Reconstruction and Analysis of Cattle Metabolic Networks in Normal and Acidosis Rumen Tissue


Simple Summary
Abstract
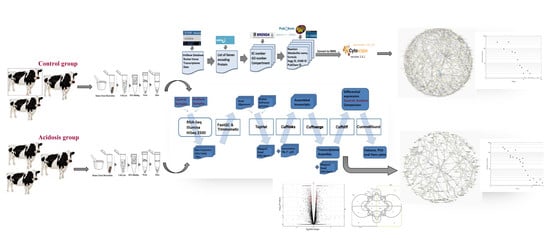
1. Introduction
2. Materials and Methods
2.1. Data Collection and Pretreatment
2.2. Reconstruction and Topological Analysis of the Metabolic Networks
2.3. Animal Management and Rumen Tissue Collection
2.4. RNA Extraction, Quantification and Sequencing
2.5. Sequence Data Processing and Differential Gene Expression Analysis
2.6. Quantitative Real-Time PCR (qRT-PCR) Validation of Selected Differentially Expressed Genes
3. Results
3.1. The Primary Metabolic Network
3.2. Transcriptome Profiling of the Rumen Epithelium
3.3. Acidosis Metabolic Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cottle, D.; Kahn, L. Beef Cattle Production and Trade; CSIRO Publishing: Clayton, Australia, 2014; p. 221. [Google Scholar]
- Degtyarenko, K.; de Matos, P.; Ennis, M.; Hastings, J.; Zbinden, M.; McNaught, A.; Alcántara, R.; Darsow, M.; Guedj, M.; Ashburner, M. ChEBI: A database and ontology for chemical entities of biological interest. Nucleic Acids Res. 2008, 36, D344–D350. [Google Scholar] [CrossRef] [PubMed]
- Steele, M.A.; Croom, J.; Kahler, M.; AlZahal, O.; Hook, S.E.; Plaizier, K.; McBride, B.W. Bovine rumen epithelium undergoes rapid structural adaptations during grain-induced subacute ruminal acidosis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R1515–R1523. [Google Scholar] [CrossRef] [PubMed]
- Steele, M.A.; Vandervoort, G.; AlZahal, O.; Hook, S.E.; Matthews, J.C.; McBride, B.W. Rumen epithelial adaptation to high-grain diets involves the coordinated regulation of genes involved in cholesterol homeostasis. Physiol. Genomics 2011, 43, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Enemark, J.M. The monitoring, prevention and treatment of sub-acute ruminal acidosis (SARA): A review. Vet. J. 2008, 176, 32–43. [Google Scholar] [CrossRef]
- Hernández, J.; Benedito, J.L.; Abuelo, A.; Castillo, C. Ruminal acidosis in feedlot: From aetiology to prevention. Sci. World J. 2014, 702572. [Google Scholar] [CrossRef]
- Li, W.; Gelsinger, S.; Edwards, A.; Riehle, C.; Koch, D. Transcriptome analysis of rumen epithelium and meta-transcriptome analysis of rumen epimural microbial community in young calves with feed induced acidosis. Sci. Rep. 2019, 9, 4744. [Google Scholar] [CrossRef]
- Jiang, X.Y.; Ni, Y.D.; Zhang, S.K.; Zhang, Y.S.; Shen, X.Z. Identification of Differentially Expressed Proteins in Liver in Response to Subacute Ruminal Acidosis (SARA) Induced by High-concentrate Diet. Asian Australas J. Anim. Sci. 2014, 27, 1181–1188. [Google Scholar] [CrossRef]
- Kleen, J.L.; Hooijer, G.A.; Rehage, J.; Noordhuizen, J.P. Subacute ruminal acidosis (SARA): A review. J. Vet. Med. A Physiol. Pathol. Clin. Med. 2003, 50, 406–414. [Google Scholar] [CrossRef]
- Xue, F.; Pan, X.; Jiang, L.; Guo, Y.; Xiong, B. GC-MS analysis of the ruminal metabolome response to thiamine supplementation during high grain feeding in dairy cows. Metabolomics 2018, 14, 67. [Google Scholar] [CrossRef]
