Genetic Diversity and Signatures of Selection in a Native Italian Horse Breed Based on SNP Data

 , , , and
, , , and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Genotyping
2.2. Effective Population Size
2.3. Runs of Homozygosity
3. Results
3.1. Genotyping Quality Control
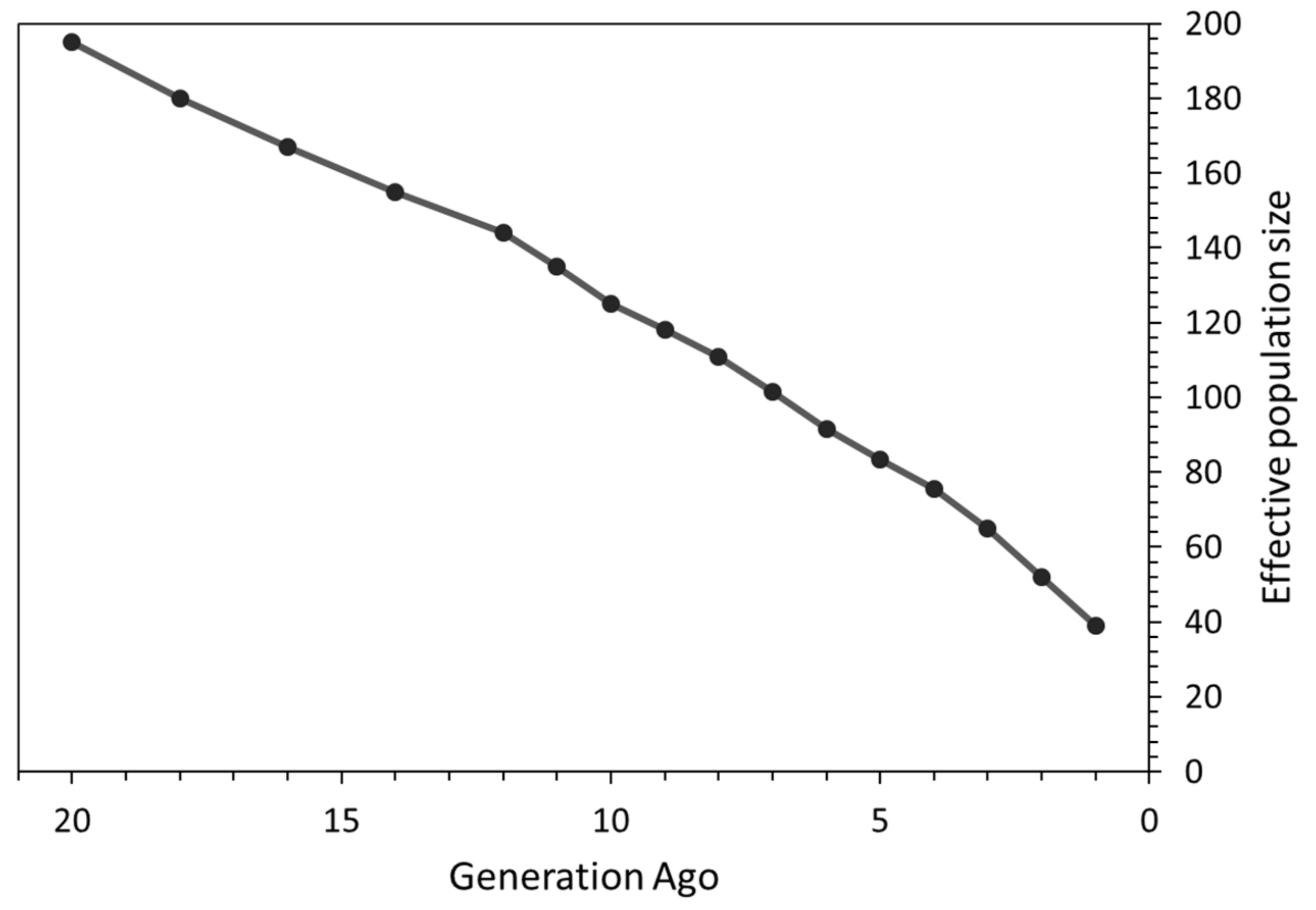
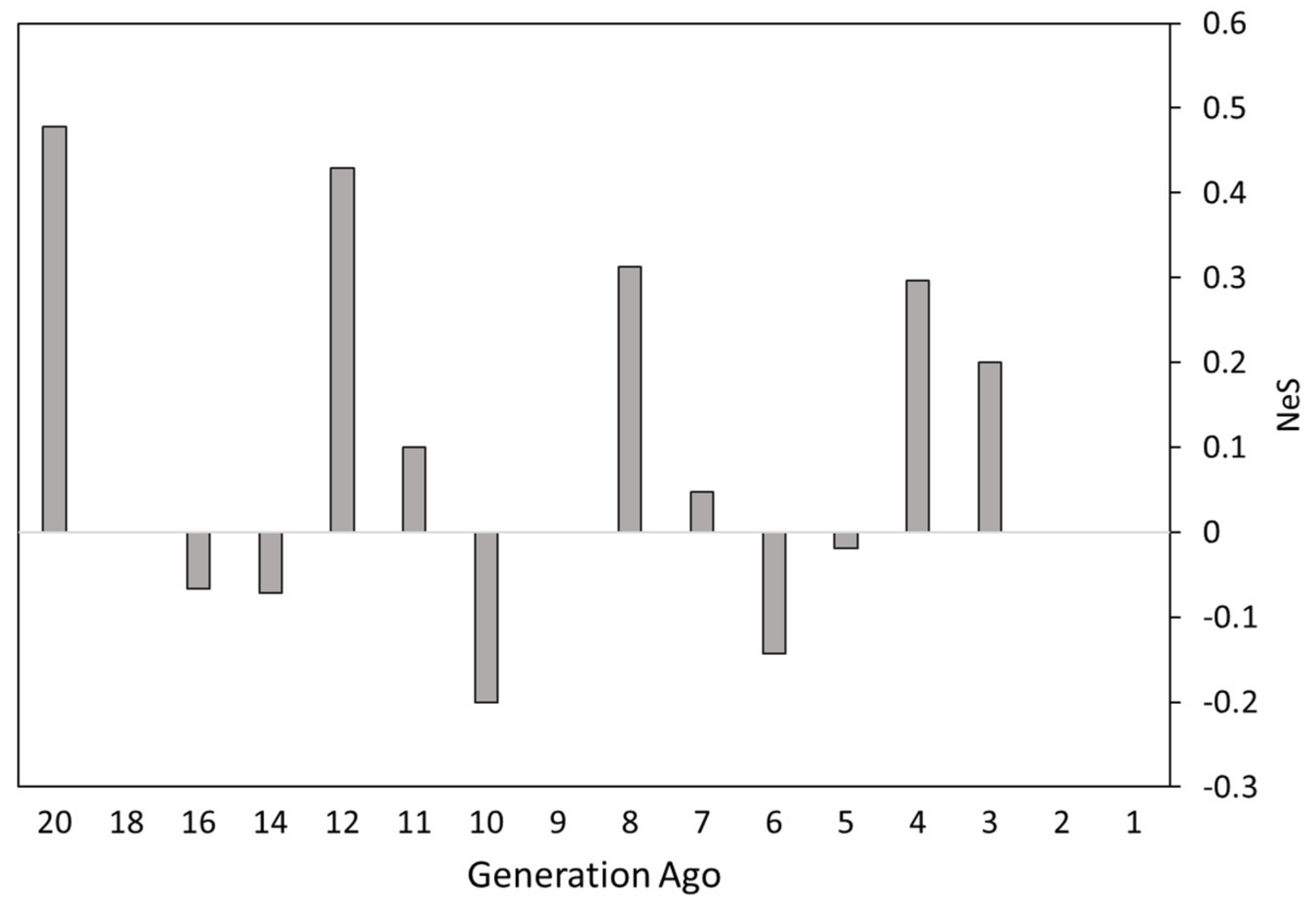
3.2. Effective Population Size
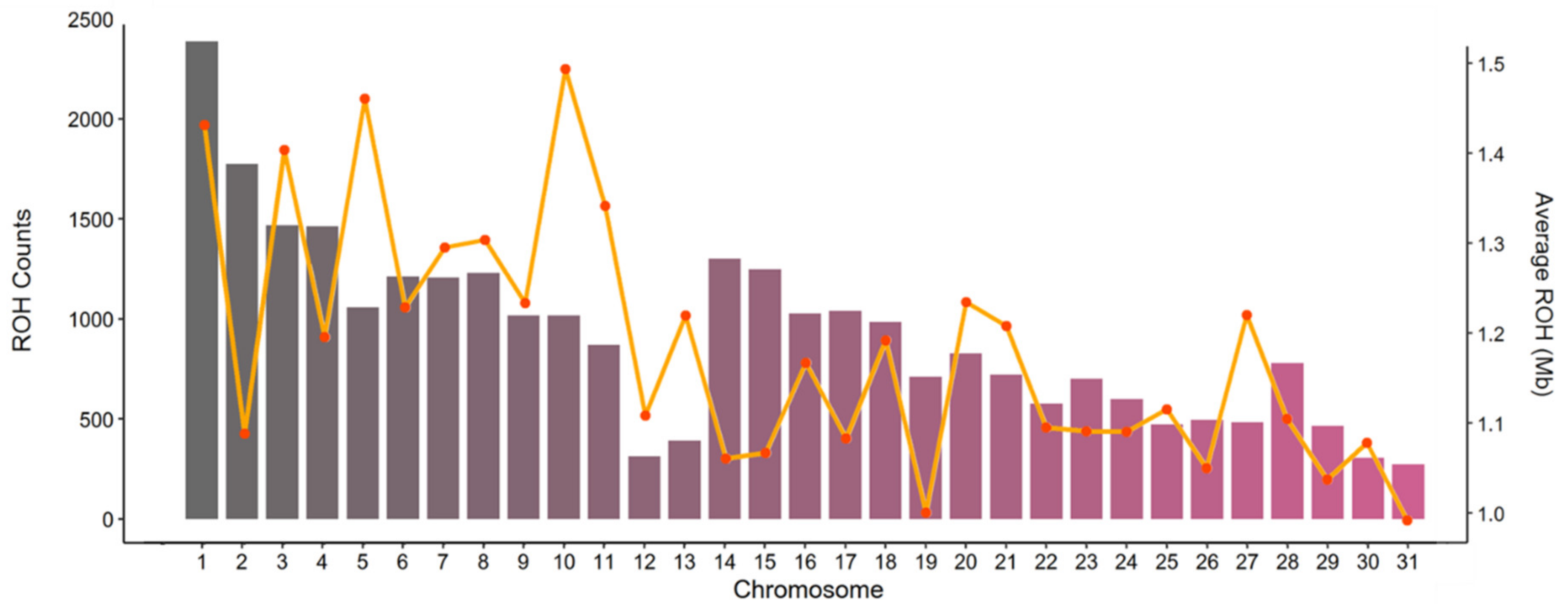
3.3. Runs of Homozygosity
3.3.1. ROH as a Measure of Inbreeding
3.3.2. ROH as a Measure to Detect Signatures of Selection
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hendricks, L.B. International Encyclopedia of Horse Breeds; University of Oklahoma Press: Norman, OK, USA, 1995. [Google Scholar]
- Ablondi, M.; Summer, A.; Vasini, M.; Simoni, M.; Sabbioni, A. Genetic parameters estimation in an Italian horse native breed to support the conversion from agricultural uses to riding purposes. J. Anim. Breed. Genet. 2020, 137, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Di Stasio, L.; Perrotta, G.; Blasi, M.; Lisa, C. Genetic characterization of the Bardigiano horse using microsatellite markers. Ital. J. Anim. Sci. 2008, 7, 243–250. [Google Scholar] [CrossRef]
- Sherf, B. World Watch List for Domestic Animal Diversity, 3rd ed.; FAO: Rome, Italy, 2000. [Google Scholar]
- Ablondi, M.; Vasini, M.; Beretti, V.; Superchi, P.; Sabbioni, A. Exploring genetic diversity in an Italian horse native breed to develop strategies for preservation and management. J. Anim. Breed. Genet. 2018, 135, 1–10. [Google Scholar] [CrossRef]
- Kim, E.S.; Cole, J.B.; Huson, H.; Wiggans, G.R.; Van Tassel, C.P.; Crooker, B.A.; Liu, G.; Da, Y.; Sonstegard, T.S. Effect of artificial selection on runs of homozygosity in U.S. Holstein cattle. PLoS ONE 2013, 8, 1–14. [Google Scholar] [CrossRef]
- Howard, J.T.; Pryce, J.E.; Baes, C.; Maltecca, C. Invited review: Inbreeding in the genomics era: Inbreeding, inbreeding depression, and management of genomic variability. J. Dairy Sci. 2017, 100, 6009–6024. [Google Scholar] [CrossRef]
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef]
- Muñoz, M.; Bozzi, R.; García-Casco, J.; Núñez, Y.; Ribani, A.; Franci, O.; García, F.; Škrlep, M.; Schiavo, G.; Bovo, S.; et al. Genomic diversity, linkage disequilibrium and selection signatures in European local pig breeds assessed with a high density SNP chip. Sci. Rep. 2019, 9, 13546. [Google Scholar] [CrossRef]
- Talenti, A.; Bertolini, F.; Pagnacco, G.; Pilla, F.; Ajmone-Marsan, P.; Rothschild, M.F.; Crepaldi, P. The Italian goat consortium the valdostana goat: A genome-wide investigation of the distinctiveness of its selective sweep regions. Mamm. Genome 2017, 28, 129. [Google Scholar] [CrossRef]
