Genetic Variability in Local and Imported Germplasm Chicken Populations as Revealed by Analyzing Runs of Homozygosity

 ,
,  , , , , , ,
, , , , , ,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
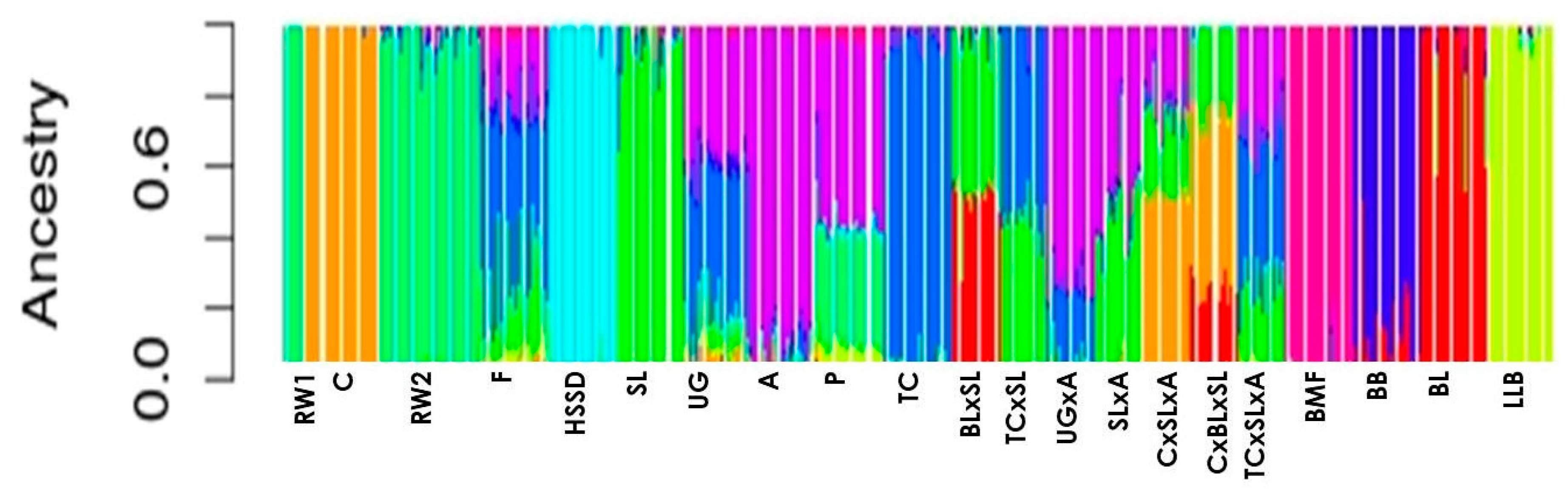
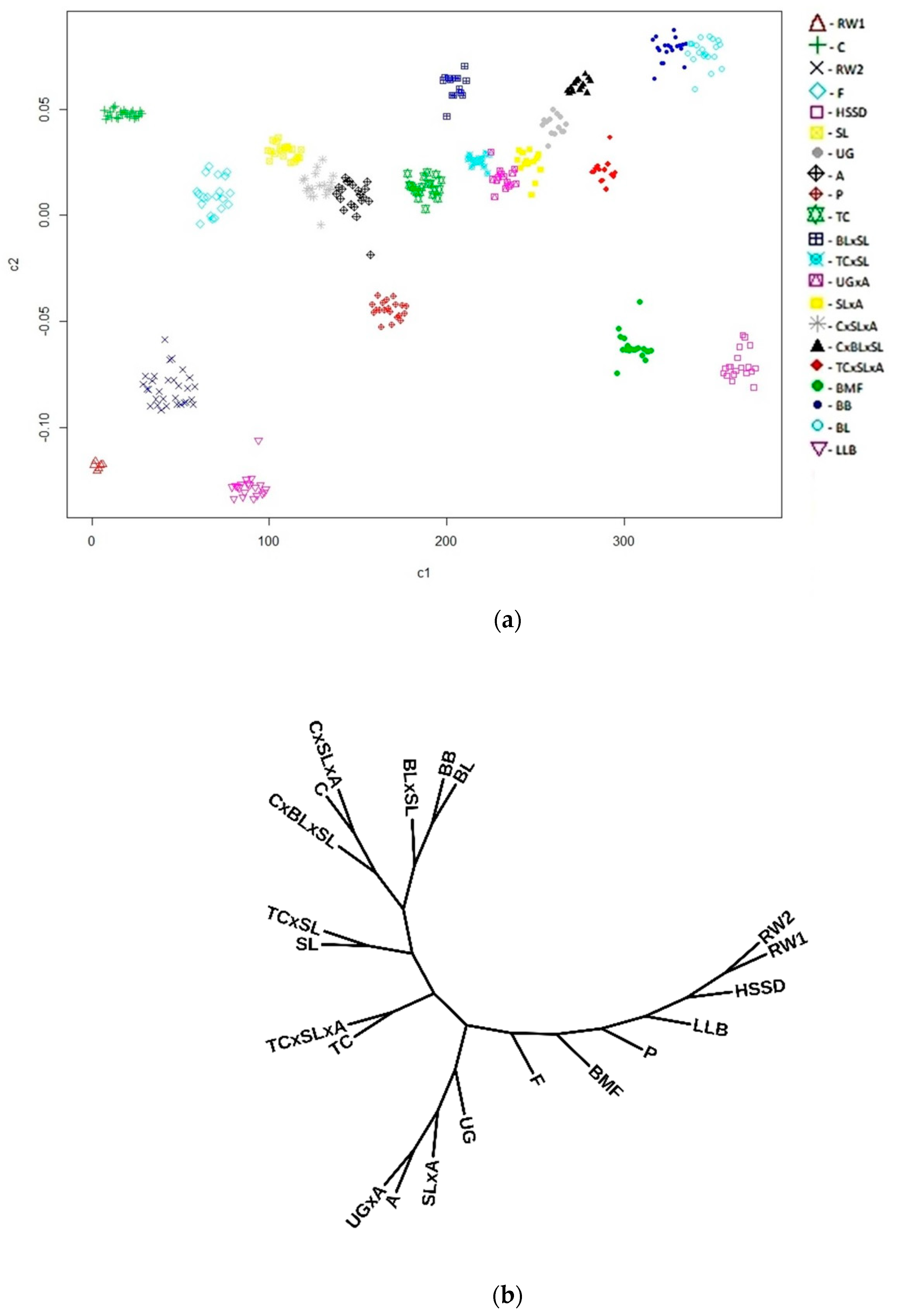
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Romanov, M.N.; Weigend, S. Genetic diversity in chicken populations based on microsatellite markers. In Proceedings of the Conference from Jay Lush to Genomics: Visions for Animal Breeding and Genetics, Ames, IA, USA, 16–18 May 1999; Dekkers, J.C.M., Lamont, S.J., Rothschild, M.F., Eds.; Iowa State University, Department of Animal Science: Ames, IA, USA, 1999; p. 174. [Google Scholar]
- Chen, L.; Wang, X.; Cheng, D.; Chen, K.; Fan, Y.; Wu, G.; You, J.; Liu, S.; Mao, H.; Ren, J. Population genetic analyses of seven Chinese indigenous chicken breeds in a context of global breeds. Anim. Genet. 2019, 50, 82–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joaquim, L.B.; Chud, T.C.S.; Marchesi, J.A.P.; Savegnago, R.P.; Buzanskas, M.E.; Zanella, R.; Cantao, M.E.; Peixoto, J.O.; Ledur, M.C.; Irgang, R.; et al. Genomic structure of a crossbred Landrace pig population. PLoS ONE 2019, 14, e0212266. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.O.; Romanov, M.N.; Plemyashov, K.V.; Dementieva, N.V.; Mitrofanova, O.V.; Barkova, O.Y.; Womack, J.E. Haplotype structure and copy number polymorphism of the beta-defensin 7 genes in diverse chicken breeds. Anim. Genet. 