
Microbial Communities of Cladonia Lichens and Their Biosynthetic Gene Clusters Potentially Encoding Natural Products

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Lichen Sampling and Environmental DNA Extraction
2.2. Microbial Community Analysis
2.3. Metagenome Sequencing, Assembly, and Analysis
2.4. Extraction and Chemical Analysis of Lichen Natural Product Compounds
3. Results
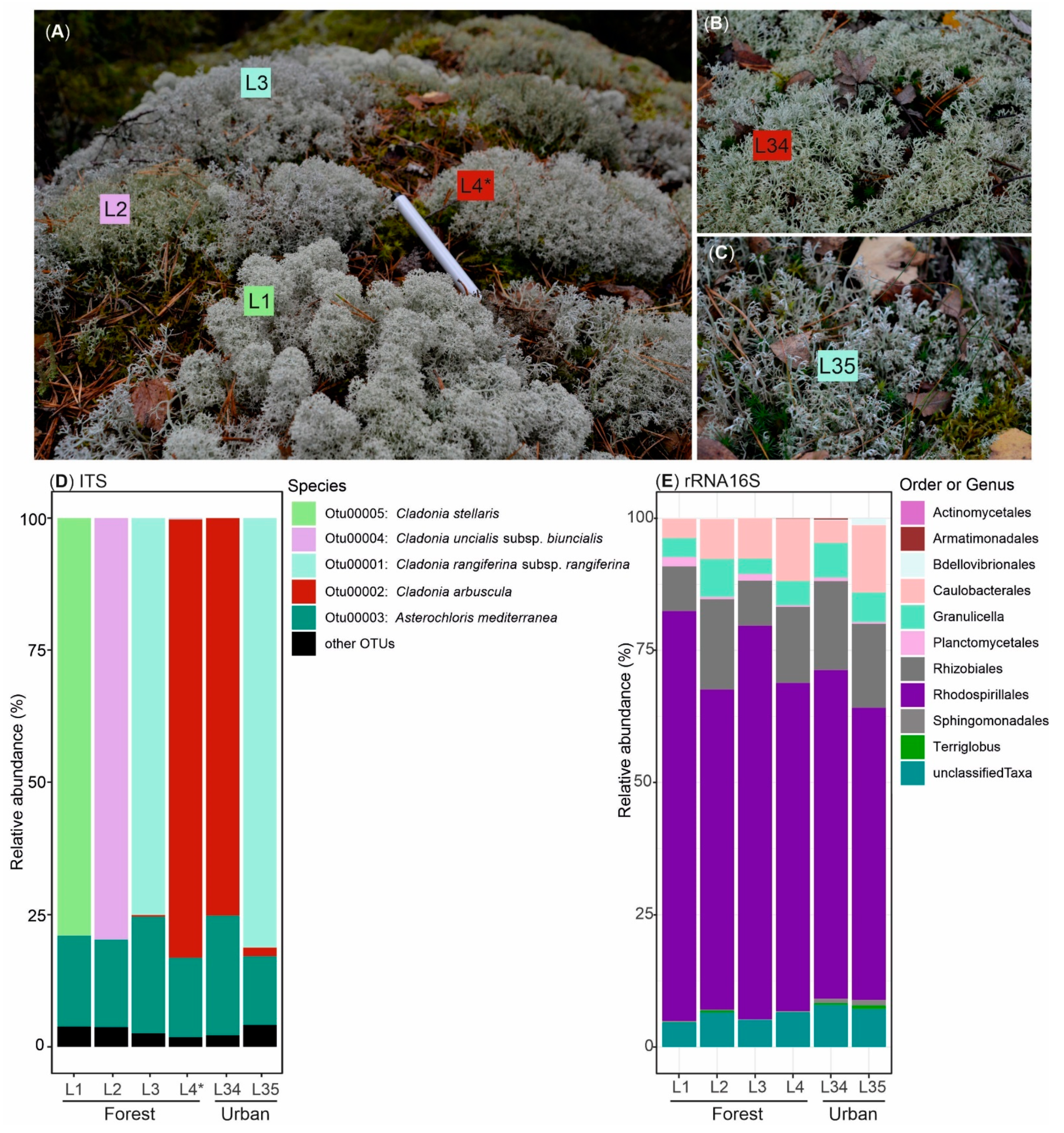
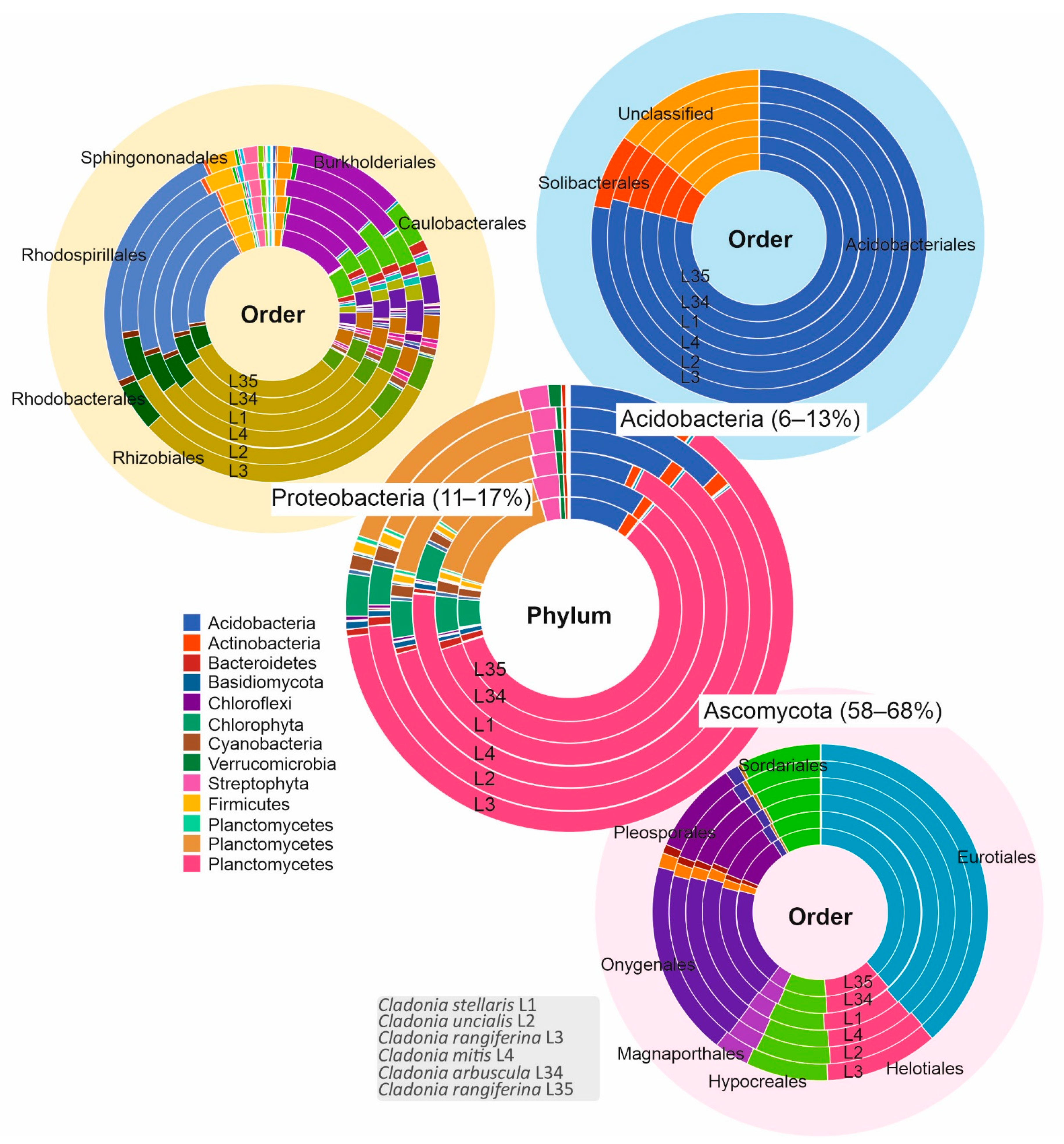
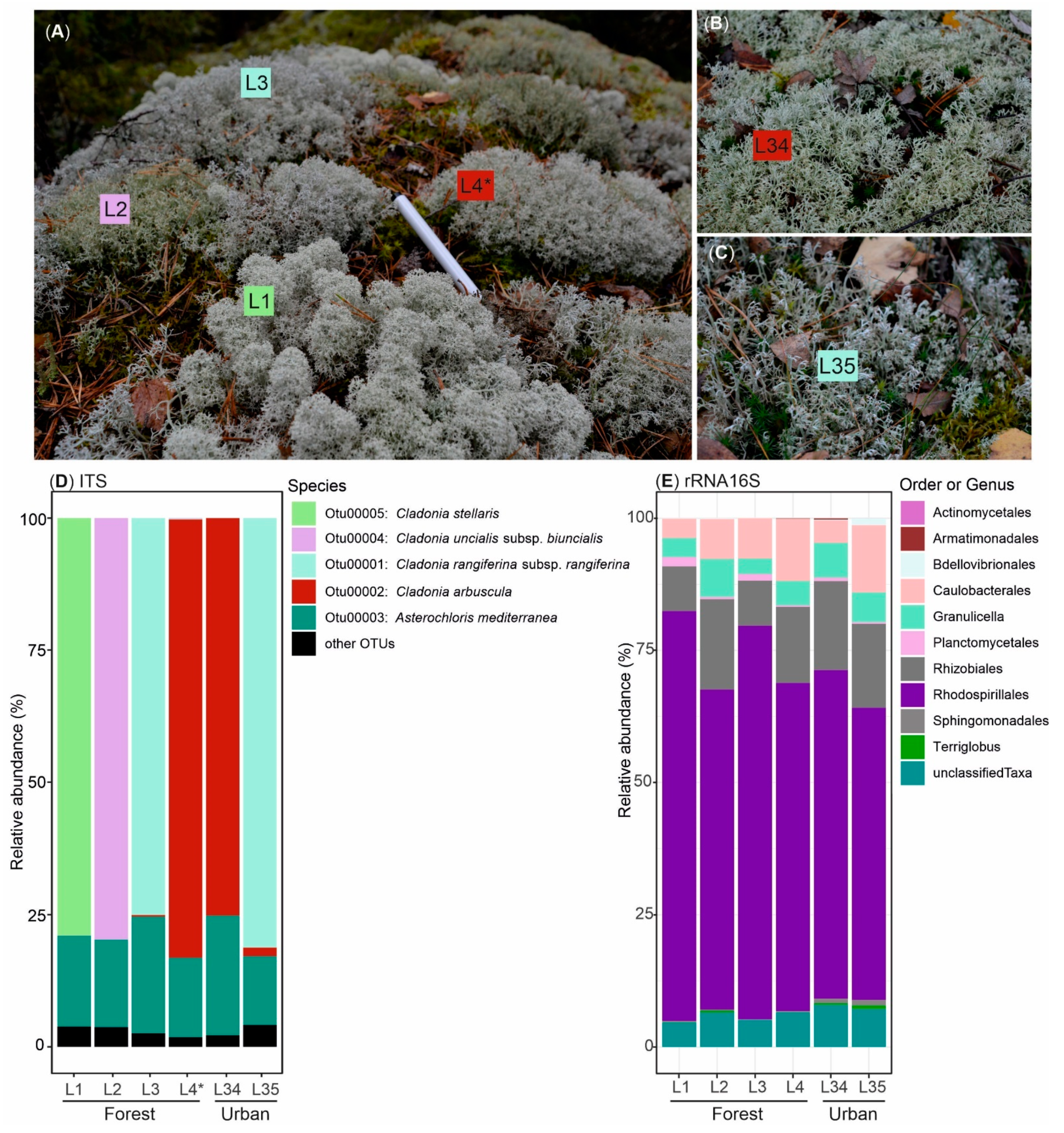
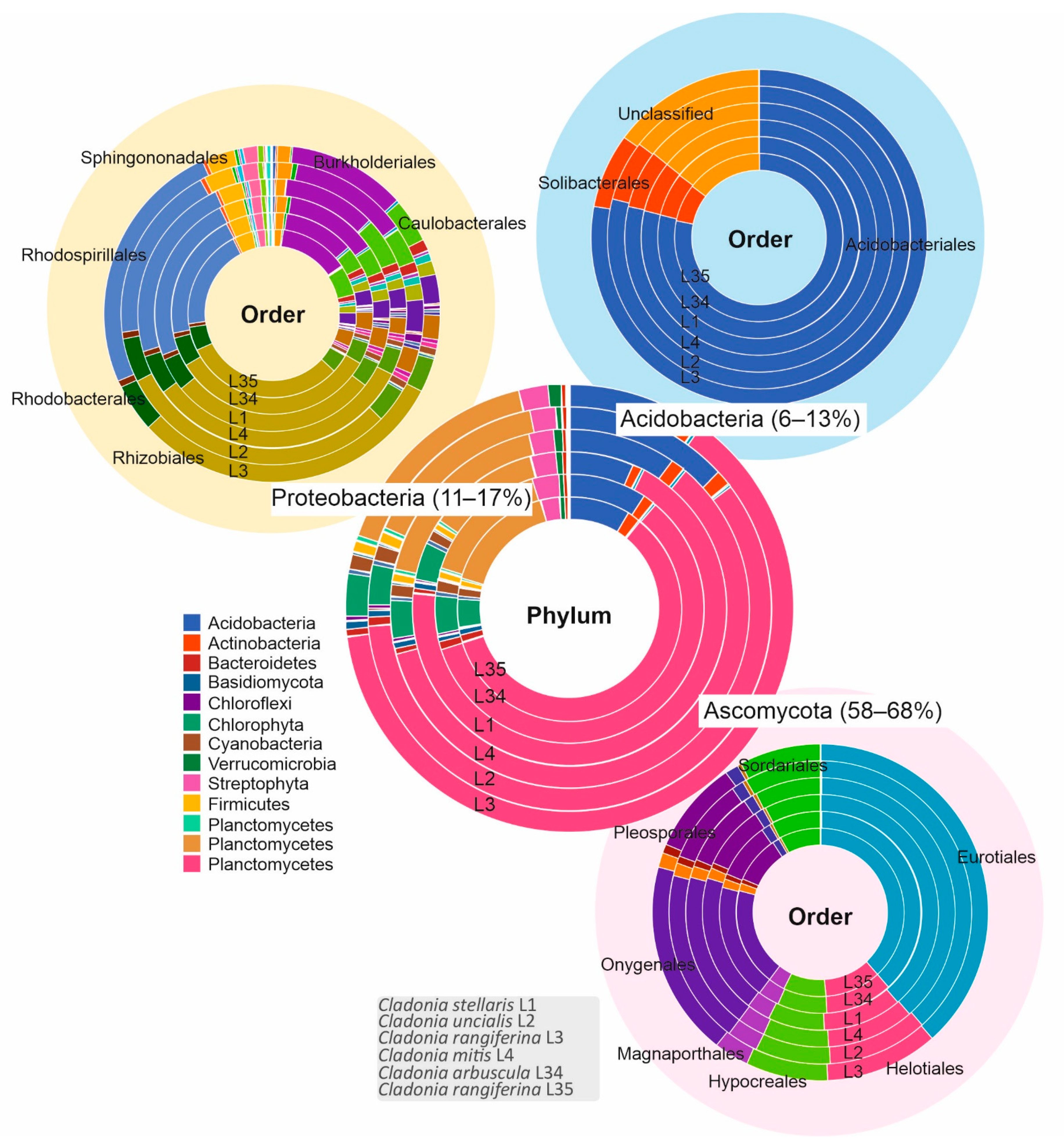
3.1. Biological Diversity within Cladonia Thalli
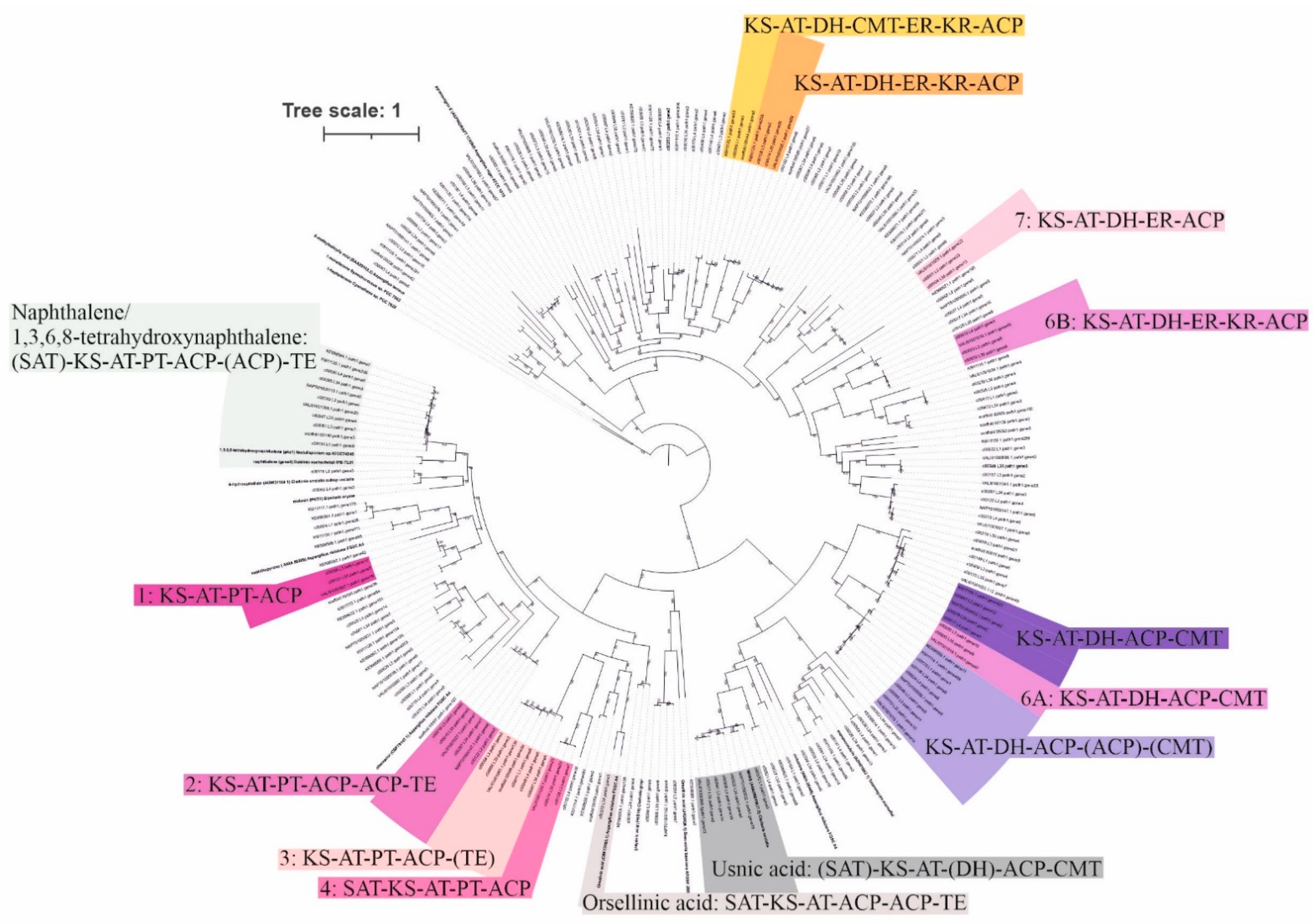
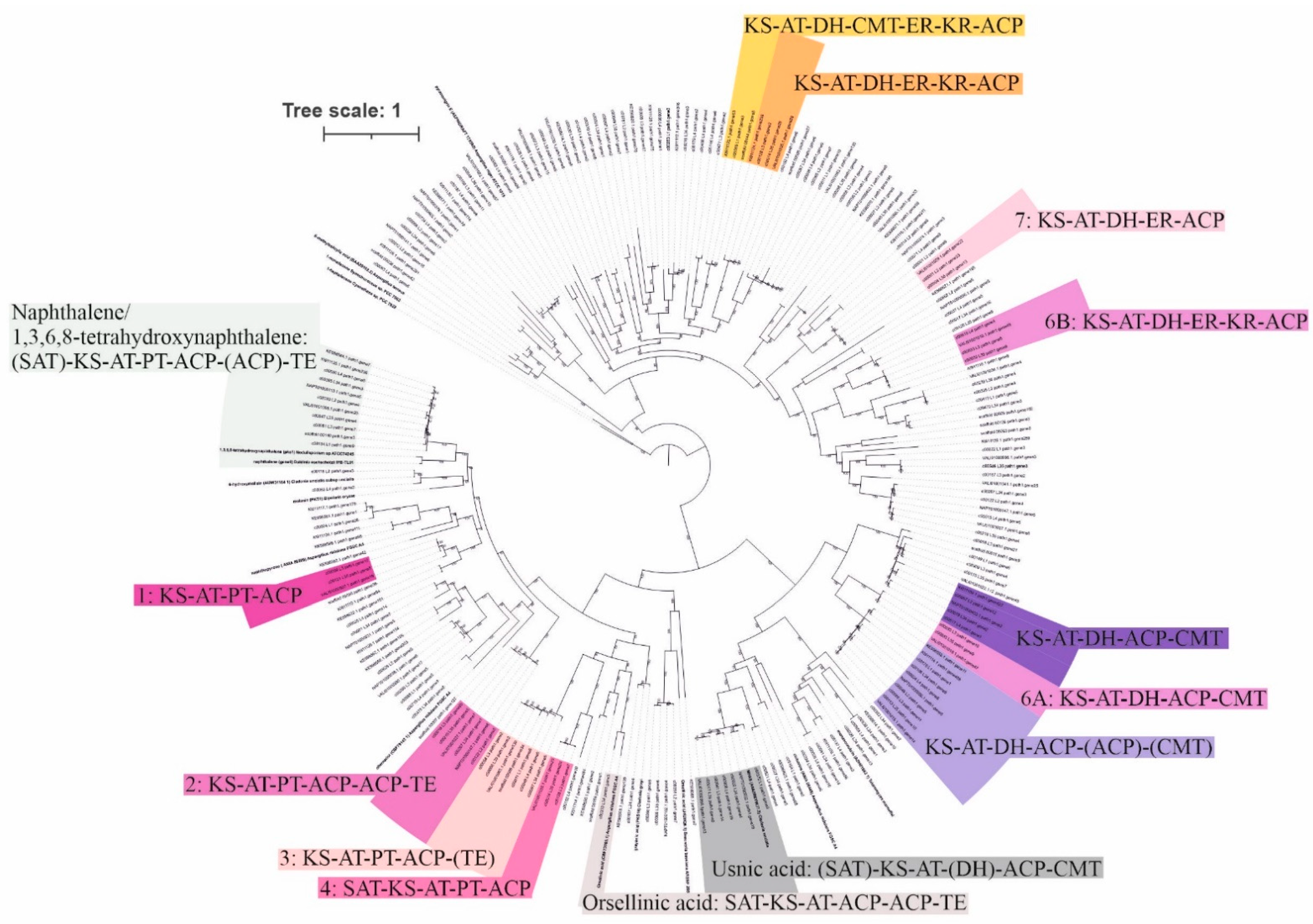
3.2. Potential for Biosynthesis of Natural Products
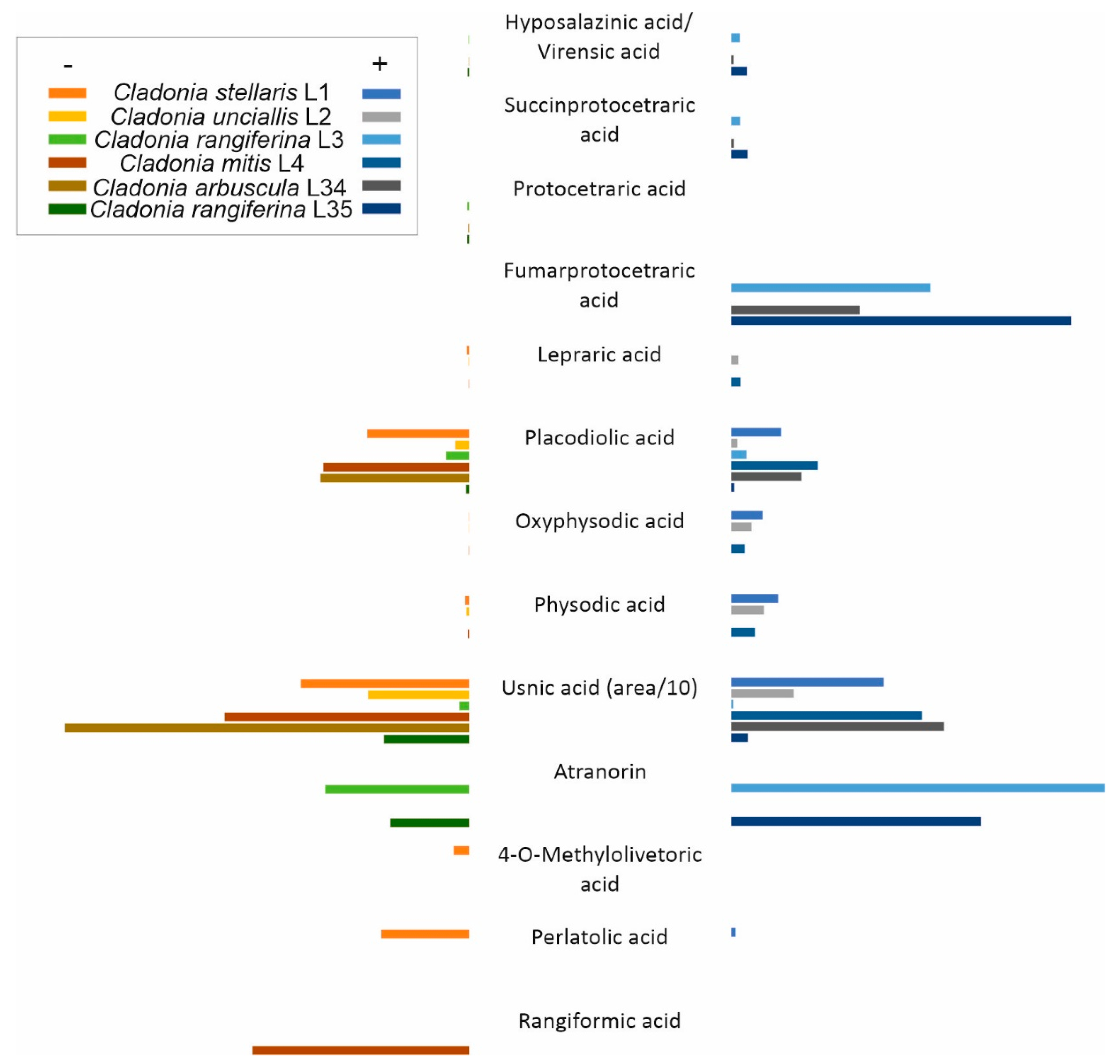
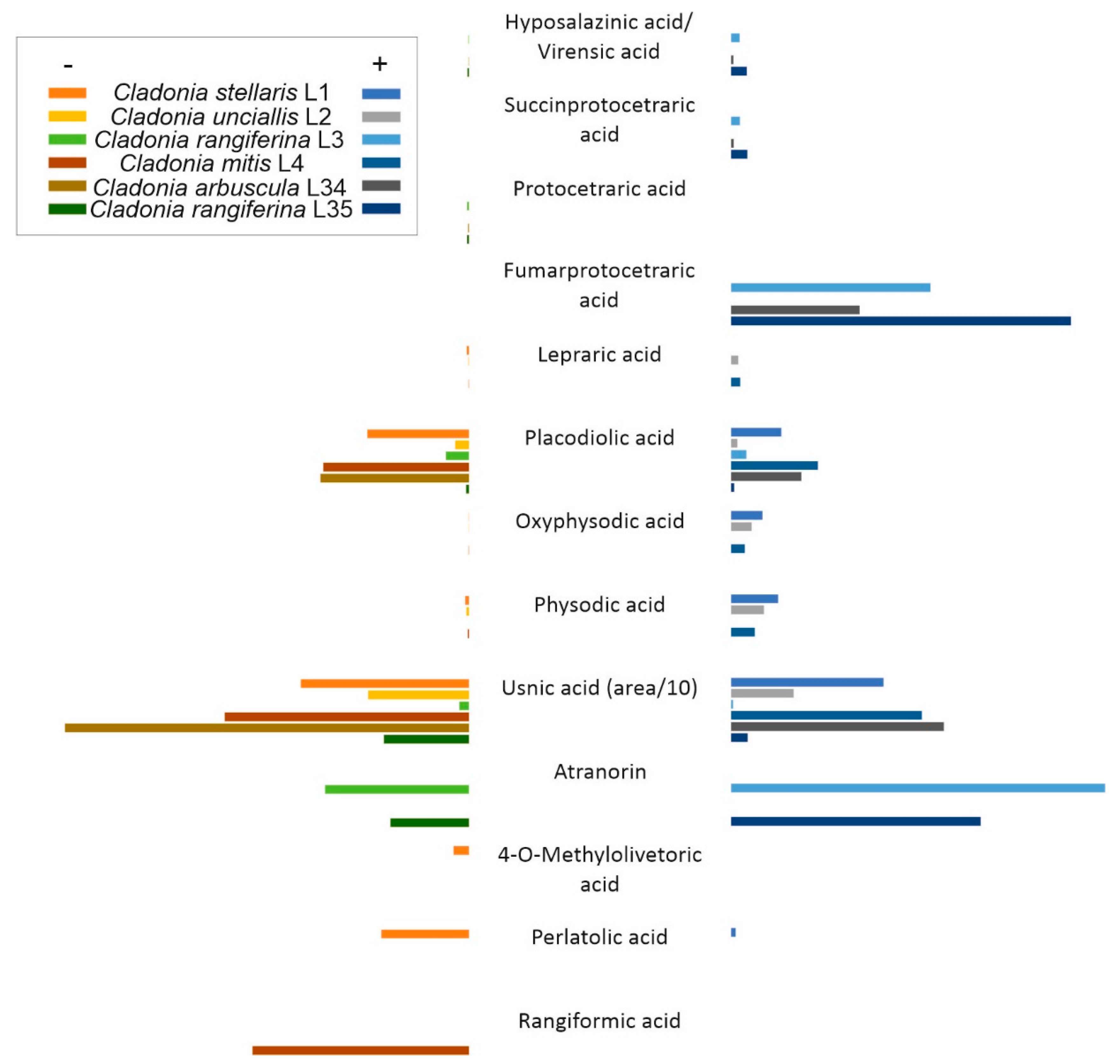
3.3. Natural Products Detected in Lichen Thalli
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hawksworth, D.L.; Grube, M. Lichens redefined as complex ecosystems. New Phytol. 2020, 227, 1281–1283. [Google Scholar] [CrossRef]
- Grimm, M.; Grube, M.; Schiefelbein, U.; Zühlke, D.; Bernhardt, J.; Riedel, K. The Lichens’ Microbiota, Still a Mystery? Front. Microbiol. 2021, 12, 623839. [Google Scholar] [CrossRef] [PubMed]
- Lumbsch, H.; Ahti, T.; Altermann, S.; De Paz, G.; Aptroot, A.; Arup, U.; Pena, A.; Bawingan, P.; Benatti, M.; Betancourt, L.; et al. One hundred new species of lichenized fungi: A signature of undiscovered global diversity. Phytotaxa 2011, 18, 1–127. [Google Scholar] [CrossRef] [Green Version]
- Ahti, T. What is a lichen? In Lichens of Finland; Stenroos, S., Velmala, S., Pykälä, J., Ahti, T., Eds.; Botanical Museum, Finnish Museum of Natural History LUOMUS: Helsinki, Finland, 2016; pp. 13–14. [Google Scholar]
- Svihus, B.; Holand, Ø. Lichen polysaccharides and their relation to reindeer/caribou nutrition. J. Range Manag. 2000, 53, 642–648. [Google Scholar] [CrossRef]
- Redzic, S.; Barudanovic, S.; Pilipovic, S. Wild mushrooms and lichens used as human food for survival in war conditions; Podrinje—Zepa Region (Bosnia and Herzegovina, W. Balkan). Hum. Ecol. Rev. 2010, 17, 175–187. [Google Scholar]
- Crawford, S.D. Lichens used in traditional medicine. In Lichen Secondary Metabolites; Ranković, B., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 27–80. [Google Scholar]
- Boustie, J.; Tomasi, S.; Grube, M. Bioactive lichen metabolites: Alpine habitats as an untapped source. Phytochem. Rev. 2011, 10, 287–307. [Google Scholar] [CrossRef]
- Basile, A.; Rigano, D.; Loppi, S.; Di Santi, A.; Nebbioso, A.; Sorbo, S.; Conte, B.; Paoli, L.; De Ruberto, F.; Molinari, A.M.; et al. Antiproliferative, antibacterial and antifungal activity of the lichen Xanthoria parietina and its secondary metabolite parietin. Int. J. Mol. Sci. 2015, 16, 7861–7875. [Google Scholar] [CrossRef]
- Kosanić, M.; Ranković, B. Lichen secondary metabolites as potential antibiotic agents. In Lichen Secondary Metabolites; Ranković, B., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 81–104. [Google Scholar]
- Cimmino, A.; Nimis, P.L.; Masi, M.; De Gara, L.; van Otterlo, W.A.L.; Kiss, K.; Evidente, A.; Lefranc, F. Have lichenized fungi delivered promising anticancer small molecules? Phytochem. Rev. 2019, 18, 1–36. [Google Scholar] [CrossRef]
