
Environmental Conditions Modulate the Transcriptomic Response of Both Caulobacter crescentus Morphotypes to Cu Stress


 and
and
Abstract
1. Introduction
2. Materials and Methods
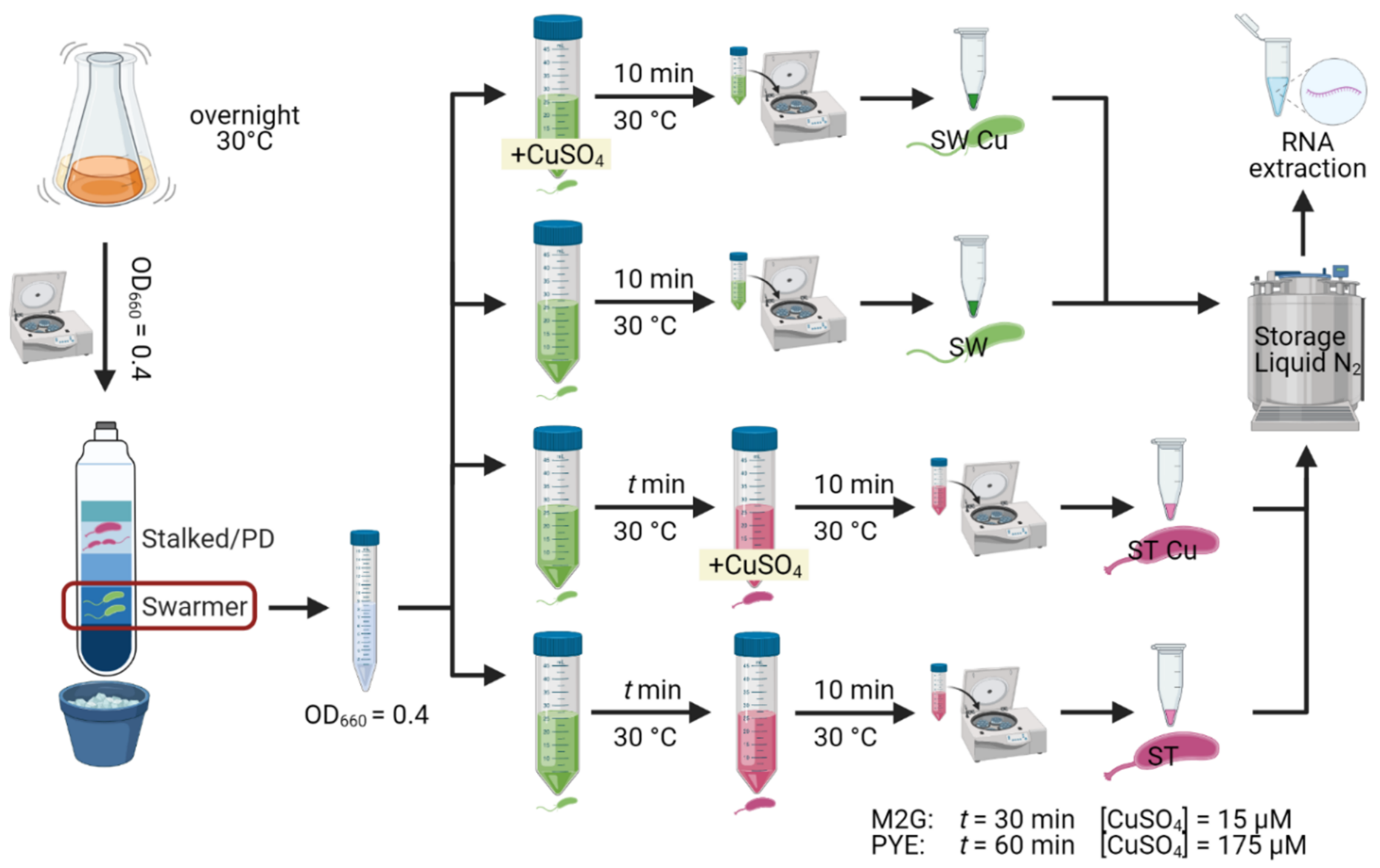
2.1. Bacterial Strains and Growth Conditions
2.2. Synchronization and RNA Extraction
2.3. Differential RNA Sequencing and Read Mapping
2.4. Differential Gene Expression Analysis
2.5. Reverse Trancription Quantitative Real-Time PCR
2.6. Data Visualization
3. Results and Discussion
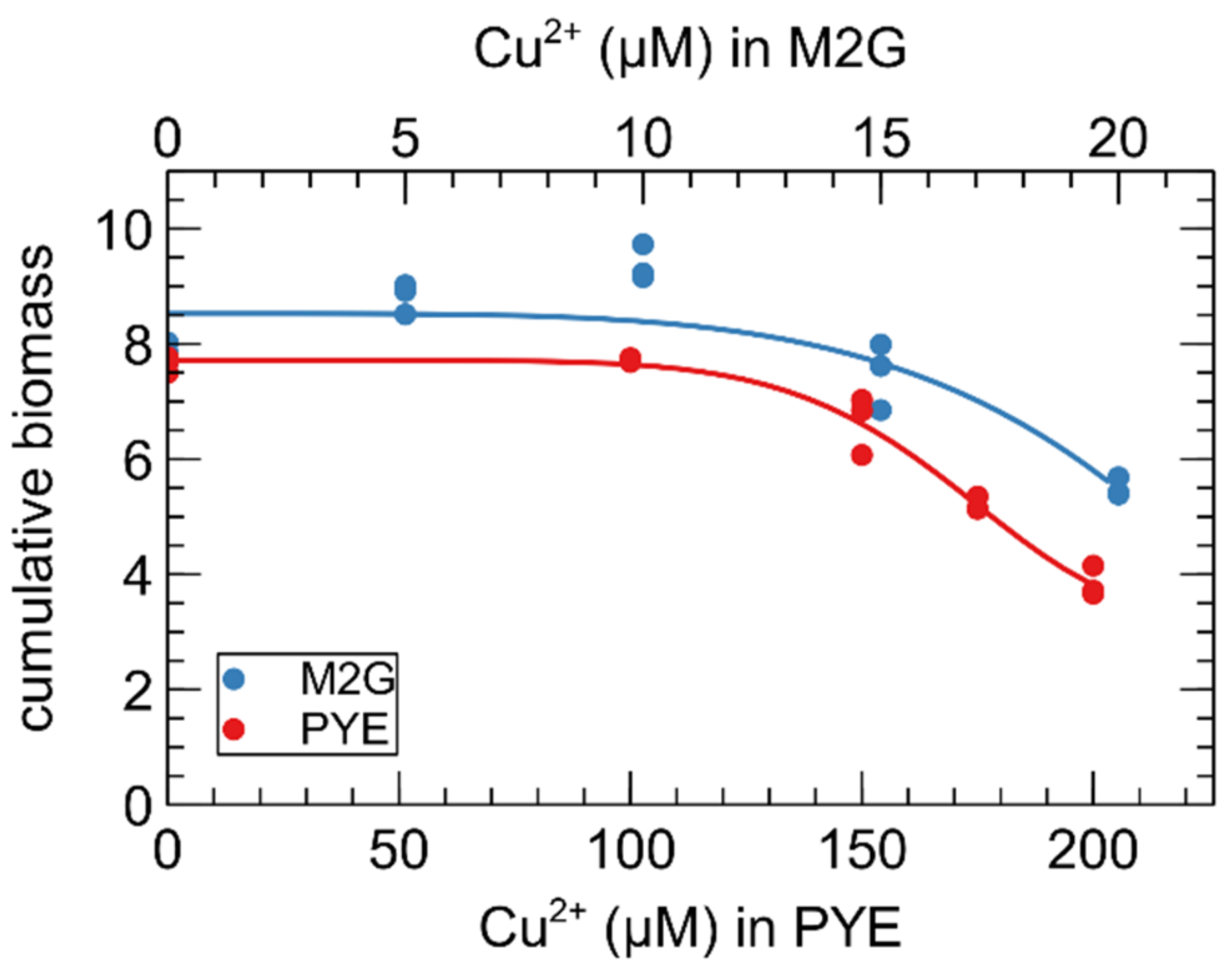
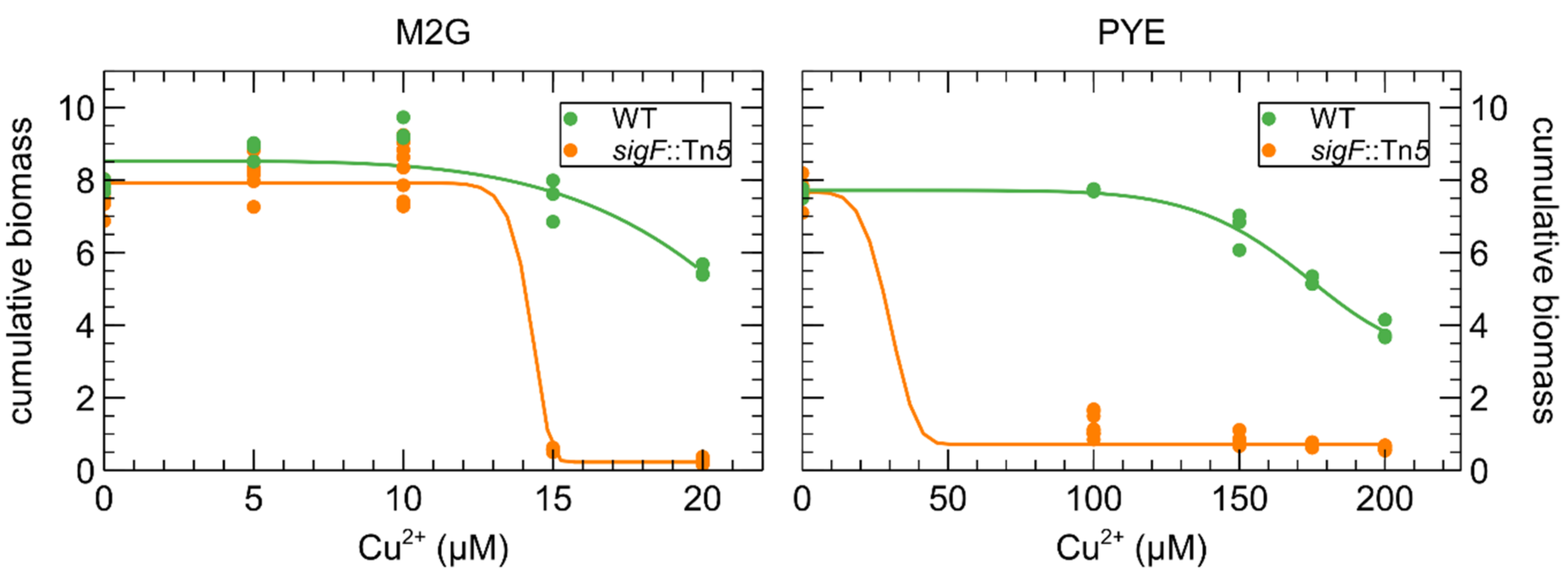
3.1. Growth of C. crescentus in the Presence of Cu
3.2. Read Coverage Analysis
3.3. Transcriptomic Response of C. crescentus to Cu Stress
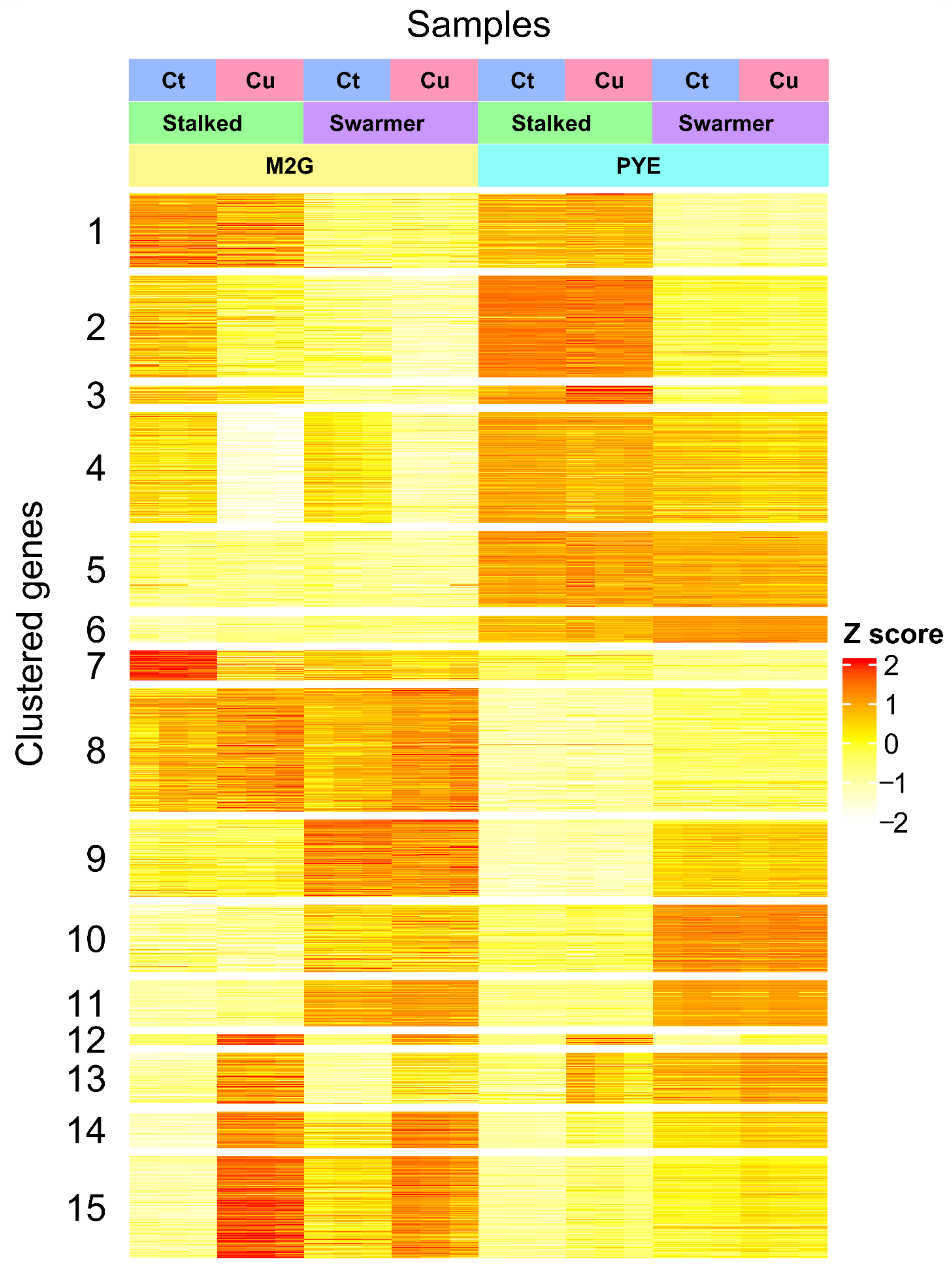
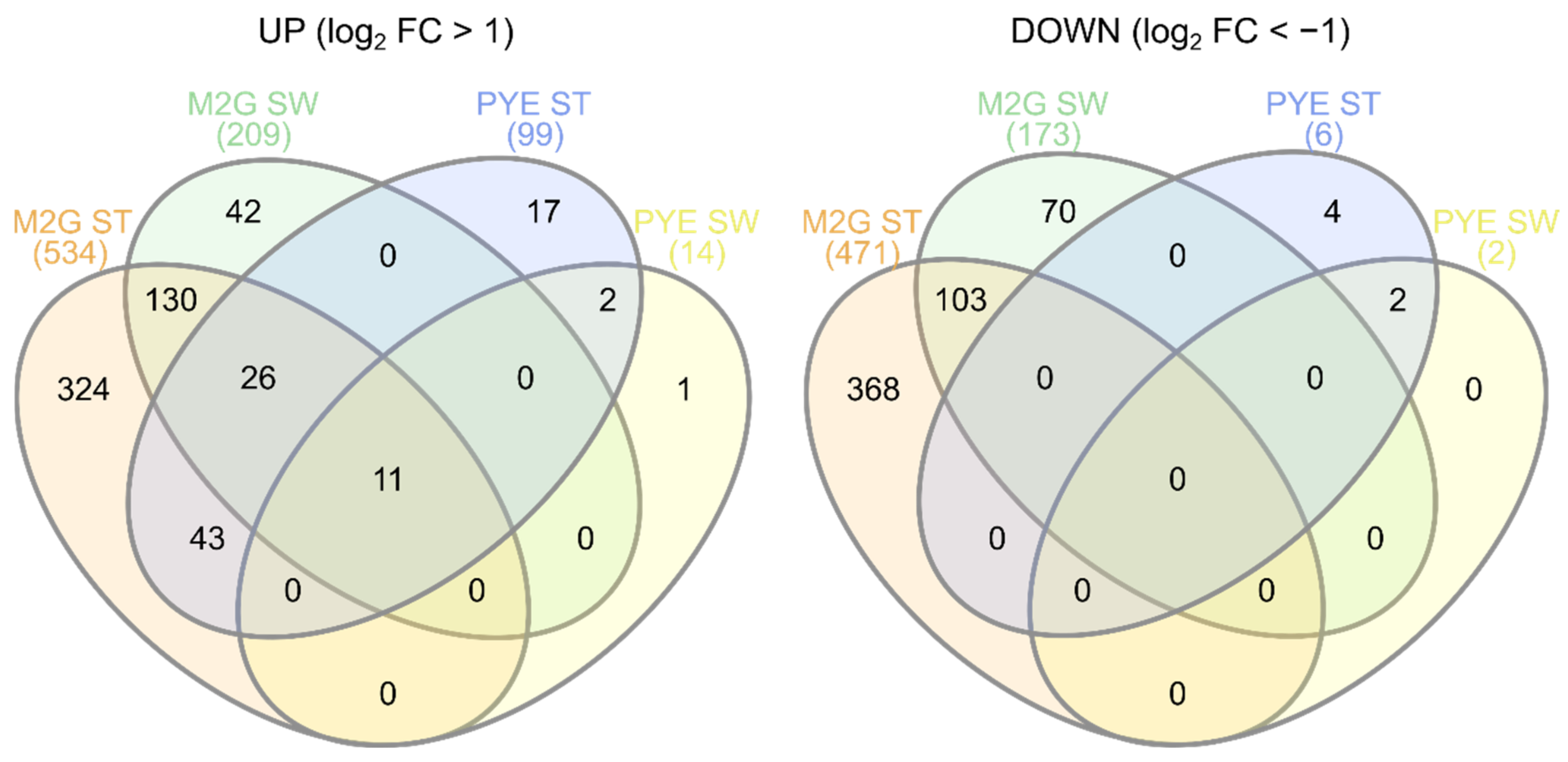
3.3.1. A Core Gene-Set in the Response to Cu
3.3.2. Oxidative Stress
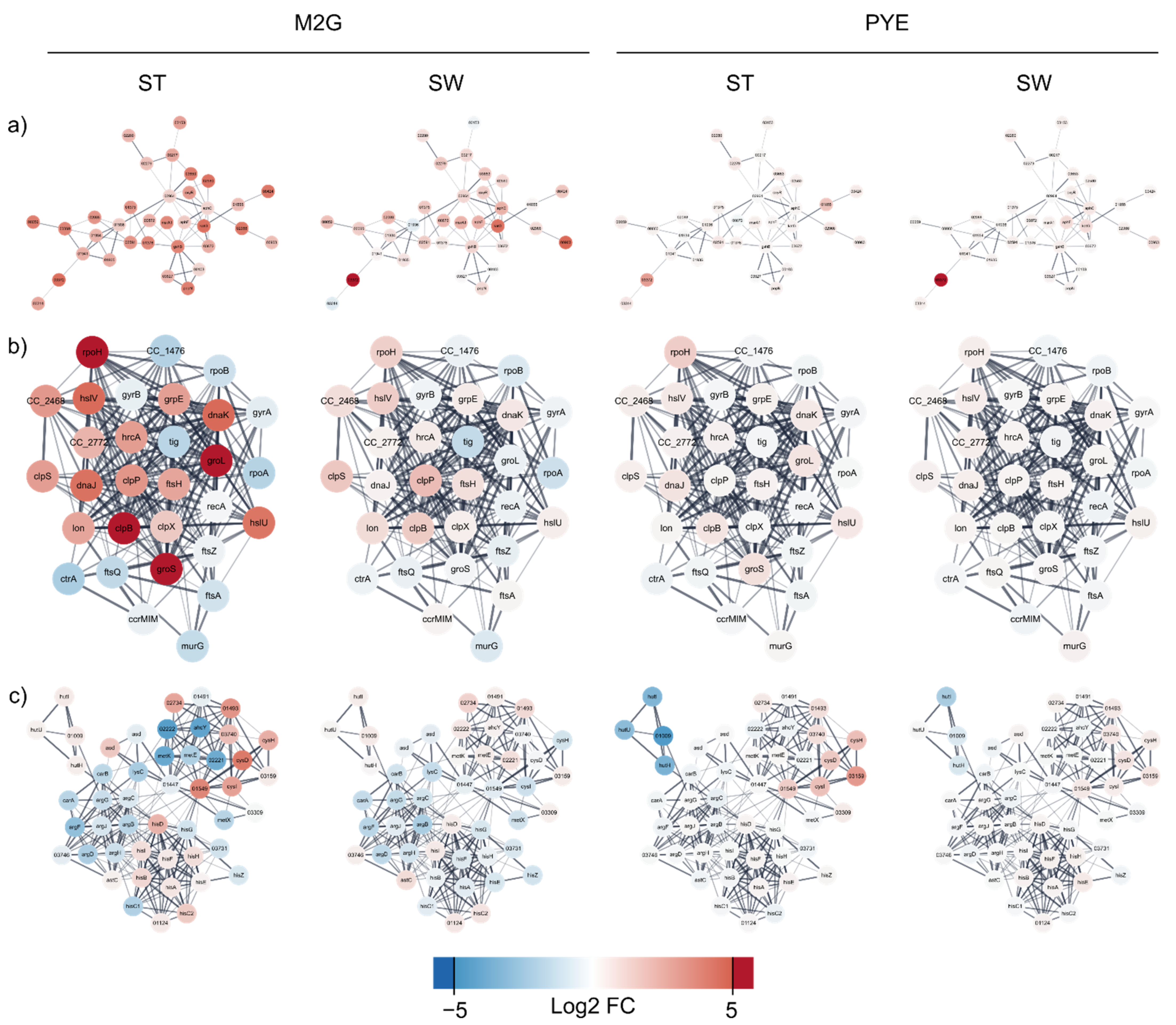
3.3.3. Protein Misfolding and Proteome Rearrangement
3.3.4. Metal Resistance and Transport Systems
3.3.5. Amino Acid Metabolism
3.3.6. Regulation by Small Regulatory RNAs
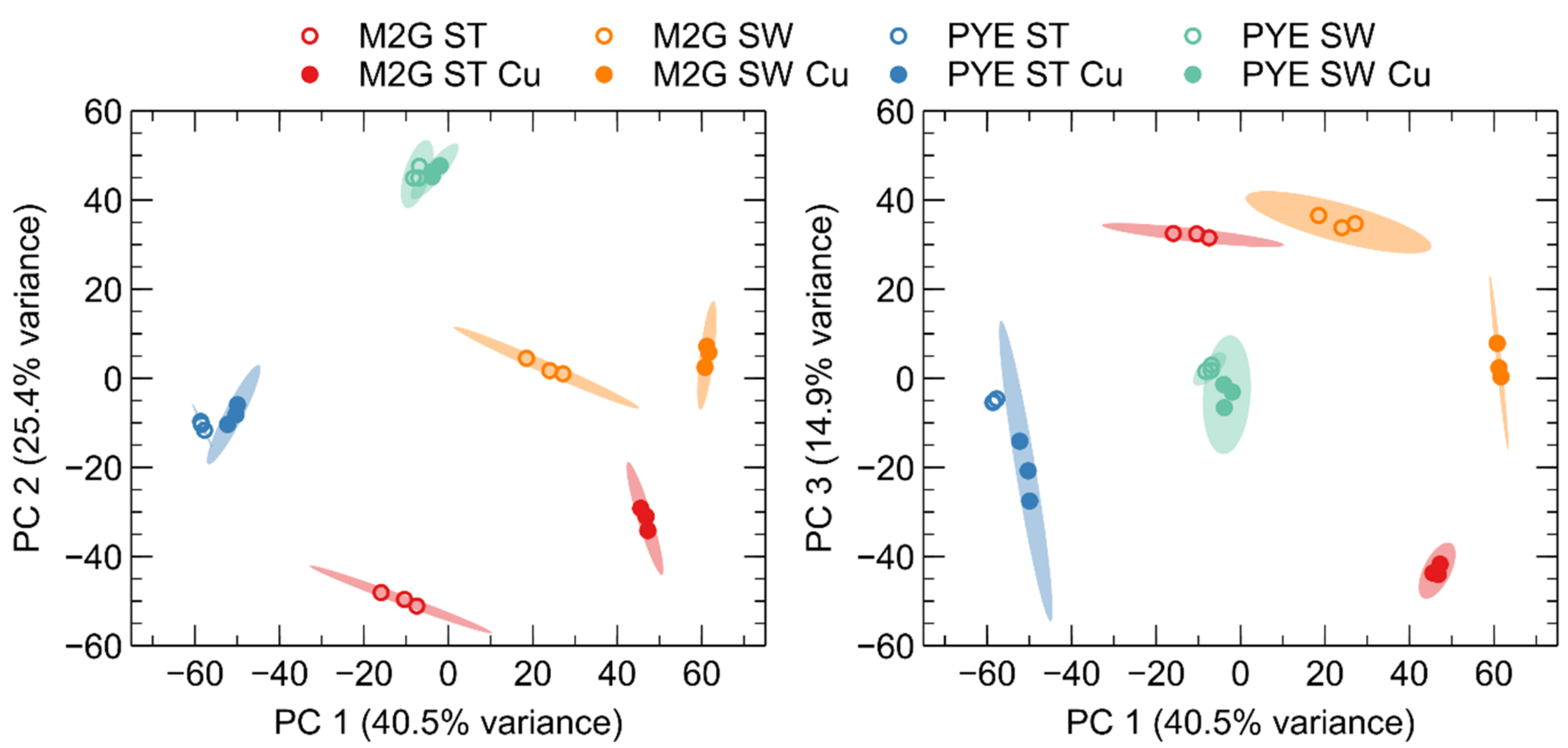
3.4. Role of Environmental Conditions in the Cu Stress Response
3.5. Cu Stress Perception by Stalked and Swarmer Cells
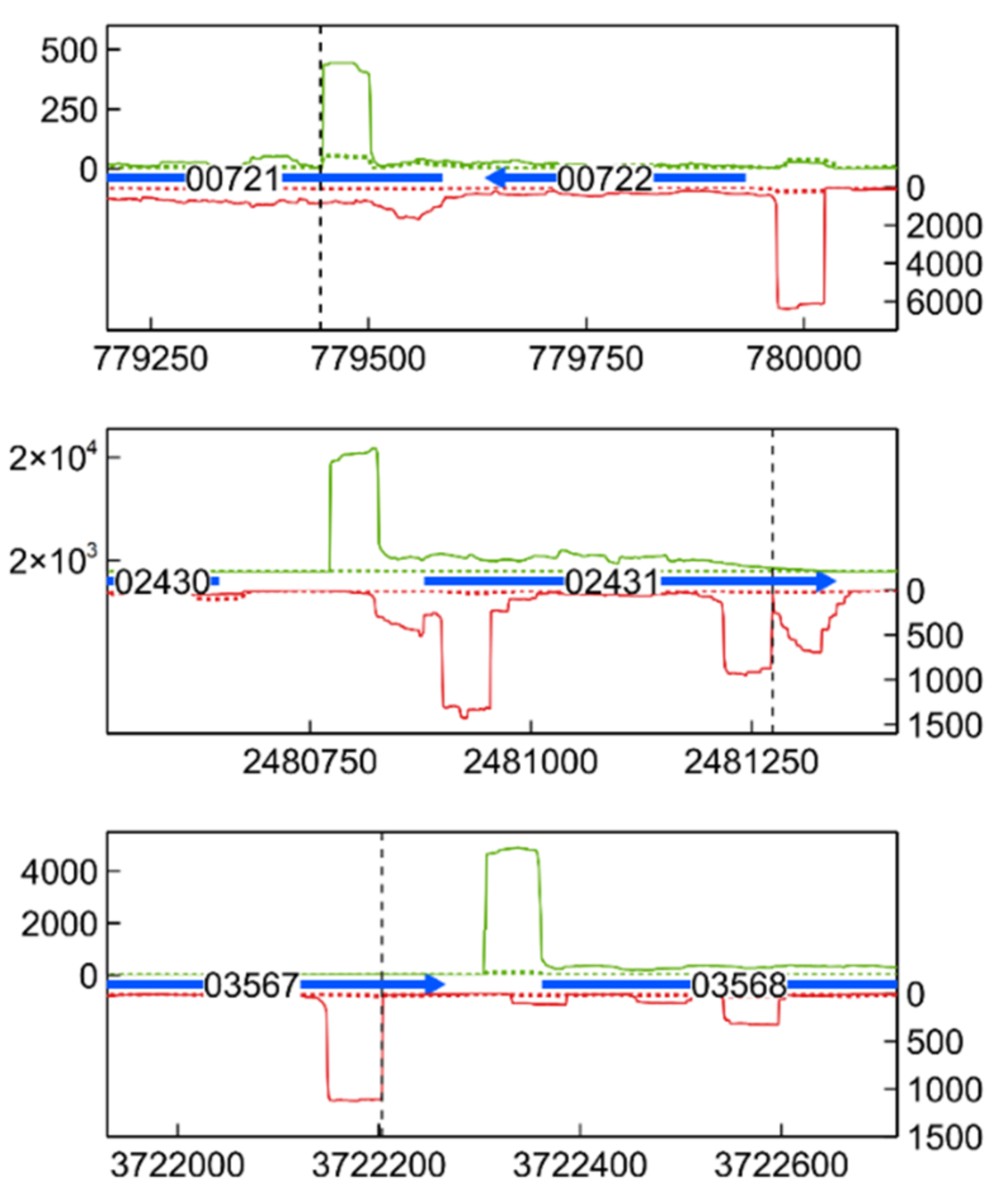
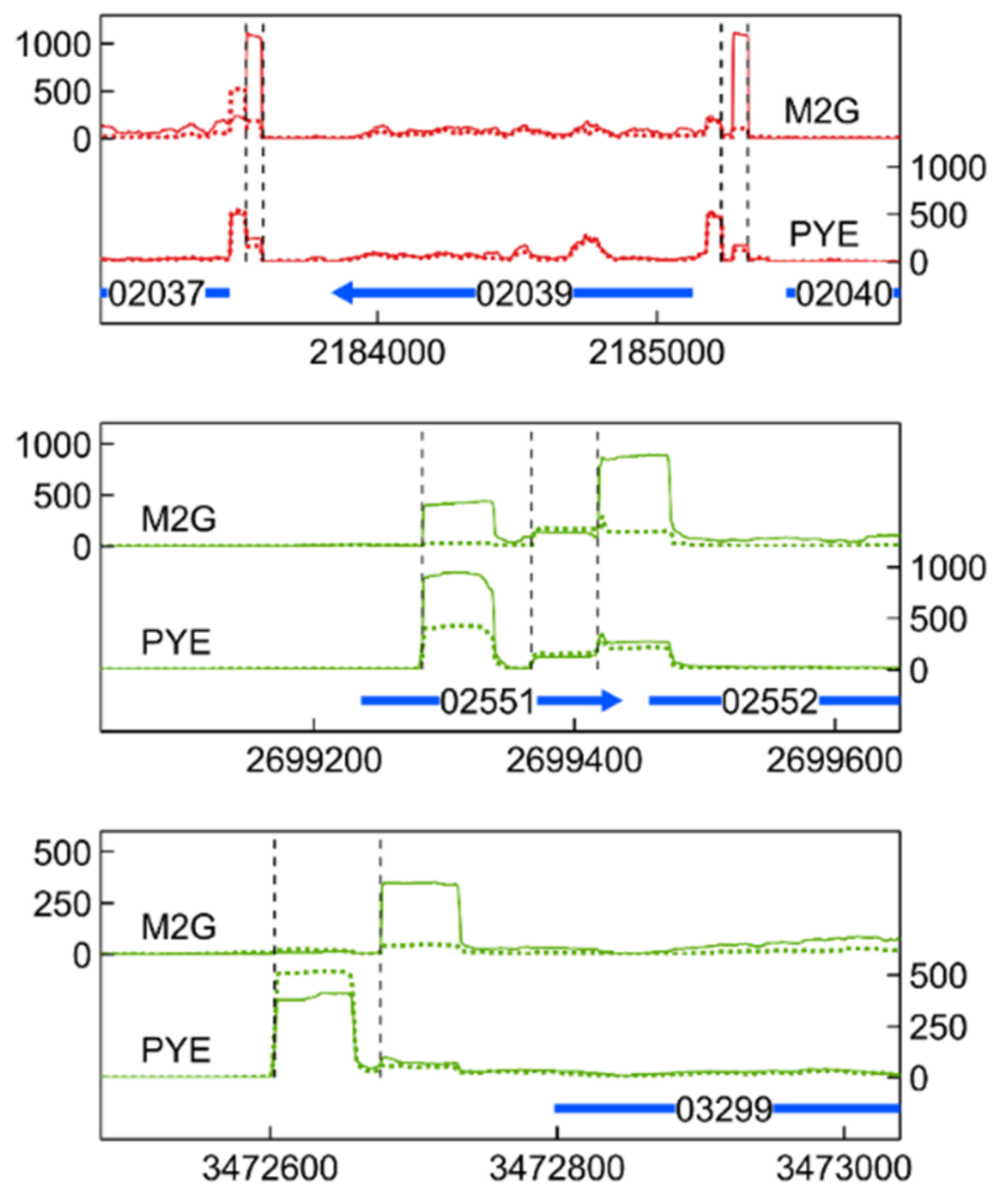
3.6. Transcription Start Site Analysis
3.7. Correspondence with Previous Transcriptomic Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Flemming, C.A.; Trevors, J.T. Copper toxicity and chemistry in the environment: A review. Water Air Soil Pollut. 1989, 44, 143–158. [Google Scholar] [CrossRef]
- Besold, A.N.; Culbertson, E.M.; Culotta, V.C. The Yin and Yang of copper during infection. JBIC J. Biol. Inorg. Chem. 2016, 21, 137–144. [Google Scholar] [CrossRef]
- Vincent, M.; Duval, R.E.; Hartemann, P.; Engels-Deutsch, M. Contact killing and antimicrobial properties of copper. J. Appl. Microbiol. 2018, 124, 1032–1046. [Google Scholar] [CrossRef]
- Giachino, A.; Waldron, K.J. Copper tolerance in bacteria requires the activation of multiple accessory pathways. Mol. Microbiol. 2020, 114, 377–390. [Google Scholar] [CrossRef]
- Peters, K.; Pazos, M.; Edoo, Z.; Hugonnet, J.E.; Martorana, A.M.; Polissi, A.; VanNieuwenhze, M.S.; Arthur, M.; Vollmer, W. Copper inhibits peptidoglycan LD-transpeptidases suppressing beta-lactam resistance due to bypass of penicillin-binding proteins. Proc. Natl. Acad. Sci. USA 2018, 115, 10786–10791. [Google Scholar] [CrossRef]
