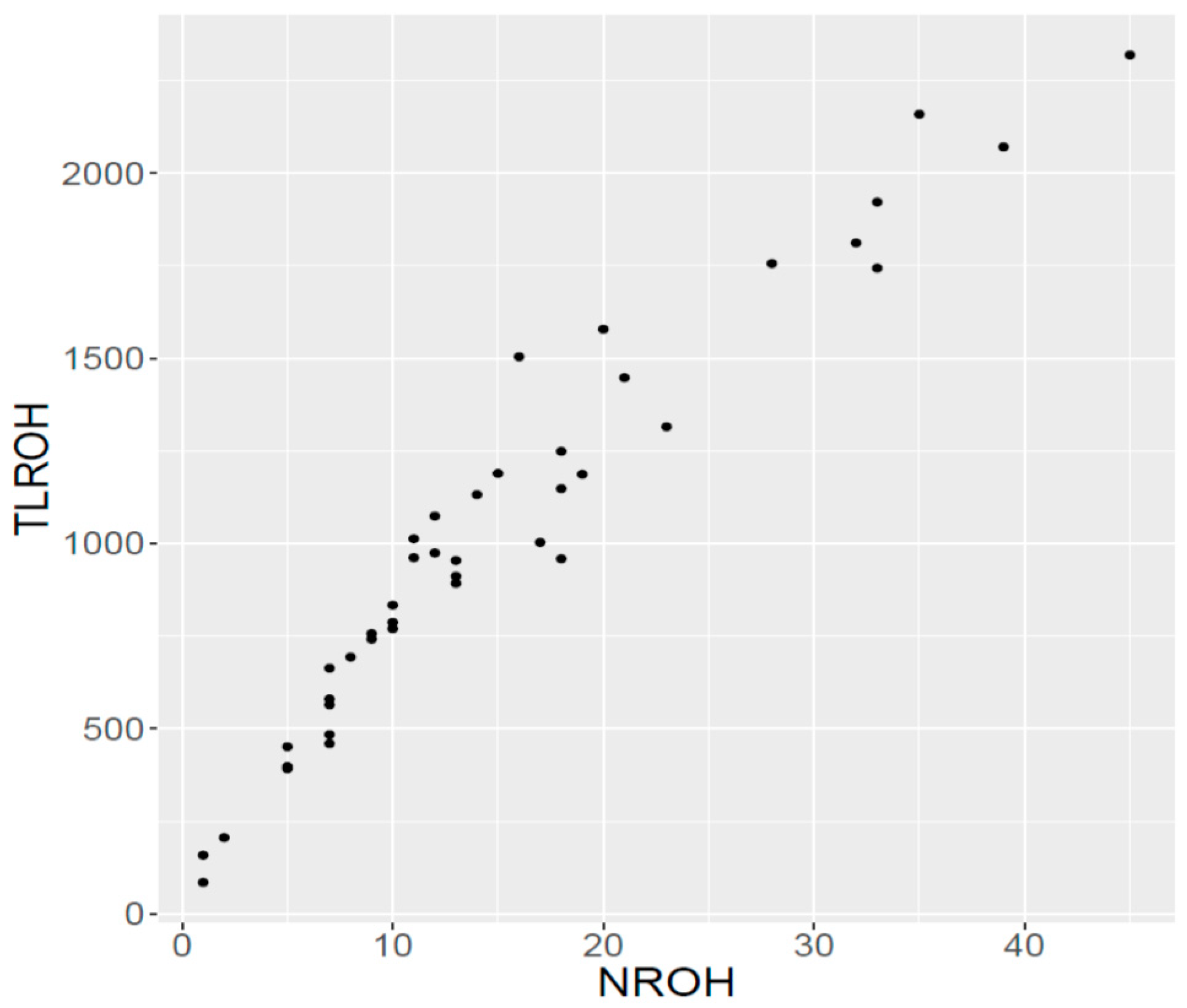
The level and time of occurrence of inbreeding was assessed by the extent of ROH segments in the sampled group [
10], as it has been shown that a strong relationship exists between pedigree and genomic inbreeding, even though pedigree inbreeding may underestimate true inbreeding [
34,
35]. When the distribution of ROH segments in our study is considered, the major feature is a relatively uniform distribution of the frequency of ROH fragments of small, intermediate and large size, with about one-third of the total number of ROH in each category. Indeed, the frequency of long ROH (larger than 16 Mb) was about 32% in our study, whereas the frequency reported for this ROH length in studies with various other breeds ranged between about 12 and 25% [
17,
34,
35]. The same pattern was observed for the mean length of ROH, which was also larger in our study, and confirm the occurrence of long ROH segments in Ramo Grande, and it is known that long ROH segments reflect the occurrence of recent inbreeding in a population, e.g., over the last three generations [
16,
34]. These results confirm that recent inbreeding has increased severely in the Ramo Grande breed, possibly as a consequence of the fragmentation of the population by various islands, which inevitably leads to reduced gene flow between groups and a higher within-island relationship.