- Cernava, T.; Erlacher, A.; Aschenbrenner, I.A.; Krug, L.; Lassek, C.; Riedel, K.; Grube, M.; Berg, G. Deciphering functional diversification within the lichen microbiota by meta-omics. Microbiome 2017, 5, 82. [Google Scholar] [CrossRef] [Green Version]
- Parrot, D.; Legrave, N.; Delmail, D.; Grube, M.; Suzuki, M.; Tomasi, S. Review—Lichen-associated bacteria as a hot spot of chemodiversity: Focus on uncialamycin, a promising compound for future medicinal applications. Planta Med. 2016, 82, 1143–1152. [Google Scholar] [CrossRef] [Green Version]
- Kampa, A.; Gagunashvili, A.N.; Gulder, T.A.; Morinaka, B.I.; Daolio, C.; Godejohann, M.; Miao, V.P.; Piel, J.; Andrésson, Ó. Metagenomic natural product discovery in lichen provides evidence for a family of biosynthetic pathways in diverse symbioses. Proc. Natl. Acad. Sci. USA 2013, 110, E3129–E3137. [Google Scholar] [CrossRef] [Green Version]
- Calcott, M.J.; Ackerley, D.F.; Knight, A.; Keyzers, R.A.; Owen, J.G. Secondary metabolism in the lichen symbiosis. Chem. Soc. Rev. 2018, 47, 1730–1760. [Google Scholar] [CrossRef]
- Huneck, S.; Yoshimura, I. Identification of Lichen Substances; Springer: Berlin/Heidelberg, Germany, 1996; pp. 11–123. [Google Scholar]
- Yamamoto, Y.; Hara, K.; Kawakami, H.; Komine, M. Lichen substances and their biological activities. In Recent Advances in Lichenology; Upreti, D., Divakar, P., Shukla, V., Bajpai, R., Eds.; Springer: New Delhi, India, 2015; pp. 181–199. [Google Scholar]
- Armaleo, D.; Sun, X.; Culberson, C. Insights from the first putative biosynthetic gene cluster for a lichen depside and depsidone. Mycologia 2011, 103, 741–754. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Hameed, M.; Bertrand, R.L.; Piercey-Normore, M.D.; Sorensen, J.L. Putative identification of the usnic acid biosynthetic gene cluster by de novo whole-genome sequencing of a lichen-forming fungus. Fungal Biol. 2016, 120, 306–316. [Google Scholar] [CrossRef]
- Abdel-Hameed, M.; Bertrand, R.L.; Piercey-Normore, M.D.; Sorensen, J.L. Identification of 6-hydroxymellein synthase and accessory genes in the lichen Cladonia uncialis. J. Nat. Prod. 2016, 79, 1645–1650. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Geng, C.; Yuan, X.; Hua, M.; Tian, F.; Li, C. Identification of a putative polyketide synthase gene involved in usnic acid biosynthesis in the lichen Nephromopsis pallescens. PLoS ONE 2018, 13, e0199110. [Google Scholar] [CrossRef]
- Pizarro, D.; Divakar, P.K.; Grewe, F.; Crespo, A.; Dal Grande, F.; Lumbsch, H.T. Genome-wide analysis of biosynthetic gene cluster reveals correlated gene loss with absence of usnic acid in lichen-forming fungi. Genome Biol. Evol. 2020, 12, 1858–1868. [Google Scholar] [CrossRef]
- Bertrand, R.L.; Abdel-Hameed, M.; Sorensen, J.L. Lichen biosynthetic gene clusters. Part, I. Genome sequencing reveals a rich biosynthetic potential. J. Nat. Prod. 2018, 81, 723–731. [Google Scholar] [CrossRef]
- Ranković, B.; Kosanić, M. Lichens as a potential source of bioactive secondary metabolites. In Lichen Secondary Metabolites; Ranković, B., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 1–6. [Google Scholar]