- Kijas, J.W.; Lenstra, J.A.; Hayes, B.; Boitard, S.; Neto, L.R.; Cristobal, M.S.; Servin, B.; McCulloch, R.; Whan, V.; Gietzen, K.; et al. Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 2012, 10. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.L.; Mickelson, J.R.; Cothran, E.G.; Andersson, L.S.; Axelsson, J.; Bailey, E.; Bannasch, D.; Binns, M.M.; Borges, A.S.; Brama, P.; et al. Genetic diversity in the modern horse illustrated from genome-wide SNP data. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Maiorano, A.M.; Lourenco, D.L.; Tsuruta, S.; Toro Ospina, A.M.; Stafuzza, N.B.; Masuda, Y.; Filho, A.E.V.; Dos Santos-Goncalves, C.J.N.; Curi, R.A.; De Vasconcelos-Silva, J.A. Assessing genetic architecture and signatures of selection of dual purpose Gir cattle populations using genomic information. PLoS ONE 2018, 13, 1–24. [Google Scholar] [CrossRef]
- Khayatzadeh, N.; Mészáros, G.; Utsunomiya, Y.T.; Garcia, J.F.; Schnyder, U.; Gredler, B.; Curik, I.; Sölkner, J. Locus-specific ancestry to detect recent response to selection in admixed Swiss Fleckvieh cattle. Anim. Genet. 2016, 47, 637–646. [Google Scholar] [CrossRef]
- Msalya, G.; Kim, E.S.; Laisser, E.L.K.; Kipanyula, M.J.; Karimuribo, E.D.; Kusiluka, L.J.M.; Chenyambuga, S.W.; Rothschild, M.F. Determination of genetic structure and signatures of selection in three strains of Tanzania shorthorn Zebu, Boran and Friesian cattle by genome-wide SNP analyses. PLoS ONE 2017, 12, 1–18. [Google Scholar] [CrossRef]
- Peripolli, E.; Munari, D.P.; Silva, M.V.G.B.; Lima, A.L.F.; Irgang, R.; Baldi, F. Runs of homozygosity: Current knowledge and applications in livestock. Anim. Genet. 2017, 48, 255–271. [Google Scholar] [CrossRef]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef]
- McQuillan, R.; Leutenegger, A.L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef]
- Curik, I.; Ferenčaković, M.; Sölkner, J. Inbreeding and runs of homozygosity: A possible solution to an old problem. Livest. Sci. 2014, 166, 26–34. [Google Scholar] [CrossRef]
- Ferenčaković, M.; Hamzić, E.; Gredler, B.; Solberg, T.R.; Klemetsdal, G.; Curik, I.; Sölkner, J. Estimates of autozygosity derived from runs of homozygosity: Empirical evidence from selected cattle populations. J. Anim. Breed. Genet. 2013, 130, 286–293. [Google Scholar] [CrossRef]
- Marras, G.; Gaspa, G.; Sorbolini, S.; Dimauro, C.; Ajmone-Marsan, P.; Valentini, A.; Williams, J.L.; Macciotta, N.P.P. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim. Genet. 2015, 46, 110–121. [Google Scholar] [CrossRef]
- Schurink, A.; Arts, D.J.G.; Ducro, B.J. Genetic diversity in the Dutch harness horse population using pedigree analysis. Livest. Sci. 2012, 143, 270–277. [Google Scholar] [CrossRef]
- Sørensen, M.K.; Sørensen, A.C.; Baumung, R.; Borchersen, S.; Berg, P. Optimal genetic contribution selection in Danish holstein depends on pedigree quality. Livest. Sci. 2008, 118, 212–222. [Google Scholar] [CrossRef]