- May, K.L.; Lehman, K.M.; Mitchell, A.M.; Grabowicz, M. A Stress Response Monitoring Lipoprotein Trafficking to the Outer Membrane. mBio 2019, 10, e00618-19. [Google Scholar] [CrossRef]
- Tan, G.; Yang, J.; Li, T.; Zhao, J.; Sun, S.; Li, X.; Lin, C.; Li, J.; Zhou, H.; Lyu, J.; et al. Anaerobic Copper Toxicity and Iron-Sulfur Cluster Biogenesis in Escherichia coli. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef]
- Djoko, K.Y.; Phan, M.-D.; Peters, K.M.; Walker, M.J.; Schembri, M.A.; McEwan, A.G. Interplay between tolerance mechanisms to copper and acid stress in Escherichia coli. Proc. Natl. Acad. Sci. USA 2017, 114, 6818–6823. [Google Scholar] [CrossRef]
- Macomber, L.; Imlay, J.A. The iron-sulfur clusters of dehydratases are primary intracellular targets of copper toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 8344–8349. [Google Scholar] [CrossRef] [PubMed]
- Lemire, J.A.; Harrison, J.J.; Turner, R.J. Antimicrobial activity of metals: Mechanisms, molecular targets and applications. Nat. Rev. Genet. 2013, 11, 371–384. [Google Scholar] [CrossRef]
- Valko, M.; Morris, H.; Cronin, M.T.D. Metals, Toxicity and Oxidative Stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef]
- Stohs, S.J.; Bagchi, D. Oxidative mechanisms in the toxicity of metal ions. Free Radic. Biol. Med. 1995, 18, 321–336. [Google Scholar] [CrossRef]
- Nies, D.H.; Silver, S. Molecular Microbiology of Heavy Metals; Microbiology Monographs Book Series; Steinbüchel, A., Ed.; Springer: Berlin, Germany, 2007. [Google Scholar]
- Argüello, J.M.; González-Guerrero, M.; Raimunda, D. Bacterial Transition Metal P1B-ATPases: Transport Mechanism and Roles in Virulence. Biochemistry 2011, 50, 9940–9949. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-H.; Nies, D.H.; McEvoy, M.M.; Rensing, C. Switch or Funnel: How RND-Type Transport Systems Control Periplasmic Metal Homeostasis. J. Bacteriol. 2011, 193, 2381–2387. [Google Scholar] [CrossRef] [PubMed]