The level of
FROH in our study, computed considering a minimum length of 4Mb for ROH, indicates that, in comparison with other studies that have used the same threshold for
FROH, Ramo Grande cattle have a higher mean level of inbreeding (
FROH = 0.09) when compared with Italian local cattle breeds with a census comparable to Ramo Grande, where the mean
FROH was about 0.05 [
17]. On the other hand, the mean
FROH in Ramo Grande was similar to that currently observed in American Holstein [
35]. When a threshold of 8 Mb is used to define
FROH, the mean for Ramo Grande was 0.079, which is close to the mean reported using the same threshold in Brown Swiss (
FROH = 0.074), but higher than the means reported for Fleckvieh, Norwegian Red and Tyrol Grey (mean
FROH ranging from 0.019 to 0.036) [
22].
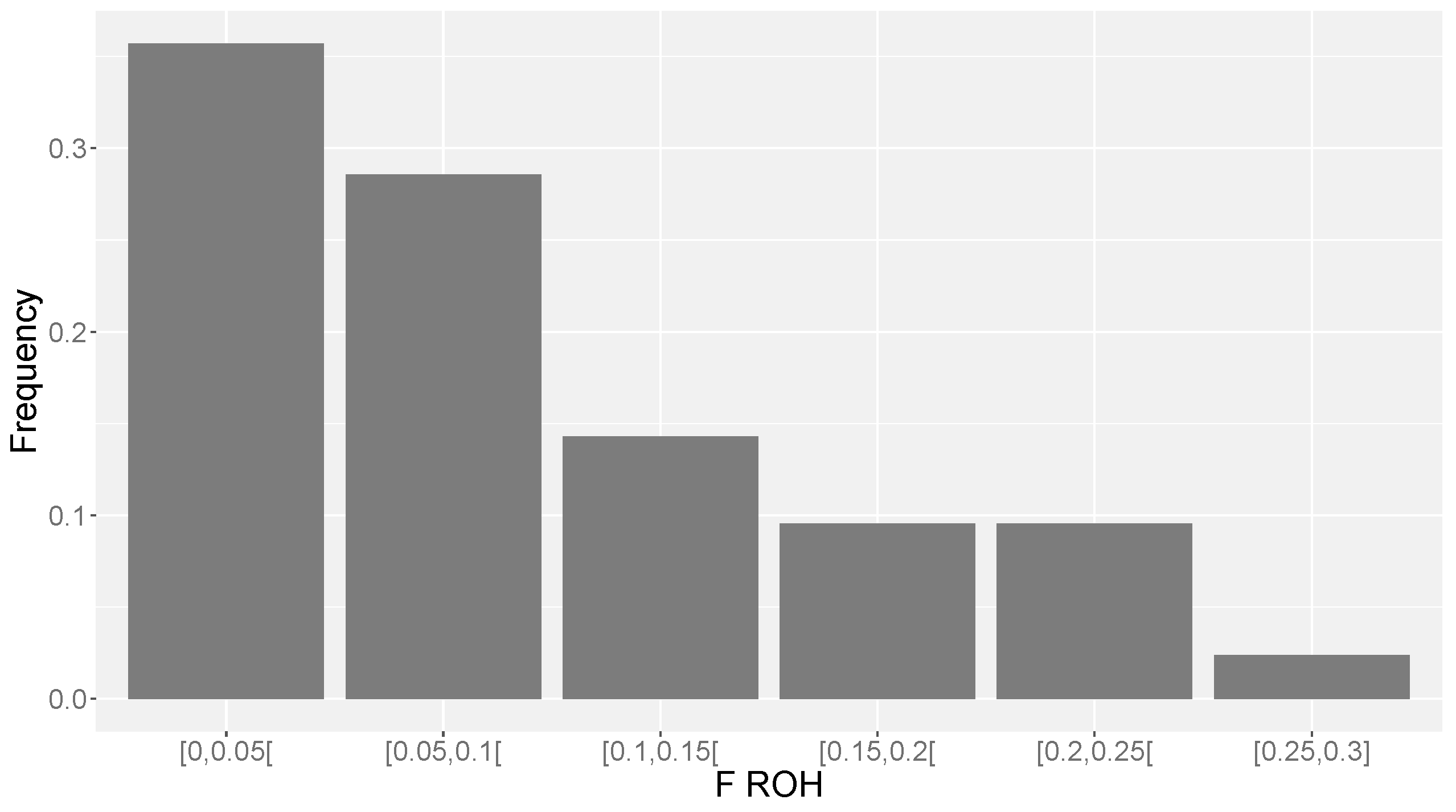
The distribution of
FROH for the studied population indicates that some individuals have extremely high levels of inbreeding (e.g., above 0.25), which may lead to inbreeding depression and loss of genetic variability in the near future [
36,
37]. This pattern could be identified with the tools of genomic analyses and would probably go unnoticed with pedigree analyses, given the shallowness of pedigrees in Ramo Grande.