- Metzger, J.; Karwath, M.; Tonda, R.; Beltran, S.; Águeda, L.; Gut, M.; Gut, I.G.; Distl, O. Runs of homozygosity reveal signatures of positive selection for reproduction traits in breed and non-breed horses. BMC Genom. 2015, 16, 764. [Google Scholar] [CrossRef] [PubMed]
- Ablondi, M.; Viklund, Å.; Lindgren, G.; Eriksson, S.; Mikko, S. Signatures of selection in the genome of Swedish warmblood horses selected for sport performance. BMC Genom. 2019, 20, 717. [Google Scholar] [CrossRef]
- Grilz-Seger, G.; Neuditschko, M.; Ricard, A.; Velie, B.; Lindgren, G.; Mesarič, M.; Cotman, M.; Horna, M.; Dobretsberger, M.; Brem, G.; et al. Genome-wide homozygosity patterns and evidence for selection in a set of European and near eastern horse breeds. Genes 2019, 10, 491. [Google Scholar] [CrossRef]
- Nolte, W.; Thaller, G.; Kuehn, C. Selection signatures in four German warmblood horse breeds: Tracing breeding history in the modern sport horse. PLoS ONE 2019, 14, 1–25. [Google Scholar] [CrossRef]
- Grilz-Seger, G.; Druml, T.; Neuditschko, M.; Dobretsberger, M.; Horna, M.; Brem, G. High-resolution population structure and runs of homozygosity reveal the genetic architecture of complex traits in the Lipizzan horse. BMC Genom. 2019, 20, 1–17. [Google Scholar] [CrossRef]
- Moravčíková, N.; Kasarda, R.; Kadlečík, O.; Trakovická, A.; Halo, M.; Candrák, J. Runs of homozygosity as footprints of selection in the norik of muran horse genome. Acta Univ. Agric. Silv. Mendel. Brun. 2019, 67, 1165–1170. [Google Scholar] [CrossRef]
- Ablondi, M.; Eriksson, S.; Tetu, S.; Sabbioni, A.; Viklund, A.; Mikko, S. Genomic divergence in swedish warmblood horses selected for equestrian disciplines. Genes 2019, 10, 976. [Google Scholar] [CrossRef]
- Grilz-Seger, G.; Mesarič, M.; Cotman, M.; Neuditschko, M.; Druml, T.; Brem, G. Runs of homozygosity and population history of three horse breeds with small population size. J. Equine Vet. Sci. 2018, 71, 27–34. [Google Scholar] [CrossRef]
- Druml, T.; Neuditschko, M.; Grilz-Seger, G.; Horna, M.; Ricard, A.; Mesarič, M.; Cotman, M.; Pausch, H.; Brem, G. Population networks associated with runs of homozygosity reveal new insights into the breeding history of the haflinger horse. J. Hered. 2018, 109, 384–392. [Google Scholar] [CrossRef]
- Ardestani, S.S.; Aminafshar, M.; Zandi-Baghche, M.M.B.; Banabazi, M.H.; Sargolzaei, M.; Miar, Y. Whole-genome signatures of selection in sport horses revealed selection footprints related to musculoskeletal system development processes. Animals 2019, 10, 53. [Google Scholar] [CrossRef]
- Grilz-Seger, G.; Druml, T.; Neuditschko, M.; Mesarič, M.; Cotman, M.; Brem, G. Analysis of ROH patterns in the Noriker horse breed reveals signatures of selection for coat color and body size. Anim. Genet. 2019, 50, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Maignel, L.; Boichard, D.; Verrier, E. Genetic variability of French dairy breeds estimated from pedigree information. Interbull Bull. 1996, 14, 49–53. [Google Scholar]