- Su, C.-C.; Yang, F.; Long, F.; Reyon, D.; Routh, M.D.; Kuo, D.W.; Mokhtari, A.K.; Van Ornam, J.D.; Rabe, K.L.; Hoy, J.A.; et al. Crystal Structure of the Membrane Fusion Protein CusB from Escherichia coli. J. Mol. Biol. 2009, 393, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Arguello, J.M.; Raimunda, D.; Padilla-Benavides, T. Mechanisms of copper homeostasis in bacteria. Front. Cell. Infect. Microbiol. 2013, 3, 73. [Google Scholar] [CrossRef] [PubMed]
- Boal, A.K.; Rosenzweig, A.C. Structural Biology of Copper Trafficking. Chem. Rev. 2009, 106, 4760–4779. [Google Scholar] [CrossRef]
- Roberts, S.A.; Weichsel, A.; Grass, G.; Thakali, K.; Hazzard, J.T.; Tollin, G.; Rensing, C.; Montfort, W.R. Crystal structure and electron transfer kinetics of CueO, a multicopper oxidase required for copper homeostasis in Escherichia coli. Proc. Natl. Acad. Sci. USA 2002, 99, 2766–2771. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Grass, G.; Rensing, C.; Montfort, W.R. Cuprous Oxidase Activity of CueO from Escherichia coli. J. Bacteriol. 2004, 186, 7815–7817. [Google Scholar] [CrossRef]
- Gillet, S.; Lawarée, E.; Matroule, J.-Y. Functional Diversity of Bacterial Strategies to Cope with Metal Toxicity. In Microbial Diversity in the Genomic Era; Elsevier BV: Amsterdam, The Netherlands, 2019; pp. 409–426. [Google Scholar]
- North, N.N.; Dollhopf, S.L.; Petrie, L.; Istok, J.D.; Balkwill, D.L.; Kostka, J.E. Change in Bacterial Community Structure during In Situ Biostimulation of Subsurface Sediment Cocontaminated with Uranium and Nitrate. Appl. Environ. Microbiol. 2004, 70, 4911–4920. [Google Scholar] [CrossRef]
- Benyehuda, G.; Coombs, J.; Ward, P.L.; Balkwill, D.; Barkay, T. Metal resistance among aerobic chemoheterotrophic bacteria from the deep terrestrial subsurface. Can. J. Microbiol. 2003, 49, 151–156. [Google Scholar] [CrossRef]
- Inagaki, F.; Takai, K.; Hirayama, H.; Yamato, Y.; Nealson, K.H.; Horikoshi, K. Distribution and phylogenetic diversity of the subsurface microbial community in a Japanese epithermal gold mine. Extremophiles 2003, 7, 307–317. [Google Scholar] [CrossRef]
- Hu, P.; Brodie, E.L.; Suzuki, Y.; McAdams, H.H.; Andersen, G.L. Whole-Genome Transcriptional Analysis of Heavy Metal Stresses in Caulobacter crescentus. J. Bacteriol. 2005, 187, 8437–8449. [Google Scholar] [CrossRef]
- Lawarée, E.; Gillet, S.; Louis, G.; Tilquin, F.; Le Blastier, S.; Cambier, P.; Matroule, J.-Y. Caulobacter crescentus intrinsic dimorphism provides a prompt bimodal response to copper stress. Nat. Microbiol. 2016, 1, 16098. [Google Scholar] [CrossRef]