2017, 48, 490–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dementieva, N.V.; Fedorova, E.S.; Krutikova, A.A.; Mitrofanova, O.V.; Stanishevskaya, O.I.; Pleshanov, N.V.; Smaragdov, M.G.; Kudinov, A.A.; Terletsky, V.P.; Romanov, M.N. Genetic variability of indels in the prolactin and dopamine receptor D2 genes and their association with the yield of allanto-amniotic fluid in Russian White laying hens. J. Agric. Sci. 2020, 26, 373–379. [Google Scholar] [CrossRef]
- Romanov, M.N.; Dementyeva, N.V.; Terletsky, V.P.; Plemyashov, K.V.; Stanishevskaya, O.I.; Kudinov, A.A.; Perinek, O.Y.; Fedorova, E.S.; Larkina, T.A.; Pleshanov, N.V. Applying SNP array technology to assess genetic diversity in Russian gene pool of chickens. In Proceedings of the International Plant and Animal Genome XXV Conference, San Diego, CA, USA, 14–18 January 2017; Scherago International: San Diego, CA, USA, 2017. Abstract P0115. [Google Scholar]
- Dementeva, N.V.; Romanov, M.N.; Kudinov, A.A.; Mitrofanova, O.V.; Stanishevskaya, O.I.; Terletsky, V.P.; Fedorova, E.S.; Nikitkina, E.V.; Plemyashov, K.V. Studying the structure of a gene pool population of the Russian White chicken breed by genome-wide SNP scan. Selskokhoziaĭstvennaia Biol. 2017, 52, 1166–1174. [Google Scholar] [CrossRef] [Green Version]
- Dementeva, N.V.; Kudinov, A.A.; Mitrofanova, O.V.; Mishina, A.I.; Smaragdov, M.G.; Yakovlev, A.F. Chicken resource population as the source of study genetic improvement of indigenous breeds. J. Anim. Sci. 2018, 96, 513. [Google Scholar] [CrossRef]