- Bertrand, R.L.; Sorensen, J.L. A comprehensive catalogue of polyketide synthase gene clusters in lichenizing fungi. J. Ind. Microbiol. Biotechnol. 2018, 45, 1067–1081. [Google Scholar] [CrossRef]
- Greshake, B.; Zehr, S.; Dal Grande, F.; Meiser, A.; Schmitt, I.; Ebersberger, I. Potential and pitfalls of eukaryotic metagenome skimming: A test case for lichens. Mol. Ecol. Resour. 2016, 16, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Meiser, A.; Otte, J.; Schmitt, I.; Grande, F.D. Sequencing genomes from mixed DNA samples—Evaluating the metagenome skimming approach in lichenized fungi. Sci. Rep. 2017, 7, 14881. [Google Scholar] [CrossRef]
- Calchera, A.; Dal Grande, F.; Bode, H.B.; Schmitt, I. Biosynthetic gene content of the ‘perfume lichens’ Evernia prunastri and Pseudevernia furfuracea. Molecules 2019, 24, 203. [Google Scholar] [CrossRef] [Green Version]
- Stenroos, S.; Pino-Bodas, R.; Hyvönen, J.; Lumbsch, H.T.; Ahti, T. Phylogeny of the family Cladoniaceae (Lecanoromycetes, Ascomycota) based on sequences of multiple loci. Cladistics 2019, 35, 351–384. [Google Scholar] [CrossRef]
- Stenroos, S.; Pino-Bodas, R.; Ahti, T. Rexiella, a new name for Rexia S. Stenroos, Pino-Bodas & Ahti (2018), non Rexia D. A. Casamatta, S.R. Gomez & J. R. Johansen (2006). Cladistics 2019, 35, 603. [Google Scholar]
- Crits-Christoph, A.; Diamond, S.; Butterfield, C.N.; Thomas, B.C.; Banfield, J.F. Novel soil bacteria possess diverse genes for secondary metabolite biosynthesis. Nature 2018, 558, 440–444. [Google Scholar] [CrossRef]
- Hultman, J.; Waldrop, M.P.; Mackelprang, R.; David, M.M.; McFarland, J.; Blazewicz, S.J.; Harden, J.; Turetsky, M.R.; McGuire, A.D.; Shah, M.B.; et al. Multi-omics of permafrost, active layer and thermokarst bog soil microbiomes. Nature 2015, 521, 208–212. [Google Scholar] [CrossRef]
- Mackelprang, R.; Waldrop, M.P.; DeAngelis, K.M.; David, M.M.; Chavarria, K.L.; Blazewicz, S.J.; Rubin, E.M.; Jansson, J.K. Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature 2011, 480, 368–371. [Google Scholar] [CrossRef] [Green Version]
- Ihrmark, K.; Bödeker, I.T.; Cruz-Martinez, K.; Friberg, H.; Kubartova, A.; Schenck, J.; Strid, Y.; Stenlid, J.; Brandström-Durling, M.; Clemmensen, K.E.; et al. New primers to amplify the fungal ITS2 region—Evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol. Ecol. 2012, 82, 666–677. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Pereira, P.A.B.; Aho, V.T.E.; Paulin, L.; Pekkonen, E.; Auvinen, P.; Scheperjans, F. Oral and nasal microbiota in Parkinson’s disease. Parkinsonism Relat. Disord. 2017, 38, 61–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aho, V.T.E.; Pereira, P.A.B.; Voutilainen, S.; Paulin, L.; Pekkonen, E.; Auvinen, P.; Scheperjans, F. Gut microbiota in Parkinson’s disease: Temporal stability and relations to disease progression. EbioMedicine 2019, 44, 691–707. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Aho, V.T.E. A Comparison of Bioinformatic Workflows for the Analysis of Fungal Amplicon Sequence Data. Master’s Thesis, University of Helsinki, Helsinki, Finland, 11 February 2014. [Google Scholar]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods. 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Chernomor, O.; von Haeseler, A.; Minh, B.Q. Terrace aware data structure for phylogenomic inference from supermatrices. Syst. Biol. 2016, 65, 997–1008. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 23, 127–128. [Google Scholar] [CrossRef]