- Bruggeman, F.J.; Westerhoff, H.V. The nature of systems biology. Trends Microbiol. 2007, 15, 45–50. [Google Scholar] [CrossRef]
- O’Shea, E.; Waters, S.M.; Keogh, K.; Kelly, A.K.; Kenny, D.A. Examination of the molecular control of ruminal epithelial function in response to dietary restriction and subsequent compensatory growth in cattle. J. Anim. Sci. Biotechnol. 2016, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, J.; Ju, Z.; Zhai, R.; Zhou, L.; Li, Q.; Li, J.; Li, R.; Huang, J.; Zhong, J. Reconstruction of metabolic network in the bovine mammary gland tissue. Mol. Biol. Rep. 2012, 39, 7311–7318. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Poolman, M.G.; Bonde, B.K.; Gevorgyan, A.; Patel, H.H.; Fell, D.A. Challenges to be faced in the reconstruction of metabolic networks from public databases. Syst. Biol. 2006, 153, 379–384. [Google Scholar] [CrossRef]
- Stein, S.E.; Heller, S.R.; Tchekhovskoi, D. An open standard for chemical structure representation: The IUPAC chemical identifier. In Proceedings of the 2003 International Chemical Information Conference, Nimes, France, 1 October 2003; pp. 131–143. [Google Scholar]
- Ma, H.; Zeng, A. Reconstruction of metabolic networks from genome data and analysis of their global structure for various organisms. Bioinformatics 2003, 19, 270–277. [Google Scholar] [CrossRef]
- Bornstein, B.J.; Keating, S.M.; Jouraku, A.; Hucka, M. LibSBML: An API library for SBML. Bioinformatics 2008, 24, 880–881. [Google Scholar] [CrossRef]
- Cline, M.S.; Smoot, M.; Cerami, E.; Kuchinsky, A.; Landys, N.; Workman, C.; Christmas, R.; Avila-Campilo, I.; Creech, M.; Gross, B.; et al. Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2007, 2, 2366–2382. [Google Scholar] [CrossRef]
- Managbanag, J.R.; Witten, T.M.; Bonchev, D.; Fox, L.A.; Tsuchiya, M.; Kennedy, B.K.; Kaeberlein, M. Shortest-path network analysis is a useful approach toward identifying genetic determinants of longevity. PLoS ONE 2008, 3, e3802. [Google Scholar] [CrossRef]
- Erdös, P.; Rényi, A. On the evolution of random graphs. Publ. Math. 1959, 6, 290–297. [Google Scholar]
- Jeong, H.; Tombor, B.; Albert, R.; Oltvai, Z.N.; Barabási, A.L. The large-scale organization of metabolic networks. Nature 2000, 407, 651–654. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kim, P.M.; Sprecher, E.; Trifonov, V.; Gerstein, M. The importance of bottlenecks in protein networks: Correlation with gene essentiality and expression dynamics. PLoS Comput. Biol. 2007, 3, e59. [Google Scholar] [CrossRef] [PubMed]
- Bi, R.; Liu, P. Sample size calculation while controlling false discovery rate for differential expression analysis with RNA-sequencing experiments. BMC Bioinform. 2016, 17, 146. [Google Scholar] [CrossRef] [PubMed]