- Kudinov, A.A.; Dementieva, N.V.; Mitrofanova, O.V.; Stanishevskaya, O.I.; Fedorova, E.S.; Larkina, T.A.; Mishina, A.I.; Plemyashov, K.V.; Griffin, D.K.; Romanov, M.N. Genome-wide association studies targeting the yield of extraembryonic fluid and production traits in Russian White chickens. BMC Genom. 2019, 20, 270. [Google Scholar] [CrossRef]
- Zhang, Q.; Guldbrandtsen, B.; Bosse, M.; Lund, M.S.; Sahana, G. Runs of homozygosity and distribution of functional variants in the cattle genome. BMC Genom. 2015, 16, 542. [Google Scholar] [CrossRef] [Green Version]
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef] [Green Version]
- Peripolli, E.; Munari, D.P.; Silva, M.V.G.B.; Lima, A.L.F.; Irgang, R.; Baldi, F. Runs of homozygosity: Current knowledge and applications in livestock. Anim. Genet. 2016, 48, 255–271. [Google Scholar] [CrossRef]
- Kirin, M.; McQuillan, R.; Franklin, C.S.; Campbell, H.; McKeigue, P.M.; Wilson, J.F. Genomic runs of homozygosity record population history and consanguinity. PLoS ONE 2010, 5, e13996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosse, M.; Megens, H.J.; Madsen, O.; Paudel, Y.; Frantz, L.A.F.; Schook, L.B.; Crooijmans, R.P.; Groenen, M.A. Regions of homozygosity in the porcine genome: Consequence of demography and the recombination landscape. BMC Genet. 2012, 8, e1003100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrero-Medrano, J.M.; Megens, H.J.; Groenen, M.A.M.; Ramis, G.; Bosse, M.; Perez-Enciso, M.; Crooijmans, R.P. Conservation genomic analysis of domestic and wild pig populations from the Iberian Peninsula. BMC Genet. 2013, 14, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef]
- Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; Sham, P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- McQuillan, R.; Leutenegger, A.-L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Hornik, K. The R FAQ (2017). Available online: https://CRAN.R-project.org/doc/FAQ/R-FAQ.html (accessed on 10 October 2020).
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, s13742-015. [Google Scholar] [CrossRef]
- Wright, S. Variability within and among Natural Populations. In Evolution and the Genetics of Populations; University of Chicago Press: Chicago, IL, USA, 1978; Volume 4. [Google Scholar]
- Patterson, N.; Price, A.L.; Reich, D. Population structure and eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef]
- Felsenstein, J. PHYLIP−Phylogeny Inference Package (Version 3.2). Cladistics. 1989, 5, 164–166. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Sokolova, A.N. Genetic and Selection Methods of Creation of a Chicken Population with an Increased Resistance to Neoplasms: Author’s Abstract. Ph.D. Thesis, RRIFAGB, Pushkin, St. Petersburg, Russia, 1999. [Google Scholar]
- Mastrangelo, S.; Ciani, E.; Sardina, M.T.; Sottile, G.; Pilla, F.; Portolano, B. Runs of homozygosity reveal genome-wide autozygosity in Italian sheep breeds. Anim. Genet. 2018, 49, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Mastrangelo, S.; Tolone, M.; Di Gerlando, R.; Fontanesi, L.; Sardina, M.T.; Portolano, B. Genomic inbreeding estimation in small populations: Evaluation of runs of homozygosity in three local dairy cattle breeds. Animal 2019, 10, 746–754. [Google Scholar] [CrossRef] [Green Version]