- Dal Grande, F.; Rolshausen, G.; Divakar, P.K.; Crespo, A.; Otte, J.; Schleuning, M.; Schmitt, I. Environment and host identity structure communities of green algal symbionts in lichens. New Phytol. 2018, 217, 277–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [Green Version]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.A.; Korobeynikov, A.; Lapidus, A.; Prjibelski, A.D.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol. 2013, 20, 714–737. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. In Gene Prediction. Methods in Molecular Biology; Kollmar, M., Ed.; Humana: New York, NY, USA, 2019; pp. 227–245. [Google Scholar]
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 2018, 34, i142–i150. [Google Scholar] [CrossRef]
- Mikheenko, A.; Saveliev, V.; Gurevich, A. MetaQUAST: Evaluation of metagenome assemblies. Bioinformatics 2016, 32, 1088–1090. [Google Scholar] [CrossRef] [Green Version]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [Green Version]
- Navarro-Muñoz, J.C.; Selem-Mojica, N.; Mullowney, M.W.; Kautsar, S.A.; Tryon, J.H.; Parkinson, E.I.; De Los Santos, E.L.C.; Yeong, M.; Cruz-Morales, P.; Abubucker, S.; et al. A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 2020, 16, 60–68. [Google Scholar] [CrossRef]
- Medlar, A.J.; Toronen, P.; Holm, L. AAI-profiler: Fast proteome-wide exploratory analysis reveals taxonomic identity, misclassification and contamination. Nucleic Acids Res. 2018, 46, W479–W485. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Kautsar, S.A.; Blin, K.; Shaw, S.; Navarro-Muñoz, J.C.; Terlouw, B.R.; van der Hooft, J.J.J.; van Santen, J.A.; Tracanna, V.; Suarez Duran, H.G.; Pascal Andreu, V.; et al. MIBiG 2.0: A repository for biosynthetic gene clusters of known function. Nucleic Acids Res. 2019, 48, D454–D458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shishido, T.K.; Popin, R.V.; Jokela, J.; Wahlsten, M.; Fiore, M.F.; Fewer, D.P.; Herfindal, L.; Sivonen, K. Dereplication of natural products with antimicrobial and anticancer activity from Brazilian cyanobacteria. Toxins 2019, 12, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feige, G.B.; Lumbsch, H.T.; Huneck, S.; Elix, J.A. The identification of lichen products by a standardized high-performance liquid chromatographic method. J. Chromatogr. 1993, 646, 417–427. [Google Scholar] [CrossRef]
- Prokopiev, I.A.; Shavarda, A.L.; Filippova, G.V.; Shein, A.A. Application of high-performance liquid chromatography to the determination of the concentration of lichen secondary metabolites. J. Anal. Chem. 2017, 72, 1178–1183. [Google Scholar] [CrossRef]
- Xu, M.; Heidmarsson, S.; Thorsteinsdottir, M.; Eiriksson, F.F.; Omarsdottir, S.; Olafsdottir, E.S. DNA barcoding and LC-MS metabolite profiling of the lichen-forming genus Melanelia: Specimen identification and discrimination focusing on Icelandic taxa. PLoS ONE 2017, 12, e0178012. [Google Scholar] [CrossRef] [Green Version]