- Ely, B. [17] Genetics of Caulobacter crescentus. Regul. Cell Death Part. A Apoptotic Mech. 1991, 204, 372–384. [Google Scholar] [CrossRef]
- Maertens, L.; Leys, N.; Matroule, J.-Y.; Van Houdt, R. The Transcriptomic Landscape of Cupriavidus metallidurans CH34 Acutely Exposed to Copper. Genes 2020, 11, 1049. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Smyth, G.K. Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics 2009, 25, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Sprouffske, K.; Wagner, A. Growthcurver: An R package for obtaining interpretable metrics from microbial growth curves. BMC Bioinform. 2016, 17, 172. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.; Weisberg, S. An R Companion to Applied Regression, 3rd ed.; Sage: Thousand Oaks, CA, USA, 2019. [Google Scholar]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Kohler, C.; Lourenço, R.F.; Avelar, G.M.; Gomes, S.L. Extracytoplasmic function (ECF) sigma factor σF is involved in Caulobacter crescentus response to heavy metal stress. BMC Microbiol. 2012, 12, 210. [Google Scholar] [CrossRef]
- Alvarez-Martinez, C.E.; Baldini, R.L.; Gomes, S.L. A Caulobacter crescentus Extracytoplasmic Function Sigma Factor Mediating the Response to Oxidative Stress in Stationary Phase. J. Bacteriol. 2006, 188, 1835–1846. [Google Scholar] [CrossRef]
- Monsieurs, P.; Moors, H.; Van Houdt, R.; Janssen, P.J.; Janssen, A.; Coninx, I.; Mergeay, M.; Leys, N. Heavy metal resistance in Cupriavidus metallidurans CH34 is governed by an intricate transcriptional network. BioMetals 2011, 24, 1133–1151. [Google Scholar] [CrossRef]
- Wiesemann, N.; Mohr, J.; Grosse, C.; Herzberg, M.; Hause, G.; Reith, F.; Nies, D.H. Influence of Copper Resistance Determinants on Gold Transformation by Cupriavidus metallidurans Strain CH34. J. Bacteriol. 2013, 195, 2298–2308. [Google Scholar] [CrossRef]
- Gennaris, A.; Ezraty, B.; Henry, C.; Agrebi, R.; Vergnes, A.; Oheix, E.; Bos, J.; Leverrier, P.; Espinosa, L.; Szewczyk, J.; et al. Repairing oxidized proteins in the bacterial envelope using respiratory chain electrons. Nat. Cell Biol. 2015, 528, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.G.; Lorenzetti, A.P.; Ribeiro, R.A.; Alves, I.R.; Leaden, L.; Galhardo, R.S.; Koide, T.; Marques, M.V. OxyR and the hydrogen peroxide stress response in Caulobacter crescentus. Gene 2019, 700, 70–84. [Google Scholar] [CrossRef]