- Radrich, K.; Tsuruoka, Y.; Dobson, P.; Gevorgyan, A.; Swainston, N.; Baart, G.; Schwartz, J.M. Integration of metabolic databases for the reconstruction of genome-scale metabolic networks. BMC Syst. Biol. 2010, 4, 114. [Google Scholar] [CrossRef]
- Kong, R.S.; Liang, G.; Chen, Y.; Stothard, P.; Guan le, L. Transcriptome profiling of the rumen epithelium of beef cattle differing in residual feed intake. BMC Genom. 2016, 17, 592. [Google Scholar] [CrossRef]
- Telesford, Q.K.; Joyce, K.E.; Hayasaka, S.; Burdette, J.H.; Laurienti, P.J. The ubiquity of small-world networks. Brain Connect. 2011, 1, 367–375. [Google Scholar] [CrossRef]
- Embar, V.; Handen, A.; Ganapathiraju, M.K. Is the average shortest path length of gene set a reflection of their biological relatedness? J. Bioinform. Comput. Biol. 2016, 14, 1660002. [Google Scholar] [CrossRef]
- Zhang, P.; Foerster, H.; Tissier, C.P.; Mueller, L.; Paley, S.; Karp, P.D.; Rhee, S.Y. MetaCyc and AraCyc. Metabolic pathway databases for plant research. Plant Physiol. 2005, 138, 27–37. [Google Scholar] [CrossRef]
- Bergman, E.N. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol. Rev. 1990, 70, 567–590. [Google Scholar] [CrossRef] [PubMed]
- Ash, R.; Baird, G.D. Activation of volatile fatty acids in bovine liver and rumen epithelium. Biochem. J. 1973, 136, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Braun, P.; Rietman, E.; Vidal, M. Networking metabolites and diseases. Proc. Natl. Acad. Sci. USA 2008, 105, 9849–9850. [Google Scholar] [CrossRef]
- Leonardi, R.; Jackowski, S. Biosynthesis of Pantothenic Acid and Coenzyme A. EcoSal Plus 2007, 2. [Google Scholar] [CrossRef] [PubMed]
- Artegoitia, V.M.; Foote, A.P.; Lewis, R.M.; Freetly, H.C. Rumen Fluid Metabolomics Analysis Associated with Feed Efficiency on Crossbred Steers. Sci. Rep. 2017, 6, 2864. [Google Scholar] [CrossRef]
- Atteia, A.; van Lis, R.; Gelius-Dietrich, G.; Adrait, A.; Garin, J.; Joyard, J.; Rolland, N.; Martin, W. Pyruvate formate-lyase and a novel route of eukaryotic ATP synthesis in Chlamydomonas mitochondria. J. Biol. Chem. 2006, 281, 9909–9918. [Google Scholar] [CrossRef]
- Abd Ellah, M.R.; Okada, K.; Yasuda, J. Oxidative stress and bovine liver diseases: Role of glutathione peroxidase and glucose 6-phosphate dehydrogenase. Jpn. J. Vet. Res. 2007, 54, 163–173. [Google Scholar]
- Moffatt, B.A.; Ashihara, H. Purine and pyrimidine nucleotide synthesis and metabolism. Arab. Book 2002, 1, e0018. [Google Scholar] [CrossRef]
- Clardy, J.; Fischbach, M.A.; Currie, C.R. The natural history of antibiotics. Curr. Biol. 2009, 19, R437–R441. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | The 1st Day ‡ | The 30th Day ‡ | The 129th Day ‡ |
|---|---|---|---|
| control | 5.71; 5.89; 5.88 | 5.57; 5.97; 5.87 | 6.03; 6.17; 6.0 |
| acidosis | 5.95; 5.23; 5.64 | 5.97; 5.17; 5.57 | 5.19; 5.10; 5.24 |
| Parameters | Values |
|---|---|
| Number of nodes | 1429 |
| Number of edges | 1771 |
| Network density | 0.002 |
| Network heterogeneity | 0.862 |
| Number of self-loops | 0 |
| Network diameter | 38 |
| Network centralization | 0.019 |