- Bortoluzzi, C.; Crooijmans, R.P.M.A.; Bosse, M.; Hiemstra, S.J.; Groenen, M.A.M.; Megens, H.-J. The effects of recent changes in breeding preferences on maintaining traditional Dutch chicken genomic diversity. Heredity 2018, 121, 564–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, T.F.; Amills, M.; Bertolini, F.; Rothschild, M.; Marras, G.; Boink, G.; Jordana, J.; Capote, J.; Carolan, S.; Hallsson, J.H.; et al. Patterns of homozygosity in insular and continental goat breeds. Genet. Sel. Evol. 2018, 50, 56. [Google Scholar] [CrossRef] [Green Version]
- Grilz-Seger, G.; Druml, T.; Neuditschko, M.; Dobretsberger, M.; Horna, M.; Brem, G. High-resolution population structure and runs of homozygosity reveal the genetic architecture of complex traits in the Lipizzan horse. BMC Genom. 2019, 20, 174. [Google Scholar] [CrossRef] [Green Version]
- Howard, J.T.; Tiezzi, F.; Huang, Y.; Gray, K.A.; Maltecca, C. Characterization and management of long runs of homozygosity in parental nucleus lines and their associated crossbred progeny. Genet. Sel. Evol. 2016, 48, 91. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
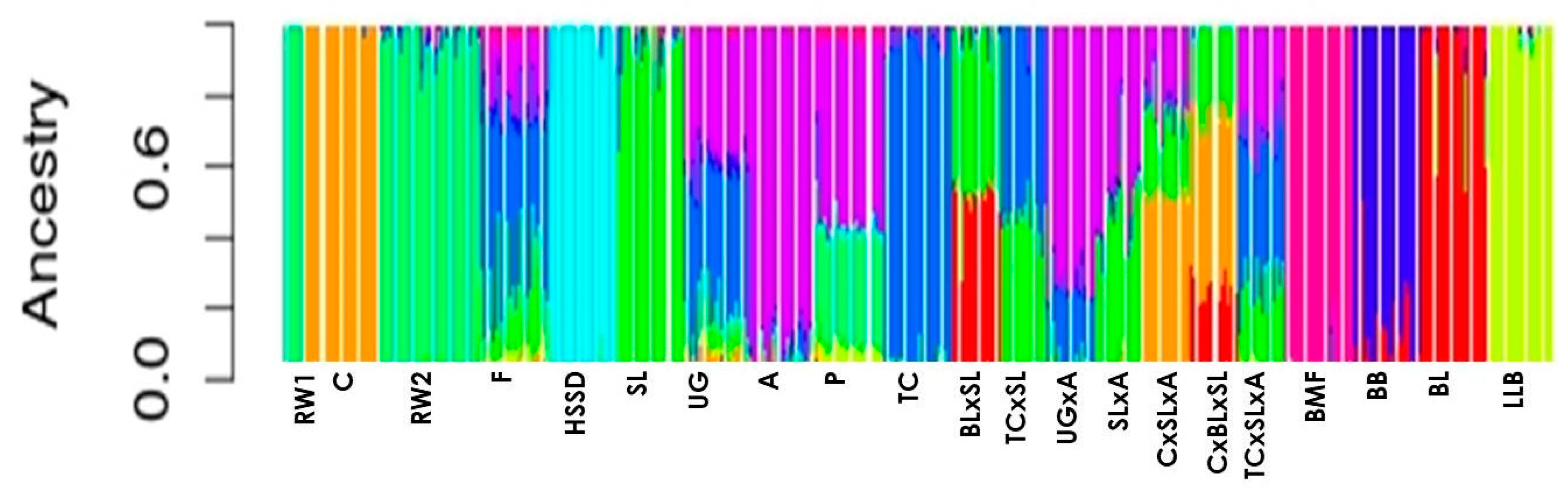{kind=link}
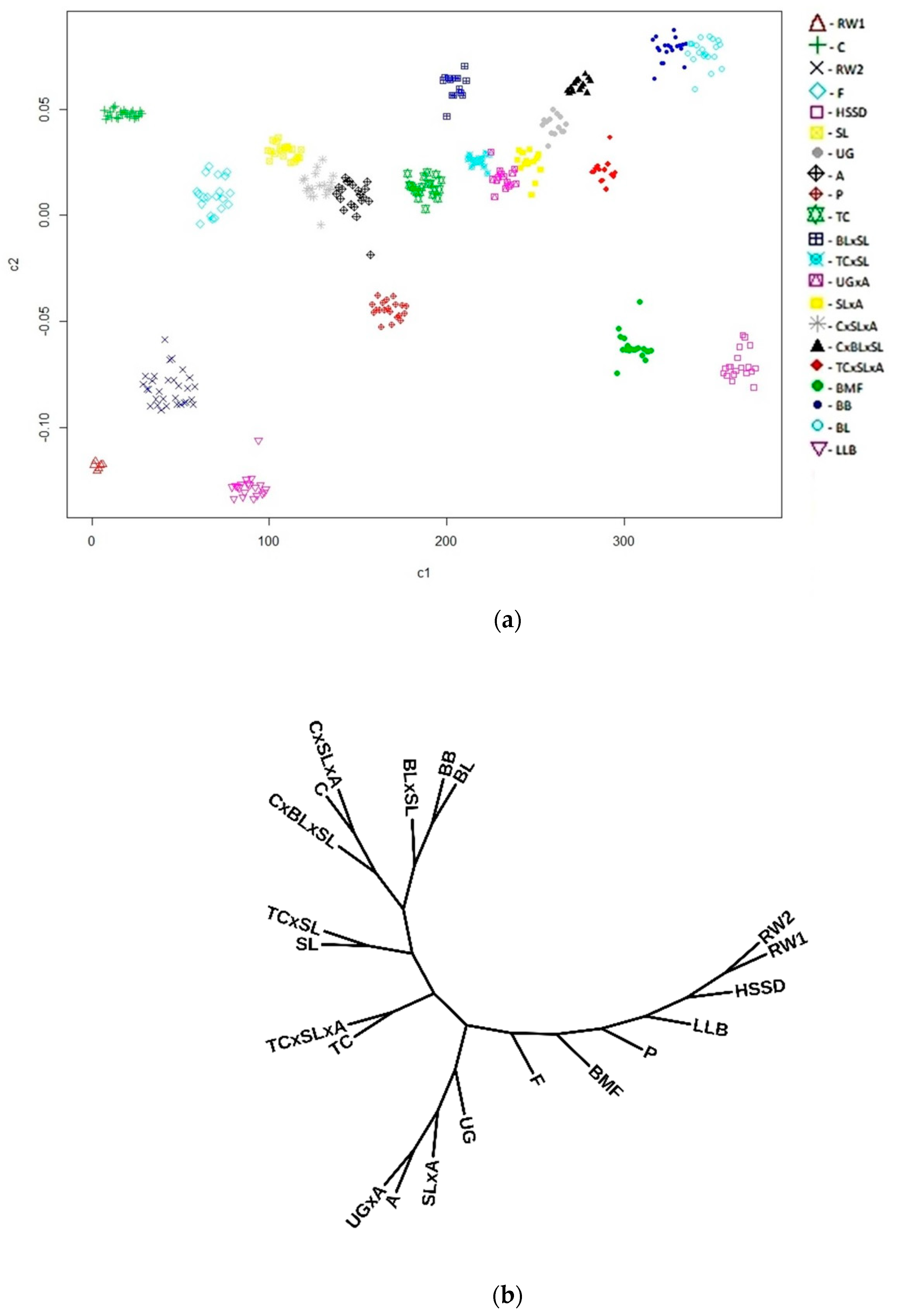{kind=link}
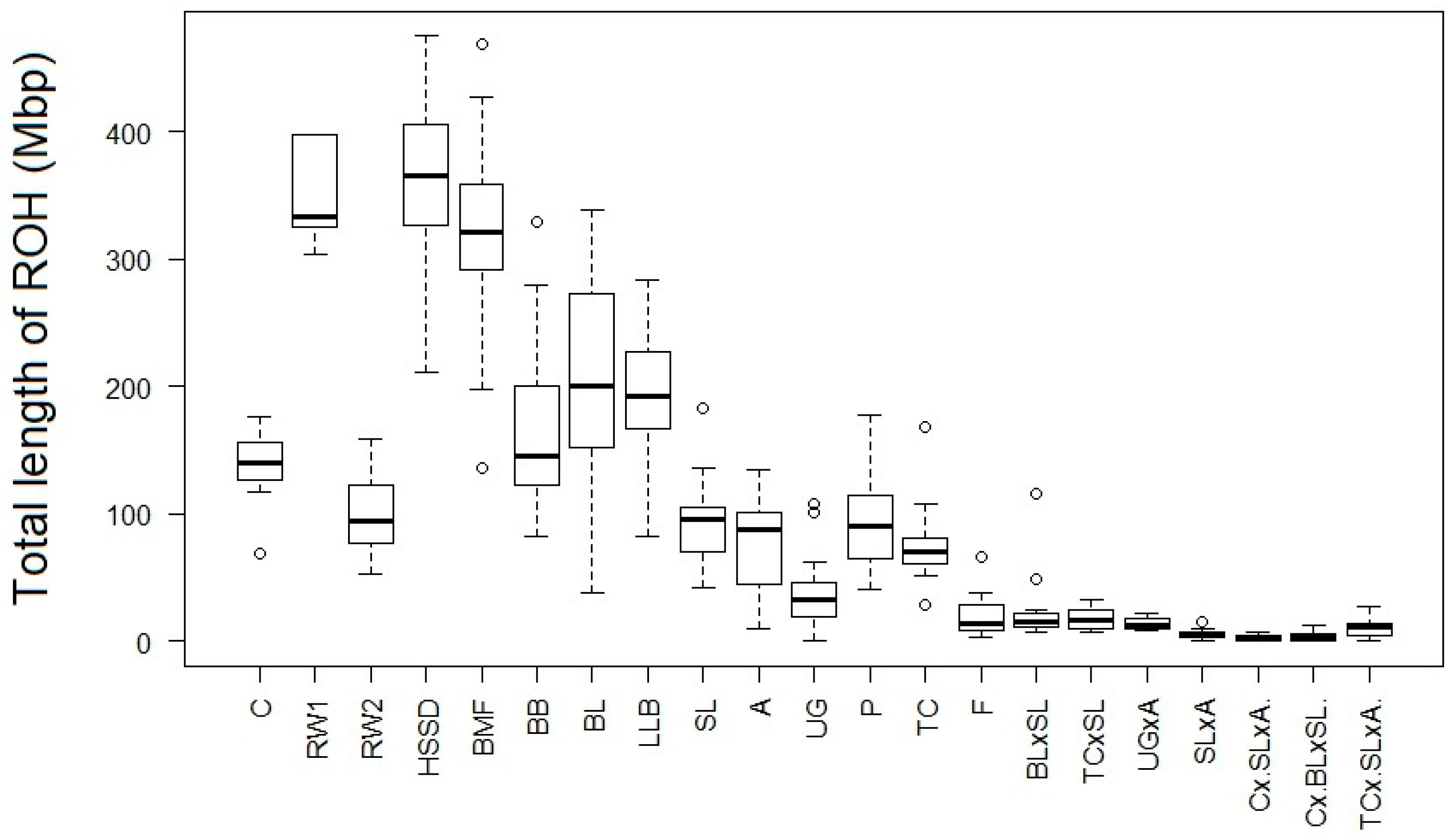
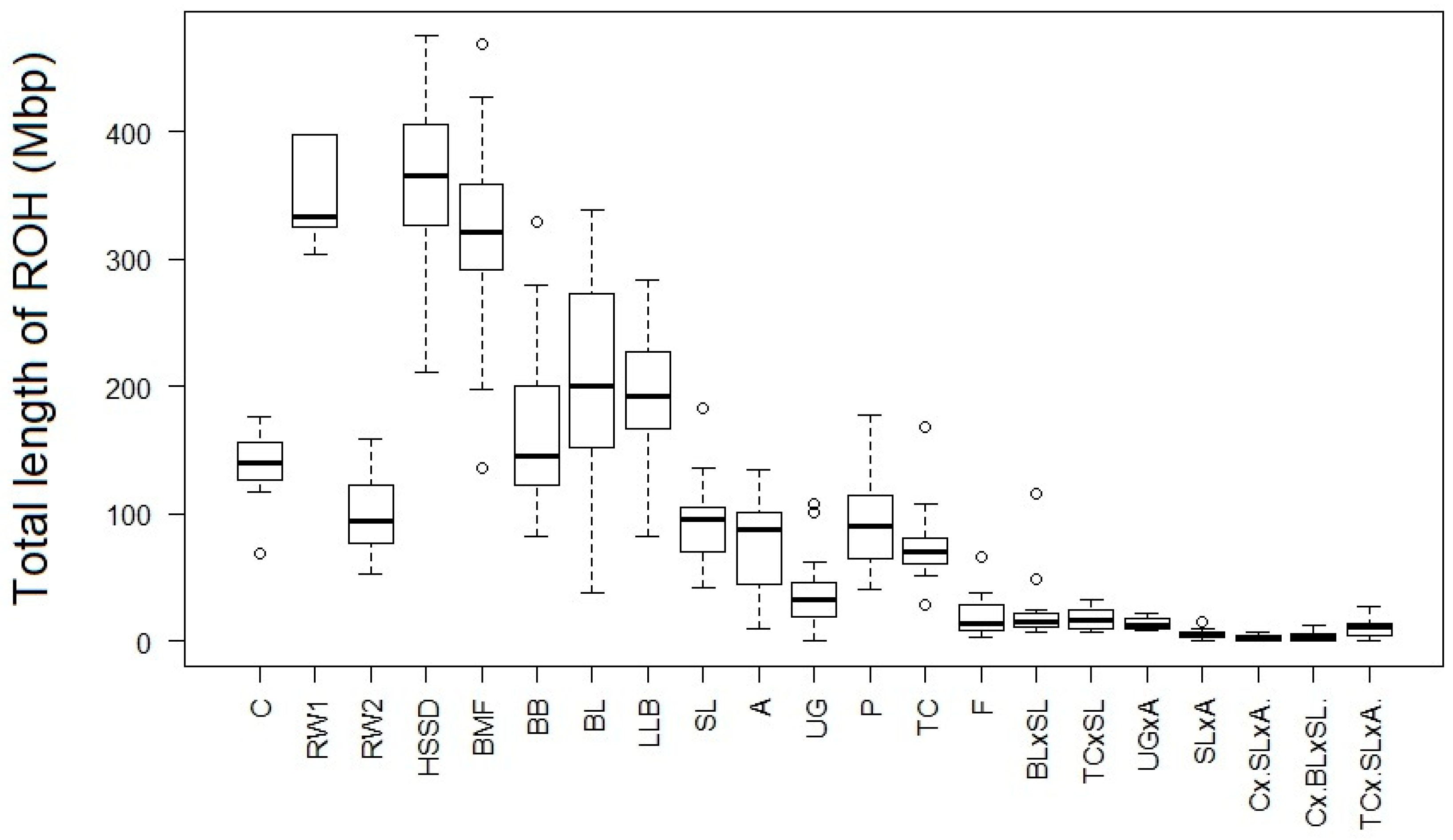
| Population | Abbreviation Code | Origin | Type | Sample Size | Number of ROHs per Individual | Length of ROH, Kb | FROH1 | LD | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | Minimum | Maximum | Mean | Minimum | Maximum | |||||||