- Neto, J.F.D.S.; Lourenço, R.F.; Marques, M.V. Global transcriptional response of Caulobacter crescentus to iron availability. BMC Genom. 2013, 14, 549. [Google Scholar] [CrossRef]
- Neto, J.F.D.S.; Braz, V.S.; Italiani, V.C.S.; Marques, M.V. Fur controls iron homeostasis and oxidative stress defense in the oligotrophic alpha-proteobacterium Caulobacter crescentus. Nucleic Acids Res. 2009, 37, 4812–4825. [Google Scholar] [CrossRef]
- Italiani, V.C.S.; Neto, J.F.D.S.; Braz, V.S.; Marques, M.V. Regulation of Catalase-Peroxidase KatG Is OxyR Dependent and Fur Independent in Caulobacter crescentus. J. Bacteriol. 2011, 193, 1734–1744. [Google Scholar] [CrossRef]
- Um, H.Y.; Kong, H.G.; Lee, H.J.; Choi, H.K.; Park, E.J.; Kim, S.T.; Murugiyan, S.; Chung, E.; Kang, K.Y.; Lee, S.-W. Altered Gene Expression and Intracellular Changes of the Viable but Nonculturable State in Ralstonia solanacearum by Copper Treatment. Plant Pathol. J. 2013, 29, 374–385. [Google Scholar] [CrossRef]
- Baker, J.; Sitthisak, S.; Sengupta, M.; Johnson, M.; Jayaswal, R.K.; Morrissey, J.A. Copper Stress Induces a Global Stress Response in Staphylococcus aureus and Represses sae and agr Expression and Biofilm Formation. Appl. Environ. Microbiol. 2009, 76, 150–160. [Google Scholar] [CrossRef]
- López, C.; Checa, S.K.; Soncini, F.C. CpxR/CpxA Controls scsABCD Transcription to Counteract Copper and Oxidative Stress in Salmonella enterica Serovar Typhimurium. J. Bacteriol. 2018, 200. [Google Scholar] [CrossRef] [PubMed]
- Svenningsen, N.B.; Damgaard, M.; Rasmussen, M.; Pérez-Pantoja, D.; Nybroe, O.; Nicolaisen, M.H. Cupriavidus pinatubonensis AEO106 deals with copper-induced oxidative stress before engaging in biodegradation of the herbicide 4-chloro-2-methylphenoxyacetic acid. BMC Microbiol. 2017, 17, 211. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, G.H.; Mukherjee, A.; Kelly, R.M. Transcriptomes of the Extremely Thermoacidophilic Archaeon Metallosphaera sedula Exposed to Metal “Shock” Reveal Generic and Specific Metal Responses. Appl. Environ. Microbiol. 2016, 82, 4613–4627. [Google Scholar] [CrossRef]
- Huang, N.; Mao, J.; Hu, M.; Wang, X.; Huo, M. Responses to copper stress in the metal-resistant bacterium Cupriavidus gilardii CR3: A whole-transcriptome analysis. J. Basic Microbiol. 2019, 59, 446–457. [Google Scholar] [CrossRef]