| Average path length | 13.142 |
| Average number of neighbors | 2.476 |
| Metabolites | Neutral Formula | KeggID | PubChemID | Compartment |
|---|---|---|---|---|
| CoA | C21H36N7O16P3S | C00010 | 49600643 | Mitochondrion |
| glutathione | C10H17N3O6S | C00051 | 3353 | Cytoplasm |
| S-adenosyl-L-homocysteine | C14H20N6O5S | C00021 | 439155 | Nucleus |
| S-adenosyl-L-methionine | C15H22N6O5S | C00019 | 34755 | Nucleus |
| 2-oxoglutarate | C5H6O5 | C00026 | 3328 | Mitochondrion |
| Acetyl-CoA | C23H38N7O17P3S | C00024 | 3326 | Cytoplasm |
| D-fructose_6-phosphate | C6H13O9P | C00085 | 3385 | Cytoplasm |
| ubiquinone | C19H26O4 | C00399 | 5280346 | Mitochondrion |
| formate | CH2O2 | C00058 | 3358 | Mitochondrion |
| electron-transferring flavoprotein | C27H30N9O15P2 | C04253 | 6918 | Mitochondrion |
| L-tryptophan | C11H12N2O2 | C00078 | 3378 | Cytoplasm |
| pyruvate | C3H4O3 | C00022 | 3324 | Mitochondrion |
| L-tyrosine | C9H11NO3 | C00082 | 3382 | Cytoplasm |
| tetrahydrofolate | C19H23N7O6 | C00101 | 3401 | Mitochondrion |
| L-serine | C3H7NO3 | C00065 | 3365 | Nucleus |
| Gene | EC Number | Enzyme Name | Reaction | P-Value |
|---|---|---|---|---|
| HSPA6 | 4.2.1.134 | very-long-chain (3R)-3-hydroxyacyl-CoA dehydratase | R10827 | 5.00E-05 |
| GUCY1B1 | 4.6.1.2 | guanylate cyclase | R00434 | 0.0001 |
| PDK2 | 5.3.99.4 | prostacyclin synthase | R02267 | 0.0019 |
| PTGDS | 5.3.99.2 | prostaglandin-D synthase | R02266 | 0.0021 |
| OXCT1 | 2.8.3.5 | succinyl-CoA transferase | R01780 | 0.0002 |
| SLC24A3 | 3.1.1.34 | lipoprotein lipase | R01369 | 0.0001 |
| PLA2G4A | 3.1.1.4 | phospholipase A2 | R01313 | 0.004 |
| PDE5A | 3.1.4.35 | cGMP phosphodiesterase | R01234 | 5.00E-05 |
| CYP2W1 | 3.1.4.17 | cyclic 3′,5′-phosphodiesterase | R03259 | 0.00085 |
| PLCD3 | 3.1.4.11 | Phosphorinositidase C | R03435 | 0.00355 |
| EPHX1 | 3.3.2.9 | microsomal epoxide hydrolase | R07627 | 0.00085 |
| DYSF | 3.6.1.5 | ATP-diphosphatase | R10443 | 5.00E-05 |
| GSTM3 | 2.5.1.18 | glutathione transferase | R03522 | 0.0003 |
| FOXF1 | 2.6.1.42 | branched-chain-amino-acid transaminase | R01090 | 0.0002 |
| PFKM | 2.7.1.11 | 6-phosphofructokinase | R00756 | 0.0015 |
| CHPT1 | 2.7.8.2 | diacylglycerol cholinephosphotransferase | R01321 | 0.0012 |
| CDS2 | 2.7.7.41 | phosphatidate cytidylyl transferase | R01799 | 0.00215 |
| MMP16 | 2.7.7.40 | CDP ribitol pyro phosphorylase | R02921 | 0.0027 |
| CEP112 | 2.7.12.2 | MAP kinase kinase | R00162 | 0.0014 |
| CHRNA3 | 1.14.13.39 | nitric-oxide synthase (NADPH) | R00557 | 5.00E-05 |
| TNS2 | 1.11.1.9 | glutathione peroxidase | R00274 | 0.0018 |
| TSPAN12 | 1.14.14.1 | flavoprotein monooxygenase | R04122 | 5.00E-05 |
| DPYD | 1.3.1.2 | dihydrouracil dehydrogenase (NADP+) | R00978 | 5.00E-05 |
| ALDH9A1 | 1.2.1.3 | aldehyde dehydrogenase (NAD+) | R00538 | 0.0001 |
| AOC3 | 1.4.3.21 | primary-amine oxidase | R01853 | 0.0001 |
| SOX5 | 1.4.3.1 | D-aspartate oxidase | R00359 | 5.00E-05 |