- Kalbfleisch, T.S.; Rice, E.S.; DePriest, M.S.; Walenz, B.P.; Hestand, M.S.; Vermeesch, J.R.; O′Connell, B.L.; Fiddes, I.T.; Vershinina, A.O.; Saremi, N.F.; et al. Improved reference genome for the domestic horse increases assembly contiguity and composition. Commun. Biol. 2018, 1, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Beeson, S.K.; Schaefer, R.J.; Mason, V.C.; McCue, M.E. Robust remapping of equine SNP array coordinates to EquCab3. Anim. Genet. 2019, 50, 114–115. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Barbato, M.; Orozco-ter, W.P.; Tapio, M.; Bruford, M.W. SNeP: A tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 2015, 6, 109. [Google Scholar] [CrossRef]
- Sved, J.A.; Feldman, M.W. Correlation and probability methods for one and two loci. Popul. Biol. 1973, 4, 129–132. [Google Scholar] [CrossRef]
- Beeson, S.K.; Mickelson, J.R.; McCue, M.E. Exploration of fine-scale recombination rate variation in the domestic horse. Genome Res. 2019, 29, 1744–1752. [Google Scholar] [CrossRef]
- Pitt, D.; Bruford, M.W.; Barbato, M.; Orozco-ter, W.P.; Martínez, R.; Sevane, N. Demography and rapid local adaptation shape creole cattle genome diversity in the tropics. Evol. Appl. 2019, 12, 105–122. [Google Scholar] [CrossRef]
- Biscarini, F.; Cozzi, P.; Gaspa, G.; Marras, G. detectRUNS: An R Package to Detect Runs of Homozygosity and Heterozygosity in Diploid Genomes; University of Sassari: Sassari, Italy, 2019. [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Development Core Team: Vienna, Austria, 2011. [Google Scholar]
- De Souza-Fonseca, P.A.; dos Santos, F.C.; Rosse, I.C.; Ventura, R.V.; Brunelli, F.Â.T.; Penna, V.M.; da Silva Verneque, R.; Machado, M.A.; da Silva, M.V.G.B.; Carvalho, M.R.S.; et al. Retelling the recent evolution of genetic diversity for Guzerá: Inferences from LD decay, runs of homozygosity and Ne over the generations. Livest. Sci. 2016, 193, 110–117. [Google Scholar] [CrossRef]
- Aken, B.L.; Ayling, S.; Barrell, D.; Clarke, L.; Curwen, V.; Fairley, S.; Fernandez Banet, J.; Billis, K.; García Girón, C.; Hourlier, T.; et al. The Ensembl gene annotation system. Database 2016, 2016, 93. [Google Scholar] [CrossRef] [PubMed]
- Karolchik, D. The UCSC table browser data retrieval tool. Nucleic Acids Res. 2004, 32, D493–D496. [Google Scholar] [CrossRef]
- Hu, Z.-L.; Park, C.A.; Reecy, J.M. Building a livestock genetic and genomic information knowledgebase through integrative developments of animal QTLdb and CorrDB. Nucleic Acids Res. 2019, 47, D701–D710. [Google Scholar] [CrossRef]