- Polovinka, M.P.; Komarova, N.I.; Korchagina, D.V.; Sokolov, D.N.; Luzina, O.A.; Vlasenko, N.G.; Malyuga, A.A.; Romanova, E.V.; Salakhutdinov, N.F. Secondary metabolites of the lichen Cladonia stellaris. Chem. Nat. Compd. 2012, 48, 392–395. [Google Scholar] [CrossRef]
- Spribille, T.; Tuovinen, V.; Resl, P.; Vanderpool, D.; Wolinski, H.; Aime, M.C.; Schneider, K.; Stabentheiner, E.; Toome-Heller, M.; Thor, G.; et al. Basidiomycete yeasts in the cortex of ascomycete macrolichens. Science 2016, 353, 488–492. [Google Scholar] [CrossRef] [Green Version]
- Armaleo, D.; Müller, O.; Lutzoni, F.; Andrésson, Ó.S.; Blanc, G.; Bode, H.B.; Collart, F.R.; Dal Grande, F.; Dietrich, F.; Grigoriev, I.V.; et al. The lichen symbiosis re-viewed through the genomes of Cladonia grayi and its algal partner Asterochloris glomerata. BMC Genom. 2019, 20, 605. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-Y.; Choi, J.; Kim, J.A.; Jeong, M.H.; Kim, S.; Lee, Y.H.; Hur, J.S. Draft genome sequence of Cladonia macilenta KoLRI003786, a lichen-forming fungus producing biruloquinone. Genome Announc. 2013, 1, e00695-13. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-Y.; Choi, J.; Lee, G.W.; Kim, J.A.; Oh, S.O.; Jeong, M.H.; Yu, N.H.; Kim, S.; Lee, Y.H.; Hur, J.S. Draft genome sequence of lichen-forming fungus Cladonia metacorallifera strain KoLRI002260. Genome Announc. 2014, 2, e01065-13. [Google Scholar] [CrossRef] [Green Version]
- Cox, R.J. Polyketides, proteins and genes in fungi: Programmed nano-machines begin to reveal their secrets. Org. Biomol. Chem. 2007, 5, 2010–2026. [Google Scholar] [CrossRef]
- Cardinale, M.; Puglia, A.M.; Grube, M. Molecular analysis of lichen-associated bacterial communities. FEMS Microbiol. Ecol. 2006, 57, 484–495. [Google Scholar] [CrossRef]
- Cardinale, M.; Vieira de Castro, J., Jr.; Müller, H.; Berg, G.; Grube, M. In situ analysis of the bacterial community associated with the reindeer lichen Cladonia arbuscula reveals predominance of Alphaproteobacteria. FEMS Microbiol. Ecol. 2008, 66, 63–71. [Google Scholar] [CrossRef]
- Grube, M.; Cardinale, M.; de Castro, J.V., Jr.; Müller, H.; Berg, G. Species-specific structural and functional diversity of bacterial communities in lichen symbioses. ISME J. 2009, 3, 1105–1115. [Google Scholar] [PubMed] [Green Version]
- Bates, S.T.; Cropsey, G.W.; Caporaso, J.G.; Knight, R.; Fierer, N. Bacterial communities associated with the lichen symbiosis. Appl. Environ. Microbiol. 2011, 77, 1309–1314. [Google Scholar] [CrossRef] [Green Version]
- Sigurbjörnsdóttir, M.A.; Andrésson, Ó.S.; Vilhelmsson, O. Analysis of the Peltigera membranacea metagenome indicates that lichen-associated bacteria are involved in phosphate solubilization. Microbiology 2015, 161, 989–996. [Google Scholar] [CrossRef]
- Lendemer, J.C.; Keepers, K.G.; Tripp, E.A.; Pogoda, C.S.; McCain, C.M.; Kane, N.C. A taxonomically broad metagenomic survey of 339 species spanning 57 families suggests cystobasidiomycete yeasts are not ubiquitous across all lichens. Am. J. Bot. 2019, 106, 1090–1095. [Google Scholar] [CrossRef]
- Mark, K.; Laanisto, L.; Bueno, C.G.; Niinemets, Ü.; Keller, C.; Scheidegger, C. Contrasting co-occurrence patterns of photobiont and cystobasidiomycete yeast associated with common epiphytic lichen species. New Phytol. 2020, 227, 1362–1375. [Google Scholar] [CrossRef]