| MAOB | 1.4.3.4 | monoamine oxidase | R11354 | 5.00E-05 |
| CILP | 1.7.1.7 | GMP reductase | R01134 | 5.00E-05 |
| AASS | 1.5.1.8 | lysine-2-oxoglutarate reductase | R00716 | 5.00E-05 |
| MTHFR | 1.5.1.20 | methylenetetrahydrofolate | R01224 | 5.00E-05 |
| GATM | 2.1.4.1 | glycine amidino transferase | R00565 | 0.00015 |
| SCN7A | 2.1.1.43 | N-methyltransferase | R03938 | 5.00E-05 |
| CPT1A | 2.3.1.21 | Palmitoyl carnitine transferase | R01923 | 5.00E-05 |
| Gene | EC Number | Enzyme Name | Reaction | P-Value |
|---|---|---|---|---|
| ASNS | 6.3.5.4 | asparagine synthase | R00578 | 5.00E-05 |
| CTPS1 | 6.3.4.2 | CTP synthase | R00573 | 5.00E-05 |
| TARS | 6.1.1.3 | threonyl-tRNA synthetase | R03663 | 0.00095 |
| WARS | 6.1.1.2 | tryptophanyl-tRNA synthetase | R03664 | 0.0013 |
| YARS | 6.1.1.1 | tyrosine-tRNA ligase | R02918 | 0.0003 |
| ISYNA1 | 5.5.1.4 | inositol-3-phosphate synthase | R07324 | 0.0005 |
| CTH | 4.4.1.1 | cystathionine gamma-lyase | R01001 | 5.00E-05 |
| SDS | 4.3.1.17 | L-serine ammonia-lyase | R00220 | 5.00E-05 |
| CA3 | 4.2.1.1 | carbonate dehydratase | R00132 | 0.00075 |
| ODC1 | 4.1.1.17 | ornithine decarboxylase | R00670 | 5.00E-05 |
| ENPP4 | 3.6.1.29 | bis(5′-adenosyl)-triphosphatase | R00187 | 0.0019 |
| PPA1 | 3.6.1.1 | inorganic diphosphatase | R00004 | 0.0001 |
| ADA | 3.5.4.4 | adenosine deaminase | R01560 | 5.00E-05 |
| ARG1 | 3.5.3.1 | arginase | R00551 | 5.00E-05 |
| GGH | 3.4.19.9 | folate gamma-glutamyl hydrolase | R04242 | 0.0033 |
| PSPH | 3.1.3.3 | phosphoserine phosphatase | R00582 | 0.0003 |
| PRDX6 | 3.1.1.4 | phospholipase A2 | R01313 | 0.00115 |
| ABHD12 | 3.1.1.23 | acyl glycerol lipase | R01351 | 0.0003 |
| PDXK | 2.7.1.35 | pyridoxal kinase | R00174 | 0.0034 |
| GK | 2.7.1.30 | glycerol kinase | R00847 | 5.00E-05 |
| PSAT1 | 2.6.1.52 | phosphoserine transaminase | R04173 | 5.00E-05 |
| GOT2 | 2.6.1.1 | aspartate transaminase | R00355 | 0.0009 |
| UPP1 | 2.4.2.3 | uridine phosphorylase | R01876 | 5.00E-05 |
| PNP | 2.4.2.1 | purine-nucleoside phosphorylase | R08368 | 0.00295 |
| C1GALT1 | 2.4.1.122 | D-galactosyltransferase | R05908 | 5.00E-05 |
| DGAT2 | 2.3.1.20 | diglyceride acyltransferase | R02251 | 0.0002 |
| TALDO1 | 2.2.1.2 | formaldehyde transketolase | R08575 | 0.0015 |
| PYCR1 | 1.5.1.2 | proline oxidase | R01248 | 5.00E-05 |
| MTHFD2 | 1.5.1.15 | methylenetetrahydrofolate dehydrogenase (NAD+) | R01218 | 5.00E-05 |
| DHCR7 | 1.3.1.21 | 7-dehydrocholesterol reductase | R01456 | 0.0012 |
| SCD | 1.14.19.1 | stearoyl-CoA 9-desaturase | R02222 | 5.00E-05 |
| LPO | 1.11.1.7 | peroxidase | R03344 | 5.00E-05 |
| PGD | 1.1.1.44 | phosphogluconate dehydrogenase | R03532 | 0.0022 |
| DCXR | 1.1.1.10 | L-xylulose reductase | R01904 | 0.00155 |
| Parameters | Values |
|---|---|
| Number of nodes | 832 |
| Number of edges | 1021 |
| Network density | 0.003 |
| Network heterogeneity | 0.733 |
| Number of self-loops | 0 |
| Network diameter | 28 |
| Network centralisation | 0.15 |