- Qian, H.; Yu, S.; Sun, Z.; Xie, X.; Liu, W.; Fu, Z. Effects of copper sulfate, hydrogen peroxide and N-phenyl-2-naphthylamine on oxidative stress and the expression of genes involved photosynthesis and microcystin disposition in Microcystis aeruginosa. Aquat. Toxicol. 2010, 99, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Bagwell, C.E.; Hixson, K.K.; Milliken, C.E.; Lopez-Ferrer, D.; Weitz, K.K. Proteomic and Physiological Responses of Kineococcus radiotolerans to Copper. PLoS ONE 2010, 5, e12427. [Google Scholar] [CrossRef] [PubMed]
- Teitzel, G.M.; Geddie, A.; De Long, S.K.; Kirisits, M.J.; Whiteley, M.; Parsek, M.R. Survival and Growth in the Presence of Elevated Copper: Transcriptional Profiling of Copper-Stressed Pseudomonas aeruginosa. J. Bacteriol. 2006, 188, 7242–7256. [Google Scholar] [CrossRef]
- Rensing, C.; Alwathnani, H.A.; McDevitt, S.F. The Copper Metallome in Prokaryotic Cells. Met. Ions Life Sci. 2016, 12, 161–173. [Google Scholar] [CrossRef]
- Leaden, L.; Silva, L.G.; Ribeiro, R.A.; Dos Santos, N.M.; Lorenzetti, A.P.R.; Alegria, T.G.P.; Schulz, M.L.; Medeiros, M.H.G.; Koide, T.; Marques, M.V. Iron Deficiency Generates Oxidative Stress and Activation of the SOS Response in Caulobacter crescentus. Front. Microbiol. 2018, 9, 2014. [Google Scholar] [CrossRef]
- Ferreira, I.G.D.C.; Rodrigues, M.M.; Neto, J.F.D.S.; Mazzon, R.R.; Marques, M.D.V. Role and regulation of ferritin-like proteins in iron homeostasis and oxidative stress survival of Caulobacter crescentus. BioMetals 2016, 29, 851–862. [Google Scholar] [CrossRef]
- Hartl, J.; Kiefer, P.; Kaczmarczyk, A.; Mittelviefhaus, M.; Meyer, F.; Vonderach, T.; Hattendorf, B.; Jenal, U.; Vorholt, J.A. Untargeted metabolomics links glutathione to bacterial cell cycle progression. Nat. Metab. 2020, 2, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Goemans, C.V.; Beaufay, F.; Wahni, K.; Van Molle, I.; Messens, J.; Collet, J.-F. An essential thioredoxin is involved in the control of the cell cycle in the bacterium Caulobacter crescentus. J. Biol. Chem. 2018, 293, 3839–3848. [Google Scholar] [CrossRef]