| Pure breeds | ||||||||||||
| Amroks Cuckoo | A | USA | dual purpose | 20 | 18.9 ± 1.5 | 3 | 31 | 4057.2 ± 198.8 | 2308.9 | 5998.3 | 0.105 ± 0.009 | 0.187 ± 0.001 |
| Brahma Buff | BB | India, USA | fancy, meat | 20 | 36.6 ± 2.1 | 24 | 56 | 4508.4 ± 194.7 | 3116.2 | 6347.6 | 0.167 ± 0.015 | 0.283 ± 0.001 |
| Brahma Light | BL | “ 2 | “ | 20 | 37.4 ± 3.0 | 10 | 56 | 5259.3 ± 193.4 | 3772.7 | 6916.0 | 0.203 ± 0.017 | 0.286 ± 0.001 |
| Bantam Mille Fleur (or Russian Korolyok) | BMF | Russia | fancy | 20 | 61.2 ± 2.1 | 31 | 78 | 5259.0 ± 234.0 | 4043.3 | 8082.3 | 0.291 ± 0.015 | 0.374 ± 0.001 |
| White Cornish (two-way hybrid C1 bred inter se) | C | England | meat | 20 | 40.4 ± 1.1 | 33 | 52 | 3609.8 ± 80.7 | 3105.0 | 4494.0 | 0.055 ± 0.007 | 0.155 ± 0.001 |
| Frizzle | F | Asia, Europe | fancy | 20 | 5.9 ± 0.1 | 1 | 20 | 3149.0 ± 234.1 | 1394.6 | 6322.4 | 0.023 ± 0.005 | 0.171 ± 0.0003 |
| Hamburg Silver Spangled Dwarf | HSSD | Holland | “ | 20 | 63.9 ± 1.5 | 42 | 76 | 5735.0 ± 164.2 | 4672.2 | 7311.6 | 0.324 ± 0.011 | 0.438 ± 0.001 |
| Light Brown Leghorn (or Italian Partridge) | LLB | Italy | egg | 19 | 47.5 ± 2.3 | 26 | 62 | 3903.8 ± 137.7 | 2949.8 | 5033.0 | 0.167 ± 0.011 | 0.288 ± 0.001 |
| Pushkin | P | Pushkin, USSR/Russia | dual purpose | 20 | 23.8 ± 1.3 | 15 | 36 | 3889.4 ± 209.7 | 2449.5 | 5569.3 | 0.112 ± 0.009 | 0.232 ± 0.001 |
| Russian White | RW1 | Pushkin, Russia | egg | 6 | 68.3 ± 2.0 | 62 | 76 | 5121.9 ± 278.3 | 4279.8 | 5850.7 | 0.307 ± 0.014 | 0.518 ± 0.001 |