| Average path length | 6.913 |
| Metabolites | Neutral Formula | KeggID | PubChemID | Compartment |
|---|---|---|---|---|
| Stearoyl-CoA | C39H70N7O17P3S | C00412 | 3702 | Endoplasmic Reticulum |
| Ferrocytochrome b5 | C36H61O11 | C00996 | 4242 | Membrane |
| glutathione | C10H17N3O6S | C00051 | 3353 | Cytoplasm |
| 2-oxoglutarate | C5H6O5 | C00026 | 3328 | Mitochondrion |
| protein-L-arginine | C7H13N5O2R2 | C00613 | 3887 | Nucleus |
| electron-transferring flavoprotein | C27H30N9O15P2 | C04253 | 6918 | Mitochondrion |
| ubiquinone | C19H26O4 | C00399 | 5280346 | Mitochondrion |
| S-adenosyl-L-methionine | C15H22N6O5S | C00019 | 34755 | Cytoplasm |
| S-adenosyl-L-homocysteine | C14H20N6O5S | C00021 | 439155 | Nucleus |
| Acetyl-CoA | C23H38N7O17P3S | C00024 | 3326 | Cytoplasm |
| L-tryptophan | C11H12N2O2 | C00078 | 3378 | Cytoplasm |
| L-arginine | C7H13N5O2R2 | C00613 | 3887 | Mitochondrion |
| L-serine | C3H7NO3 | C00065 | 3365 | Cytoplasm |
| L-Lactate | C3H6O3 | C00186 | 3486 | Cytoplasm |
| Xanthine | C10H13N4O9P | C00655 | 73323 | Nucleus |
| Significant Up-expression enzymes | Fatty acid metabolism | Butyrate metabolism (ec00650) Arachidonic acid metabolism (ec00590) Linoleic acid metabolism (ec00591) Fatty acid degradation (ec00071) Pantothenate and CoA biosynthesis (ec00770) |
| Biosynthesis of amino acids | Valine, leucine and isoleucine metabolism (ec00280) Glycine, serine and threonine metabolism (ec00260) | |
| Pyruvate and carbon metabolism | Glycolysis/Gluconeogenesis (ec00010) Pentose phosphate (ec00030) Pyruvate metabolism (ec00620) Glutathione metabolism (ec00480) Fructose and mannose metabolism (ec00051) Galactose metabolism (ec00052) Synthesis and degradation of ketone bodies (ec00072) Pentose and glucuronate interconversions (ec00040) | |
| Significant Down-expression enzymes | Nucleotide metabolism | Purine metabolism (ec00230) Pyrimidine metabolism (ec00240) |
| Other metabolisms | Vitamin B6 metabolism (ec00750) Biosynthesis of antibiotics (ec01130) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gholizadeh, M.; Fayazi, J.; Asgari, Y.; Zali, H.; Kaderali, L. Reconstruction and Analysis of Cattle Metabolic Networks in Normal and Acidosis Rumen Tissue. Animals 2020, 10, 469. https://doi.org/10.3390/ani10030469
Gholizadeh M, Fayazi J, Asgari Y, Zali H, Kaderali L. Reconstruction and Analysis of Cattle Metabolic Networks in Normal and Acidosis Rumen Tissue. Animals. 2020; 10(3):469. https://doi.org/10.3390/ani10030469
Chicago/Turabian StyleGholizadeh, Maryam, Jamal Fayazi, Yazdan Asgari, Hakimeh Zali, and Lars Kaderali. 2020. "Reconstruction and Analysis of Cattle Metabolic Networks in Normal and Acidosis Rumen Tissue" Animals 10, no. 3: 469. https://doi.org/10.3390/ani10030469
APA StyleGholizadeh, M., Fayazi, J., Asgari, Y., Zali, H., & Kaderali, L. (2020). Reconstruction and Analysis of Cattle Metabolic Networks in Normal and Acidosis Rumen Tissue. Animals, 10(3), 469. https://doi.org/10.3390/ani10030469