- Kachur, A.V.; Koch, C.J.; Biaglow, J.E. Mechanism of Copper-Catalyzed Oxidation of Glutathione. Free Radic. Res. 1998, 28, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, A.C.; Simão, R.C.; Susin, M.F.; Baldini, R.L.; Avedissian, M.; Gomes, S.L. Downregulation of the heat shock response is independent of DnaK and sigma32 levels in Caulobacter crescentus. Mol. Microbiol. 2003, 49, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Schramm, F.D.; Heinrich, K.; Thüring, M.; Bernhardt, J.; Jonas, K. An essential regulatory function of the DnaK chaperone dictates the decision between proliferation and maintenance in Caulobacter crescentus. PLoS Genet. 2017, 13, e1007148. [Google Scholar] [CrossRef] [PubMed]
- Susin, M.F.; Baldini, R.L.; Gueiros-Filho, F.; Gomes, S.L. GroES/GroEL and DnaK/DnaJ Have Distinct Roles in Stress Responses and during Cell Cycle Progression in Caulobacter crescentus. J. Bacteriol. 2006, 188, 8044–8053. [Google Scholar] [CrossRef]
- Goemans, C.V.; Beaufay, F.; Arts, I.S.; Agrebi, R.; Vertommen, D.; Collet, J.-F. The Chaperone and Redox Properties of CnoX Chaperedoxins Are Tailored to the Proteostatic Needs of Bacterial Species. mBio 2018, 9, e01541-18. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Inaba, K. The disulfide bond formation (Dsb) system. Curr. Opin. Struct. Biol. 2008, 18, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Subedi, P.; Paxman, J.J.; Wang, G.; Ukuwela, A.A.; Xiao, Z.; Heras, B. The Scs disulfide reductase system cooperates with the metallochaperone CueP in Salmonella copper resistance. J. Biol. Chem. 2019, 294, 15876–15888. [Google Scholar] [CrossRef]
- Yung, M.C.; Ma, J.; Salemi, M.R.; Phinney, B.S.; Bowman, G.R.; Jiao, Y. Shotgun proteomic analysis unveils survival and detoxification strategies by Caulobacter crescentus during exposure to uranium, chromium, and cadmium. J. Proteome Res. 2014, 13, 1833–1847. [Google Scholar] [CrossRef] [PubMed]
- Jonas, K.; Liu, J.; Chien, P.; Laub, M.T. Proteotoxic Stress Induces a Cell-Cycle Arrest by Stimulating Lon to Degrade the Replication Initiator DnaA. Cell 2013, 154, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Fischer, B.; Rummel, G.; Aldridge, P.; Jenal, U. The FtsH protease is involved in development, stress response and heat shock control in Caulobacter crescentus. Mol. Microbiol. 2002, 44, 461–478. [Google Scholar] [CrossRef]