- Moya, P.; Škaloud, P.; Chiva, S.; García-Breijo, F.J.; Reig-Armiñana, J.; Vančurová, L.; Barreno, E. Molecular phylogeny and ultrastructure of the lichen microalga Asterochloris mediterranea sp. nov. from Mediterranean and Canary Islands ecosystems. Int. J. Syst. Evol. Microbiol. 2015, 65, 1838–1854. [Google Scholar] [CrossRef]
- Fernández-Mendoza, F.; Fleischhacker, A.; Kopun, T.; Grube, M.; Muggia, L. ITS1 metabarcoding highlights low specificity of lichen mycobiomes at a local scale. Mol. Ecol. 2017, 26, 4811–4830. [Google Scholar] [CrossRef] [PubMed]
- West, N.J.; Parrot, D.; Fayet, C.; Grube, M.; Tomasi, S.; Suzuki, M.T. Marine cyanolichens from different littoral zones are associated with distinct bacterial communities. Peer J. 2018, 6, e5208. [Google Scholar] [CrossRef] [Green Version]
- Banchi, E.; Stankovic, D.; Fernández-Mendoza, F.; Gionechetti, F.; Pallavicini, A.; Muggia, L. ITS2 metabarcoding analysis complements lichen mycobiome diversity data. Mycol Prog. 2018, 17, 1049–1066. [Google Scholar] [CrossRef] [Green Version]
- Noh, H.J.; Lee, Y.M.; Park, C.H.; Lee, H.K.; Cho, J.C.; Hong, S.G. Microbiome in Cladonia squamosa is vertically stratified according to microclimatic conditions. Front Microbiol. 2020, 11, 268. [Google Scholar] [CrossRef]
- Sierra, M.A.; Danko, D.C.; Sandoval, T.A.; Pishchany, G.; Moncada, B.; Kolter, R.; Mason, C.E.; Zambrano, M.M. The microbiomes of seven lichen genera reveal host specificity, a reduced core community and potential as source of antimicrobials. Front Microbiol. 2020, 11, 398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noh, H.-J.; Park, Y.; Hong, S.G.; Lee, Y.M. Diversity and physiological characteristics of Antarctic lichens-associated bacteria. Microorganisms 2021, 9, 607. [Google Scholar] [CrossRef]
- Alonso-García, M.; Villarreal, J.C. Geography, not host identity, shapes bacterial community in reindeer lichens. bioRxiv 2021. (preprint: bioRxiv 2021.01.30.428927). [Google Scholar] [CrossRef]
- Pino-Bodas, R.; Martín, M.P.; Burgaz, A.R.; Lumbsch, H.T. Species delimitation in Cladonia (Ascomycota): A challenge to the DNA barcoding philosophy. Mol. Ecol. Resour. 2013, 13, 1058–1068. [Google Scholar]
- Myllys, L.; Stenroos, S.; Thell, A.; Ahti, T. Phylogeny of bipolar Cladonia arbuscula and Cladonia mitis (Lecanorales, Euascomycetes). Mol. Phylogenet. Evol. 2003, 27, 58–69. [Google Scholar] [CrossRef]
- Pino-Bodas, R.; Stenroos, S. Global biodiversity patterns of the photobionts associated with the genus Cladonia (Lecanorales, Ascomycota). Microb. Ecol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Osyczka, P.; Lenart-Boroń, A.; Boroń, P.; Rola, K. Lichen-forming fungi in postindustrial habitats involve alternative photobionts. Mycologia 2021, 113, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Casano, L.M.; del Campo, E.M.; García-Breijo, F.J.; Reig-Armiñana, J.; Gasulla, F.; Del Hoyo, A.; Guéra, A.; Barreno, E. Two Trebouxia algae with different physiological performances are ever-present in lichen thalli of Ramalina farinacea. Coexistence versus competition? Environ. Microbiol. 