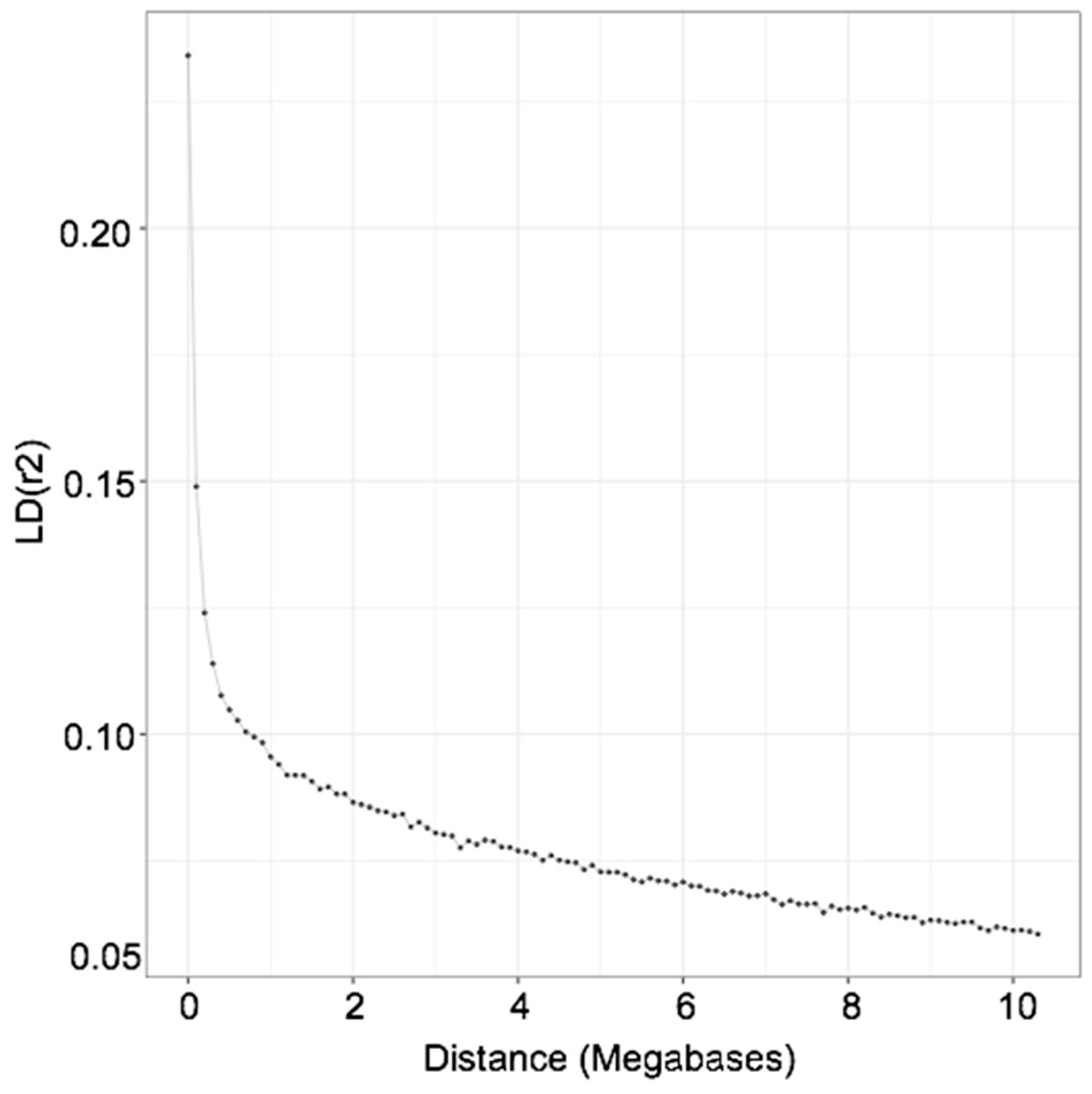
The extent of LD was used to estimate current and past effective population size (
Ne), and pairwise values of
r2 were averaged and plotted against genomic distance. We observed that the extent of LD in Ramo Grande is higher in comparison with other European local breeds with similar census [
17] but similar to the LD decay of Italian Holstein [
17] and German Holstein [
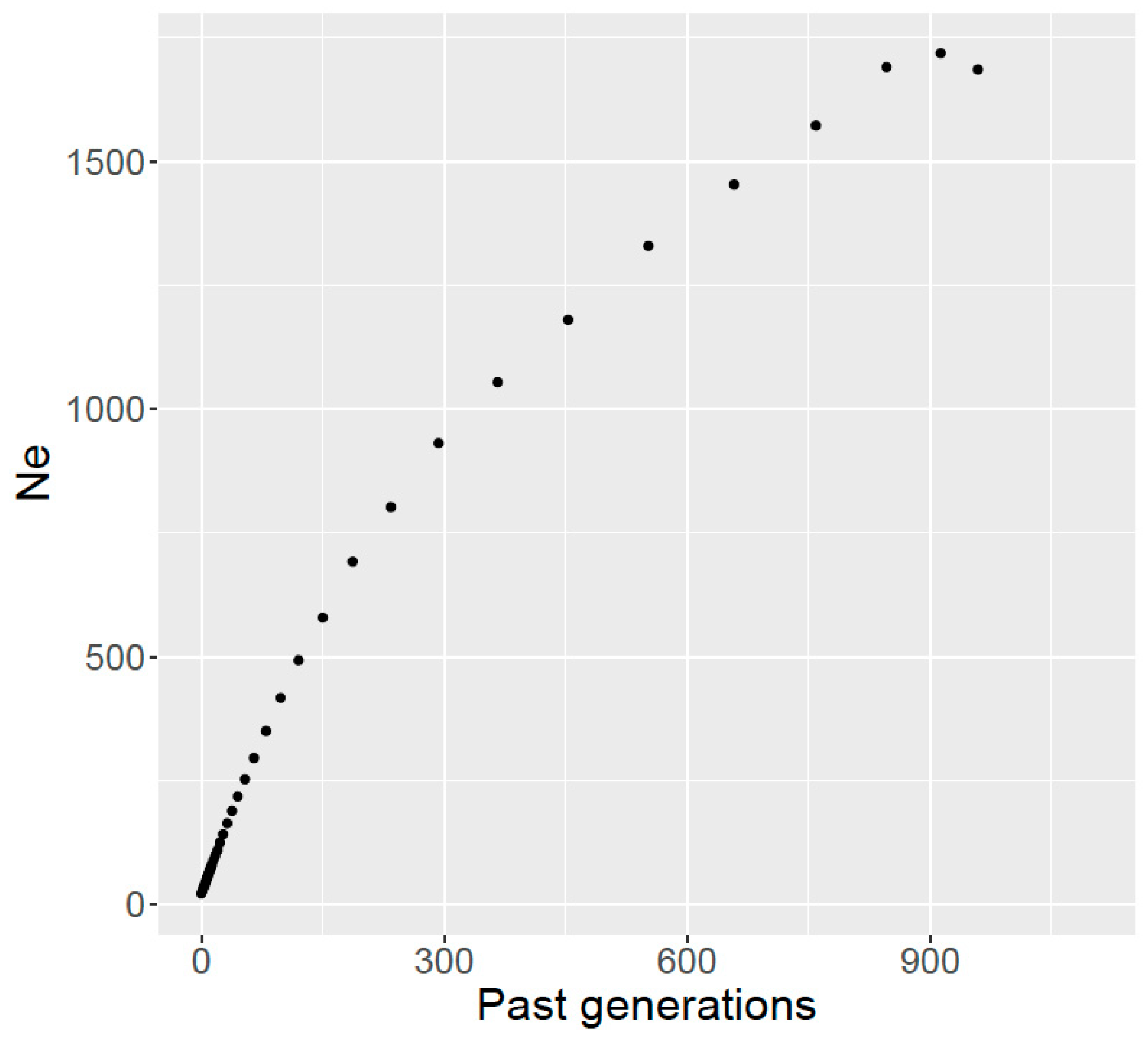
38]. Therefore, it can be expected that Ne in Ramo Grande is of similar magnitude to that of Holstein in Europe but higher than for other European local breeds. In our case, a steep increase in Ne of Ramo Grande was observed as we go back in time, which may be due to the fact that the Ramo Grande breed was developed in the archipelago of Azores by colonization using Portuguese local mainland breeds since the 16th century, with occasional inflow of germplasm of cosmopolitan breeds in more recent years, which would inflate Ne. Indeed, the introgression of exotic germplasm, especially of Holstein genes, into Ramo Grande, has been shown by analyses with microsatellite markers [