| Russian White | RW2 | “ | “ | 170 | 25.9 ± 1.1 | 16 | 36 | 3749.6 ± 111.3 | 2431.3 | 4737.0 | 0.083 ± 0.006 | 0.218 ± 0.001 |
| Sussex Light | SL | England | dual purpose | 20 | 23.1 ± 1.5 | 11 | 35 | 4075.4 ± 155.8 | 3030.5 | 5773.5 | 0.127 ± 0.003 | 0.263 ± 0.001 |
| Tsarskoe Selo (Tsarskoselskaya) | TC | Pushkin, Russia | “ | 20 | 18.6 ± 1.2 | 8 | 30 | 4098.5 ± 193.1 | 2765.8 | 6303.2 | 0.102 ± 0.009 | 0.201 ± 0.001 |
| Uzbek Game | UG | Uzbekistan | game | 19 | 9.0 ± 1.3 | 0 | 21 | 3838.4 ± 349.5 | 0 | 6215.3 | 0.095 ± 0.007 | 0.178 ± 0.001 |
| F1 crossbreds | ||||||||||||
| Brahma Light × Sussex Light | BL × SL | Pushkin, Russia | meat | 12 | 4.3 ± 0.5 | 1 | 7 | 3602.2 ± 378.5 | 2347.8 | 7266.8 | 0.051 ± 0.008 | 0.215 ± 0.001 |
| Sussex Light × Amroks Cuckoo | SL × A | “ | “ | 14 | 2.2 ± 0.3 | 0 | 5 | 2905.1 ± 291.9 | 0 | 5126.7 | 0.033 ± 0.008 | 0.188 ± 0.001 |
| Tsarskoye Selo × Sussex Light | TC × SL | “ | “ | 14 | 6.0 ± 0.7 | 3 | 10 | 3032.8 ± 188.2 | 2050.0 | 4181.1 | 0.040 ± 0.009 | 0.215 ± 0.001 |
| Uzbek Game × Amroks Cuckoo | UG × A | “ | “ | 14 | 4.9 ± 0.4 | 3 | 8 | 3023.5 ± 170.8 | 2062.4 | 4098.3 | 0.039 ± 0.008 | 0.206 ± 0.001 |
| White Cornish × (Brahma Light × Sussex Light) | C × BL × SL | “ | “ | 14 | 0.9 ± 0.3 | 0 | 3 | 1622.8 ± 457.5 | 0 | 4427.7 | 0.024 ± 0.008 | 0.206 ± 0.001 |
| White Cornish × (Sussex Light × Amroks Cuckoo) | C × SL × A | “ | “ | 14 | 1.4 ± 0.4 | 0 | 4 | 1840.6 ± 425.2 | 0 | 4641.6 | 0.033 ± 0.008 | 0.193 ± 0.001 |