- Schurink, A.; Wolc, A.; Ducro, B.J.; Frankena, K.; Garrick, D.J.; Dekkers, J.C.M.; Van Arendonk, J.A.M. Genome-wide association study of insect bite hypersensitivity in two horse populations in The Netherlands. Genet. Sel. Evol. 2012, 44, 1. [Google Scholar] [CrossRef]
- Shrestha, M.; Solé, M.; Ducro, B.J.; Sundquist, M.; Thomas, R.; Schurink, A.; Eriksson, S.; Lindgren, G. Genome-wide association study for insect bite hypersensitivity susceptibility in horses revealed novel associated loci on chromosome 1. J. Anim. Breed. Genet. 2020, 137, 223–233. [Google Scholar] [CrossRef]
- Metzger, J.; Ohnesorge, B.; Distl, O. Genome-wide linkage and association analysis identifies major gene loci for guttural pouch tympany in Arabian and German warmblood horses. PLoS ONE 2012, 7, e41640. [Google Scholar] [CrossRef]
- Haase, B.; Signer-Hasler, H.; Binns, M.M.; Obexer-Ruff, G.; Hauswirth, R.; Bellone, R.R.; Burger, D.; Rieder, S.; Wade, C.M.; Leeb, T. Accumulating mutations in series of haplotypes at the KIT and MITF loci are major determinants of white markings in franches-montagnes horses. PLoS ONE 2013, 8, 1–10. [Google Scholar] [CrossRef]
- Tetens, J.; Widmann, P.; Kühn, C.; Thaller, G. A genome-wide association study indicates LCORL/NCAPG as a candidate locus for withers height in german warmblood horses. Anim. Genet. 2013, 44, 467–471. [Google Scholar] [CrossRef]
- Thomer, A.; Gottschalk, M.; Christmann, A.; Naccache, F.; Jung, K.; Hewicker-Trautwein, M.; Distl, O.; Metzger, J. An epistatic effect of KRT25 on SP6 is involved in curly coat in horses. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Makvandi-Nejad, S.; Hoffman, G.E.; Allen, J.J.; Chu, E.; Gu, E.; Chandler, A.M.; Loredo, A.I.; Bellone, R.R.; Mezey, J.G.; Brooks, S.A.; et al. Four loci explain 83% of size variation in the horse. PLoS ONE 2012, 7, 1–6. [Google Scholar] [CrossRef]
- Skujina, I.; Winton, C.L.; Hegarty, M.J.; McMahon, R.; Nash, D.M.; Davies Morel, M.C.G.; McEwan, N.R. Detecting genetic regions associated with height in the native ponies of the British Isles by using high density SNP genotyping. Genome 2018, 61, 767–770. [Google Scholar] [CrossRef] [PubMed]
- Vostrá-Vydrová, H.; Vostrý, L.; Hofmanová, B.; Krupa, E.; Zavadilová, L. Pedigree analysis of the endangered old kladruber horse population. Livest. Sci. 2016, 185, 17–23. [Google Scholar] [CrossRef]
- Fabbri, M.C.; de Rezende, M.P.G.; Dadousis, C.; Biffani, S.; Negrini, R.; Carneiro, P.L.S.; Bozzi, R. Population structure and genetic diversity of Italian beef breeds as a tool for planning conservation and selection strategies. Animals 2019, 9, 880. [Google Scholar] [CrossRef] [PubMed]
- Mariani, E.; Summer, A.; Ablondi, M.; Sabbioni, A. Genetic variability and management in nero di parma swine breed to preserve local diversity. Animals 2020, 10, 538. [Google Scholar] [CrossRef] [PubMed]