2011, 13, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Moya, P.; Molins, A.; Martínez-Alberola, F.; Muggia, L.; Barreno, E. Unexpected associated microalgal diversity in the lichen Ramalina farinacea is uncovered by pyrosequencing analyses. PLoS ONE 2017, 12, e0175091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onuț-Brännström, I.; Benjamin, M.; Scofield, D.G.; Heiðmarsson, S.; Andersson, M.G.I.; Lindström, E.S.; Johannesson, H. Sharing of photobionts in sympatric populations of Thamnolia and Cetraria lichens: Evidence from high-throughput sequencing. Sci. Rep. 2018, 8, 4406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, F.; Otte, J.; Schmitt, I.; Dal Grande, F. Comparing Sanger sequencing and high-throughput metabarcoding for inferring photobiont diversity in lichens. Sci. Rep. 2018, 8, 8624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, H.B.; Dal Grande, F.; Muggia, L.; Keuler, R.; Divakar, P.K.; Grewe, F.; Schmitt, I.; Lumbsch, H.T.; Leavitt, S.D. Metagenomic data reveal diverse fungal and algal communities associated with the lichen symbiosis. Symbiosis 2020, 82, 133–147. [Google Scholar] [CrossRef]
- Pankratov, T.A. Bacterial complexes of Khibiny Mountains lichens revealed in Cladonia uncialis, C. portentosa, Alectoria ochroleuca, and Nephroma arcticum. Microbiology 2018, 87, 79. [Google Scholar] [CrossRef]
- Danilova, O.V.; Belova, S.E.; Gagarinova, I.V.; Dedysh, S.N. Microbial community composition and methanotroph diversity of a subarctic wetland in Russia. Mikrobiologiia 2016, 85, 545–554. [Google Scholar] [CrossRef]
- Grube, M.; Cernava, T.; Soh, J.; Fuchs, S.; Aschenbrenner, I.; Lassek, C.; Wegner, U.; Becher, D.; Riedel, K.; Sensen, C.W.; et al. Exploring functional contexts of symbiotic sustain within lichen-associated bacteria by comparative omics. ISME J. 2015, 9, 412–424. [Google Scholar] [CrossRef] [Green Version]
- Schneider, T.; Schmid, E.; de Castro, J.V., Jr.; Cardinale, M.; Eberl, L.; Grube, M.; Berg, G.; Riedel, K. Structure and function of the symbiosis partners of the lung lichen (Lobaria pulmonaria L. Hoffm.) analyzed by metaproteomics. Proteomics 2011, 11, 2752–2756. [Google Scholar] [CrossRef] [PubMed]
- Hodkinson, B.P.; Gottel, N.R.; Schadt, C.W.; Lutzoni, F. Photoautotrophic symbiont and geography are major factors affecting highly structured and diverse bacterial communities in the lichen microbiome. Environ. Microbiol. 2012, 14, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Aschenbrenner, I.A.; Cernava, T.; Berg, G.; Grube, M. Understanding microbial multi-species symbioses. Front Microbiol. 2016, 7, 180. [Google Scholar] [CrossRef] [PubMed]
- Erlacher, A.; Cernava, T.; Cardinale, M.; Soh, J.; Sensen, C.W.; Grube, M.; Berg, G. Rhizobiales as functional and endosymbiontic members in the lichen symbiosis of Lobaria pulmonaria L. Front Microbiol. 2015, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, R.L.; Abdel-Hameed, M.; Sorensen, J.L. Lichen biosynthetic gene clusters Part II: Homology mapping suggests a functional diversity. J. Nat. Prod. 2018, 81, 732–748. [Google Scholar] [CrossRef] [PubMed]
- Studzinska-Sroka, E.; Galanty, A.; Bylka, W. Atranorin—An interesting lichen secondary metabolite. Mini Rev. Med. Chem. 2017, 17, 1633–1645. [Google Scholar] [CrossRef] [PubMed]