| Tsarskoye Selo × (Sussex Light × Amroks Cuckoo) | TC × SL × A | “ | “ | 14 | 3.8 ± 0.6 | 0 | 8 | 2715.2 ± 383.2 | 0 | 5410.2 | 0.046 ± 0.008 | 0.195 ± 0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dementieva, N.V.; Kudinov, A.A.; Larkina, T.A.; Mitrofanova, O.V.; Dysin, A.P.; Terletsky, V.P.; Tyshchenko, V.I.; Griffin, D.K.; Romanov, M.N. Genetic Variability in Local and Imported Germplasm Chicken Populations as Revealed by Analyzing Runs of Homozygosity. Animals 2020, 10, 1887. https://doi.org/10.3390/ani10101887
Dementieva NV, Kudinov AA, Larkina TA, Mitrofanova OV, Dysin AP, Terletsky VP, Tyshchenko VI, Griffin DK, Romanov MN. Genetic Variability in Local and Imported Germplasm Chicken Populations as Revealed by Analyzing Runs of Homozygosity. Animals. 2020; 10(10):1887. https://doi.org/10.3390/ani10101887
Chicago/Turabian StyleDementieva, Natalia V., Andrei A. Kudinov, Tatiana A. Larkina, Olga V. Mitrofanova, Artyom P. Dysin, Valeriy P. Terletsky, Valentina I. Tyshchenko, Darren K. Griffin, and Michael N. Romanov. 2020. "Genetic Variability in Local and Imported Germplasm Chicken Populations as Revealed by Analyzing Runs of Homozygosity" Animals 10, no. 10: 1887. https://doi.org/10.3390/ani10101887
APA StyleDementieva, N. V., Kudinov, A. A., Larkina, T. A., Mitrofanova, O. V., Dysin, A. P., Terletsky, V. P., Tyshchenko, V. I., Griffin, D. K., & Romanov, M. N. (2020). Genetic Variability in Local and Imported Germplasm Chicken Populations as Revealed by Analyzing Runs of Homozygosity. Animals, 10(10), 1887. https://doi.org/10.3390/ani10101887