- Giontella, A.; Pieramati, C.; Silvestrelli, M.; Sarti, F.M. Analysis of founders and performance test effects on an autochthonous horse population through pedigree analysis: Structure, genetic variability and inbreeding. Animal 2018, 13, 1–10. [Google Scholar] [CrossRef]
- Flury, C.; Tapio, M.; Sonstegard, T.; Drögemüller, C.; Leeb, T.; Simianer, H.; Hanotte, O.; Rieder, S. Effective population size of an indigenous Swiss cattle breed estimated from linkage disequilibrium. J. Anim. Breed. Genet. 2010, 127, 339–347. [Google Scholar] [CrossRef]
- Goyache, F.; Álvarez, I.; Fernández, I.; Pérez-Pardal, L.; Royo, L.J.; Lorenzo, L. Usefulness of molecular-based methods for estimating effective population size in livestock assessed using data from the endangered black-coated Asturcón pony. J. Anim. Sci. 2011, 89, 1251–1259. [Google Scholar] [CrossRef]
- Sadeghi, R.; Moradi-Shahrbabak, M.; Ashtiani, S.R.M.; Schlamp, F.; Cosgrove, E.J.; Antczak, D.F. Genetic diversity of Persian arabian horses and their relationship to other native iranian horse breeds. J. Hered. 2019, 110, 173–182. [Google Scholar] [CrossRef]
- Druml, T.; Curik, I.; Baumung, R.; Aberle, K.; Distl, O.; Sölkner, J. Individual-based assessment of population structure and admixture in Austrian, Croatian and German draught horses. Heredity 2007, 98, 114–122. [Google Scholar] [CrossRef]
- Gutiérrez, J.P.; Cervantes, I.; Molina, A.; Valera, M.; Goyache, F. Individual increase in inbreeding allows estimating effective sizes from pedigrees. Genet. Sel. Evol. 2008, 40, 359–378. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, J.P.; Cervantes, I.; Goyache, F. Improving the estimation of realized effective population sizes in farm animals. J. Anim. Breed. Genet. 2009, 126, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Kamiński, S.; Hering, D.M.; Jaworski, Z.; Zabolewicz, T.; Ruść, A. Assessment of genomic inbreeding in Polish konik horses. Pol. J. Vet. Sci. 2017, 20, 603–605. [Google Scholar] [CrossRef] [PubMed]
- Schurink, A.; Shrestha, M.; Eriksson, S.; Bosse, M.; Bovenhuis, H.; Back, W.; Johansson, A.M.; Ducro, B.J. The genomic makeup of nine horse populations sampled in The Netherlands. Genes 2019, 10, 480. [Google Scholar] [CrossRef]
- Velie, B.D.; Solé, M.; Fegraeus, K.J.; Rosengren, M.K.; Røed, K.H.; Ihler, C.F.; Strand, E.; Lindgren, G. Genomic measures of inbreeding in the Norwegian-Swedish coldblooded trotter and their associations with known QTL for reproduction and health traits. Genet. Sel. Evol. 2019, 51, 1–10. [Google Scholar] [CrossRef]
- Solé, M.; Gori, A.S.; Faux, P.; Bertrand, A.; Farnir, F.; Gautier, M.; Druet, T. Age-based partitioning of individual genomic inbreeding levels in Belgian blue cattle. Genet. Sel. Evol. 2017, 49, 1–18. [Google Scholar] [CrossRef]
- Pemberton, T.J.; Absher, D.; Feldman, M.W.; Myers, R.M.; Rosenberg, N.A.; Li, J.Z. Genomic patterns of homozygosity in worldwide human populations. Am. J. Hum. Genet. 2012, 91, 275–292. [Google Scholar] [CrossRef]