- Pereira, E.C.; da Silva, N.H.; Buril, M.L.L.; Martins, M.C.B.; Silva, H.A.M.F.; Falcão, E.P.S.; Oliveira, H.P.; da Costa, M.M.; Legaz, M.E.; Santiago, R.; et al. Bioactive compounds from Brazilian lichens and their biotechnological applications. In Plant-Derived Bioactives; Swamy, M., Ed.; Springer: Singapore, 2020; pp. 209–238. [Google Scholar]
- Elshobary, M.E.; Becker, M.G.; Kalichuk, J.L.; Chan, A.C.; Belmonte, M.F.; Piercey-Normore, M.D. Tissue-specific localization of polyketide synthase and other associated genes in the lichen, Cladonia rangiferina, using laser microdissection. Phytochemistry 2018, 156, 142–150. [Google Scholar] [CrossRef]
- Harvey, C.J.B.; Tang, M.; Schlecht, U.; Horecka, J.; Fischer, C.R.; Lin, H.C.; Li, J.; Naughton, B.; Cherry, J.; Miranda, M.; et al. HEx: A heterologous expression platform for the discovery of fungal natural products. Sci. Adv. 2018, 4, eaar5459. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | T1PKS | T3PKS | Terpene | NRPS | NRPS–T1PKS | T3PKS–T1PKS | Indole | Hybrid/T1PKS–Indole/Terpene | Siderophore | Other | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Metagenomes | |||||||||||
| Cladonia arbuscula L34 | 20 | 2 | 3 | 7 | 3 | 0 | 2 | 1 | 0 | 0 | 38 |
| Cladonia mitis L4 | 28 | 2 | 2 | 5 | 2 | 0 | 0 | 1 | 1 | 0 | 41 |
| Cladonia rangiferina L3 | 19 | 1 | 4 | 6 | 3 | 1 | 0 | 0 | 0 | 0 | 34 |
| Cladonia rangiferina L35 | 17 | 1 | 6 | 4 | 2 | 1 | 0 | 1 | 0 | 0 | 32 |
| Cladonia stellaris L1 | 12 | 0 | 6 | 5 | 4 | 1 | 0 | 2 | 0 | 1 | 31 |
| Cladonia uncialis L2 | 15 | 0 | 4 | 5 | 3 | 1 | 0 | 0 | 0 | 0 | 28 |
| Genomes | |||||||||||
| Cladonia grayi | 12 | 0 | 2 | 9 | 1 | 0 | 0 | 0 | 0 | 1 | 25 |
| Cladonia macilenta KoLRI003786 | 18 | 0 | 3 | 6 | 3 | 1 | 0 | 0 | 0 | 0 | 31 |
| Cladonia metacorallifera KoLRI002260 | 19 | 0 | 5 | 8 | 1 | 1 | 0 | 1 | 0 | 1 | 36 |
| Cladonia rangiferina ATCC 18275 | 18 | 0 | 2 | 6 | 2 | 1 | 0 | 1 | 0 | 0 | 30 |
| Cladonia uncialis Normore8774 | 14 | 0 | 3 | 2 | 3 | 1 | 0 | 0 | 0 | 0 | 23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shishido, T.K.; Wahlsten, M.; Laine, P.; Rikkinen, J.; Lundell, T.; Auvinen, P. Microbial Communities of Cladonia Lichens and Their Biosynthetic Gene Clusters Potentially Encoding Natural Products. Microorganisms 2021, 9, 1347. https://doi.org/10.3390/microorganisms9071347
Shishido TK, Wahlsten M, Laine P, Rikkinen J, Lundell T, Auvinen P. Microbial Communities of Cladonia Lichens and Their Biosynthetic Gene Clusters Potentially Encoding Natural Products. Microorganisms. 2021; 9(7):1347. https://doi.org/10.3390/microorganisms9071347
Chicago/Turabian StyleShishido, Tânia Keiko, Matti Wahlsten, Pia Laine, Jouko Rikkinen, Taina Lundell, and Petri Auvinen. 2021. "Microbial Communities of Cladonia Lichens and Their Biosynthetic Gene Clusters Potentially Encoding Natural Products" Microorganisms 9, no. 7: 1347. https://doi.org/10.3390/microorganisms9071347
APA StyleShishido, T. K., Wahlsten, M., Laine, P., Rikkinen, J., Lundell, T., & Auvinen, P. (2021). Microbial Communities of Cladonia Lichens and Their Biosynthetic Gene Clusters Potentially Encoding Natural Products. Microorganisms, 9(7), 1347. https://doi.org/10.3390/microorganisms9071347