5], and crossbreeding will inevitably change the pattern of LD and the estimated Ne of a population [
39].
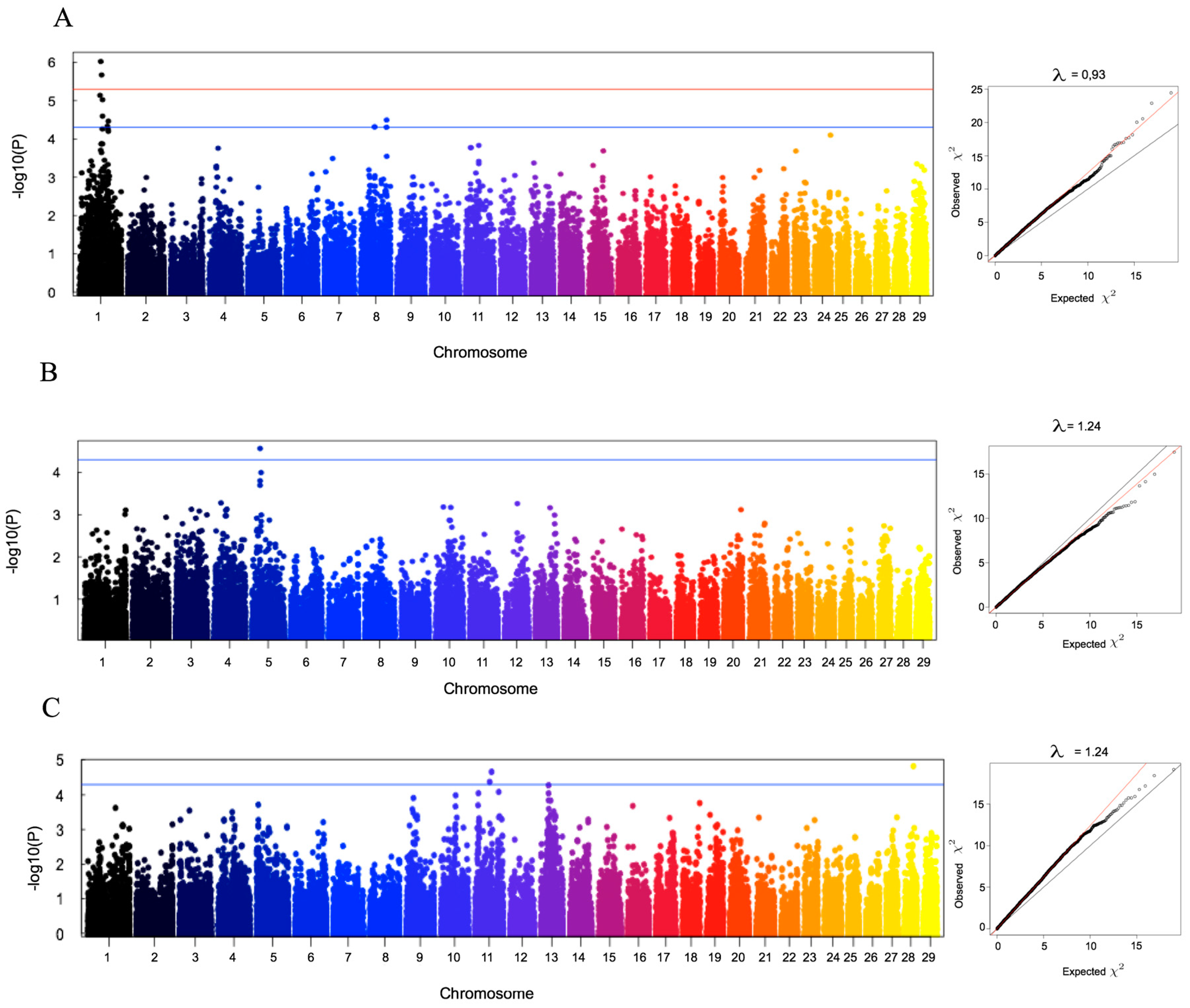
We have further investigated the possibility to identify SNP markers that were significantly associated with production traits for which this breed is currently under selection. Even though the number of animals genotyped was not high, the strategy followed in our study of using deregressed EBV allowed us to take advantage of a large number of phenotypic records collected in up to about 4000 animals, thus enhancing the reliability of the detected associations.
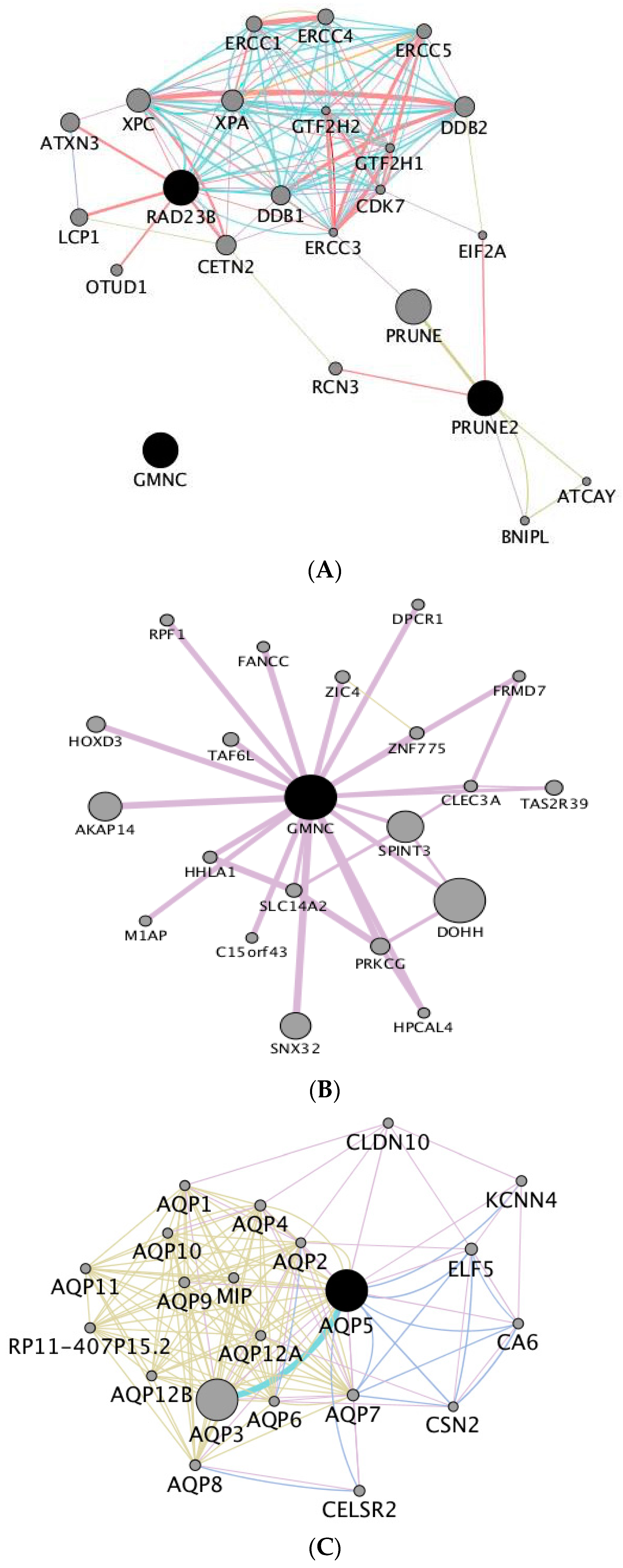
We have identified candidate SNP markers for longevity, for age at first calving and for direct genetic effects on calf weight at four months. Longevity corresponds to the length of productive life and it is, therefore, one of the most important traits in selection of local populations adapted to marginal conditions, where hardiness is crucial. However, longevity is a trait of low heritability, with an estimate in Ramo Grande of 0.07 and estimates in the literature ranging from 0.05–0.14 [