- Takasuga, A. PLAG1 and NCAPG-LCORL in livestock. Anim. Sci. J. 2016, 87, 159–167. [Google Scholar] [CrossRef]
- Allen, L.H.; Estrada, K.; Lettre, G.; Berndt, S.I.; Weedon, M.N.; Rivadeneira, F.; Willer, C.J.; Jackson, A.U.; Vedantam, S.; Raychaudhuri, S.; et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 2010, 467, 832–838. [Google Scholar] [CrossRef]
- Gudbjartsson, D.F.; Walters, G.B.; Thorleifsson, G.; Stefansson, H.; Halldorsson, B.V.; Zusmanovich, P.; Sulem, P.; Thorlacius, S.; Gylfason, A.; Steinberg, S.; et al. Many sequence variants affecting diversity of adult human height. Nat. Genet. 2008, 40, 609–615. [Google Scholar] [CrossRef]
- Weedon, M.N.; Lango, H.; Lindgren, C.M.; Wallace, C.; Evans, D.M.; Mangino, M.; Freathy, R.M.; Perry, J.R.B.; Stevens, S.; Hall, A.S.; et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nat. Genet. 2008, 40, 575–583. [Google Scholar] [CrossRef]
- Flori, L.; Fritz, S.; Jaffrézic, F.; Boussaha, M.; Gut, I.; Heath, S.; Foulley, J.-L.; Gautier, M. The genome response to artificial selection: A case study in dairy cattle. PLoS ONE 2009, 4, e6595. [Google Scholar] [CrossRef] [PubMed]
- Eberlein, A.; Takasuga, A.; Setoguchi, K.; Pfuhl, R.; Flisikowski, K.; Fries, R.; Klopp, N.; Fürbass, R.; Weikard, R.; Kühn, C. Dissection of genetic factors modulating fetal growth in cattle indicates a substantial role of the non-SMC condensin I complex, subunit G (NCAPG) gene. Genetics 2009, 183, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Loughran, G.; Huigsloot, M.; Kiely, P.A.; Smith, L.M.; Floyd, S.; Ayllon, V.; O’Connor, R. Gene expression profiles in cells transformed by overexpression of the IGF-I receptor. Oncogene 2005, 24, 6185–6193. [Google Scholar] [CrossRef] [PubMed]
- Peeters, L.M.; Janssens, S.; Coussé, A.; Buys, N. Insect bite hypersensitivity in Belgian warmblood horses: Prevalence and risk factors. Vlaams Diergeneeskd. Tijdschr. 2014, 83, 240–249. [Google Scholar]
- Hauswirth, R.; Haase, B.; Blatter, M.; Brooks, S.A.; Burger, D.; Drögemüller, C.; Gerber, V.; Henke, D.; Janda, J.; Jude, R.; et al. Mutations in MITF and PAX3 cause “splashed white” and other white spotting phenotypes in horses. PLoS Genet. 2012, 8, e1002653. [Google Scholar] [CrossRef]
- Rieder, S.; Taourit, S.; Mariat, D.; Langlois, B.; Guérin, G. Mutations in the agouti (ASIP), the extension (MCIR), and the brown (TYRP1) loci and their association to coat color phenotypes in horses (Equus caballus). Mamm. Genome 2001, 12, 450–455. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ROH | |||||
|---|---|---|---|---|---|
| Length Class (Mbp) | N. 1 | NROH 2 | Percentage 3 | SROH 4 | LROH 5 |
| 0.5–1 | 88 | 19,746 | 69.5% | 224.4 | 0.70 |
| 1–2 | 88 | 6073 | 21.4% | 69.0 | 1.33 |
| 2–4 | 88 | 1587 | 5.6% | 18.0 | 2.74 |
| 4–8 | 88 | 664 | 2.3% | 7.5 | 5.42 |
| >8 | 84 | 353 | 1.2% | 4.2 | 13.40 |
| Inbreeding Based on ROH (FROH) | ||||
|---|---|---|---|---|
| Length Class (Mbp) | Mean | Min. 1 | Max. 2 | SD 3 |
| 0.5–1 | 0.07 | 0.06 | 0.08 | 0.004 |
| 1–2 | 0.04 | 0.03 | 0.05 | 0.005 |
| 2–4 | 0.02 | 0.01 | 0.04 | 0.006 |
| 4–8 | 0.02 | 0.002 | 0.04 | 0.008 |
| >8 | 0.02 | 0.00 | 0.09 | 0.02 |
| Total | 0.17 | 0.12 | 0.26 | 0.03 |
| ECA 1 | Start (bp) 2 | End (bp) 2 | Length (Kb) | Annotated Genes | % of Horses |
|---|---|---|---|---|---|
| 3 | 35,477,778 | 36,012,699 | 535 | ZNF469, ZFPM1,ZC3H18, IL17C, CYBA, MVD, SNAI3, RNF166, CTU2, PIEZO1 | 93% |
| 3 | 36,131,080 | 37,590,699 | 1460 | CBFA2T3, ACSF3, CDH15, SLC22A31, ANKRD11, SPG7, RPL13, CPNE7, DPEP1, CHMP1A, SPATA33, CDK10, SPATA2L, VPS9D1, ZNF276, FANCA, SPIRE2, TCF25, MC1R,TUBB3, DEF8, DBNDD1, GAS8, PRDM7, CENPE, BDH2, SLC9B2 | 92% |
| 3 | 106,769,095 | 107,709,841 | 941 | LCORL, NCAPG, DCAF16, FAM184B, | 92% |
| 10 | 24,965,648 | 25,501,456 | 536 | IL11, TMEM190, TMEM238, RPL28, UBE2S, SHISA7, ISOC2, C19orf85, ZNF628, NAT14, SSC5D, SBK2, SBK3, ZNF579, FIZ1, ZNF524, ZNF865, ZNF784, ZNF580, ZNF581, CCDC106, ZNF835, U2AF2, EPN1, RFPL4A, EQUCABV1R902, EQUCABV1R903, NLRP4, NLRP13, NLRP5 | 76% |
| 10 | 25,636,022 | 26,106,403 | 470 | EDDM13, ZNF667, ZNF583, ZNF582, SMIM17, ZNF471, ZFP28, ZNF470, ZNF71 | 76% |
| 11 | 22,957,922 | 24,177,108 | 1219 | CDK12, MED1, STAC2, CACNB1, ARL5C, PLXDC1, FBXO47, LINC00672, LASP1, RPL23, C17orf98, CWC25, PIP4K2B, PSMB3, MLLT6, PCGF2, CISD3, EPOP, SRCIN1, ARHGAP23, SOCS7, GPR179, MRPL45, NPEPPS, KPNB1, TBKBP1, TBX21, OSBPL7, MRPL10, LRRC46, SCRN2, SP6, SP2, PNPO | 73% |
| 15 | 67,045,471 | 67,539,463 | 494 | LCLAT1, LBH, YPEL5 | 73% |
| 19 | 55,113,101 | 55,782,316 | 669 | // | 77% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ablondi, M.; Dadousis, C.; Vasini, M.; Eriksson, S.; Mikko, S.; Sabbioni, A. Genetic Diversity and Signatures of Selection in a Native Italian Horse Breed Based on SNP Data. Animals 2020, 10, 1005. https://doi.org/10.3390/ani10061005
Ablondi M, Dadousis C, Vasini M, Eriksson S, Mikko S, Sabbioni A. Genetic Diversity and Signatures of Selection in a Native Italian Horse Breed Based on SNP Data. Animals. 2020; 10(6):1005. https://doi.org/10.3390/ani10061005
Chicago/Turabian StyleAblondi, Michela, Christos Dadousis, Matteo Vasini, Susanne Eriksson, Sofia Mikko, and Alberto Sabbioni. 2020. "Genetic Diversity and Signatures of Selection in a Native Italian Horse Breed Based on SNP Data" Animals 10, no. 6: 1005. https://doi.org/10.3390/ani10061005
APA StyleAblondi, M., Dadousis, C., Vasini, M., Eriksson, S., Mikko, S., & Sabbioni, A. (2020). Genetic Diversity and Signatures of Selection in a Native Italian Horse Breed Based on SNP Data. Animals, 10(6), 1005. https://doi.org/10.3390/ani10061005