40,
41]. Therefore, the identification of bona fide markers for genomic selection is of upmost importance, and we have identified new markers associated with this trait. The most significant association was found in BTA1, upstream of
GMNC gene that is involved in the regulation of DNA replication. The investigation of the interactions of
GMNC gene with other genes showed a significant enrichment of genes related with cell cycle and cell development, and with regulation of gene expression. SNP variants located in
GMNC or upstream of this gene have been associated with neuronal diseases in humans, that affect mostly elderly subjects [
42,
43,
44]. To our knowledge, no significant associations between this gene and cattle traits have been reported in the literature. Furthermore we identified other markers located in BTA8 associated with longevity, namely upstream of
PRUNE2 gene and in the intron regions of gene
RAD23B. Gene
PRUNE2 is involved in the regulation of cell differentiation and survival, and is an important tumour suppressor [
45]. In humans,
PRUNE2 has also been associated with neuronal diseases in elderly subjects [
42,
43,
44], while in Nelore cattle the
PRUNE2 gene has been associated with feed intake [
46,
47]. Gene
RAD23B is involved in global genome nucleotide excision repair and in humans it has been associated with breast cancer [
48] but significant associations in cattle are unknown until now. While considering the gene network generated using these three genes, we observed enrichment of genes involved in DNA repair and in gene expression pathways. A more efficient process of gene repair and control of DNA replication and of gene expression is relevant for a trait such as longevity and therefore more research should be devoted to the role of the identified variants and their corresponding candidate genes in a trait of major importance in all cattle breeds, such as longevity.
We have further identified, although with a lower degree of significance, SNPs significantly associated with age at first calving and with direct genetic effects for calf weight at four months. In relation with age at first calving, our estimate of heritability (0.20) is within the wide range of heritability estimates that have been reported, between 0.02 and 0.47 [
49,
50,
51], and we found a significant association with a SNP located upstream of gene
AQP5, in a potential regulatory region. The protein encoded by this gene forms a water specific channel [
52], playing an important role in water homeostasis which is important for proper functioning of all body fluids, including reproductive structures. Aquaporin 5 protein, along with other aquaporins, namely aquaporin 1 and aquaporin 9 (also involved in
AQP5 gene interaction network) are crucial proteins required to ensure the proper follicular development of the ovary [
53], which is crucial for reproductive development and the ability to produce offspring at an early stage. In cattle QTLdb [
54] we found a significant association between age at first calving and SNPs located in
STAT5A [
55], and this gene is involved in the same pathway as
ZNF395 [
56], which in co-expressed with
AQP5 [
57]. This is the first study that identifies
AQP5 as being significantly associated with a trait related with reproductive development and that is functionally associated with a previously identified QTL. Finally, we have studied the trait direct genetic effects for calf weight at four months, for which our estimate of heritability was 0.22, in close agreement with the mean heritability estimate of 0.24 for direct genetic effects for weaning weight [
58]. We have further identified significant associations between this trait and SNPs located in introns of two genes, i.e.,
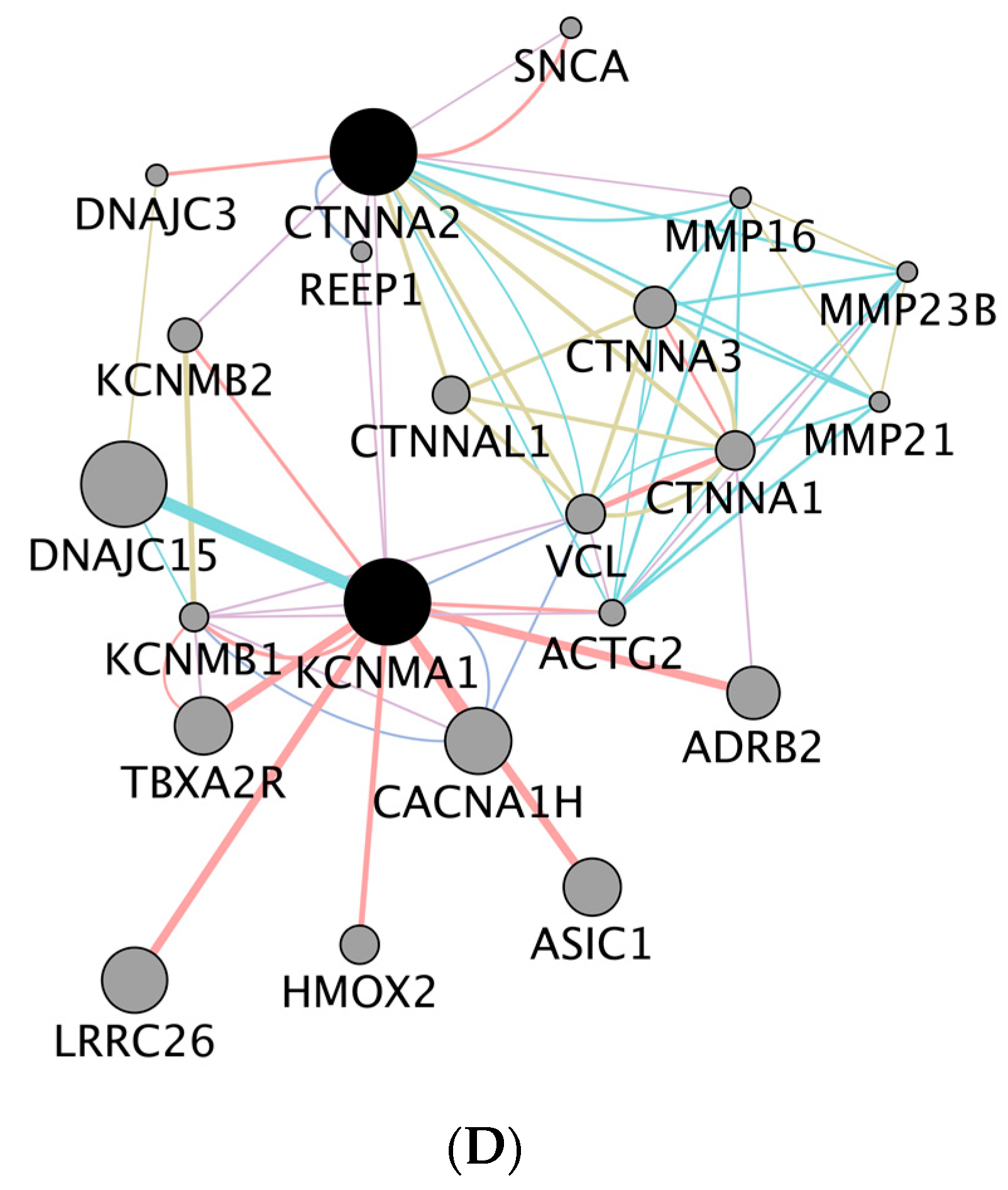
KCNMA1 and
CTNNA2. While
KCNMA1 gene encodes for a protein that forms the potassium channel [
59],
CTNNA2 encodes for a protein that may function as a linker between cadherin adhesion receptors and the cytoskeleton to regulate cell-cell adhesion and differentiation in the nervous system [
60]. In humans, variants located in
KCNMA1 have consistently been associated with obesity [
61,
62,
63], and
CTNNA2 has been associated with plasma levels of vitamin K [
64], that plays a crucial role in heart diseases and with cardiovascular measures of the autonomic tone [
65]. The functional analysis of the significant associations found for age at first calving and for direct genetic effects on calf weight at four months, along with the associations reported in the literature, require further analyses for these variants in the future.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}