- Simão, R.C.G.; Susin, M.F.; Alvarez-Martinez, C.E.; Gomes, S.L. Cells lacking ClpB display a prolonged shutoff phase of the heat shock response inCaulobacter crescentus. Mol. Microbiol. 2005, 57, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Bhat, N.H.; Vass, R.H.; Stoddard, P.R.; Shin, D.K.; Chien, P. Identification of ClpP substrates in Caulobacter crescentus reveals a role for regulated proteolysis in bacterial development. Mol. Microbiol. 2013, 88, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Francis, L.I.; Jonas, K.; Laub, M.T.; Chien, P. ClpAP is an auxiliary protease for DnaA degradation in Caulobacter crescentus. Mol. Microbiol. 2016, 102, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Vass, R.H.; Zeinert, R.D.; Chien, P. Protease regulation and capacity during Caulobacter growth. Curr. Opin. Microbiol. 2016, 34, 75–81. [Google Scholar] [CrossRef]
- Zeinert, R.D.; Baniasadi, H.; Tu, B.P.; Chien, P. The Lon Protease Links Nucleotide Metabolism with Proteotoxic Stress. Mol. Cell 2020, 79, 758–767.e6. [Google Scholar] [CrossRef]
- Gora, K.G.; Cantin, A.; Wohlever, M.L.; Joshi, K.K.; Perchuk, B.S.; Chien, P.; Laub, M.T. Regulated proteolysis of a transcription factor complex is critical to cell cycle progression inCaulobacter crescentus. Mol. Microbiol. 2013, 87, 1277–1289. [Google Scholar] [CrossRef] [PubMed]
- Parvatiyar, K.; Alsabbagh, E.M.; Ochsner, U.A.; Stegemeyer, M.A.; Smulian, A.G.; Hwang, S.H.; Jackson, C.R.; McDermott, T.R.; Hassett, D.J. Global analysis of cellular factors and responses involved in Pseudomonas aeruginosa resistance to arsenite. J. Bacteriol. 2005, 187, 4853–4864. [Google Scholar] [CrossRef]
- Perez, J.M.; Calderón, I.L.; Arenas, F.A.; Fuentes, D.E.; Pradenas, G.A.; Fuentes, E.L.; Sandoval, J.M.; Castro, M.E.; Elías, A.O.; Vásquez, C.C. Bacterial Toxicity of Potassium Tellurite: Unveiling an Ancient Enigma. PLoS ONE 2007, 2, e211. [Google Scholar] [CrossRef]
- Valencia, E.Y.; Braz, V.S.; Guzzo, C.; Marques, M.V. Two RND proteins involved in heavy metal efflux in Caulobacter crescentus belong to separate clusters within proteobacteria. BMC Microbiol. 2013, 13, 79. [Google Scholar] [CrossRef]
- Park, D.M.; Overton, K.W.; Liou, M.J.; Jiao, Y. Identification of a U/Zn/Cu responsive global regulatory two-component system inCaulobacter crescentus. Mol. Microbiol. 2017, 104, 46–64. [Google Scholar] [CrossRef]
- Fröhlich, K.S.; Förstner, K.U.; Gitai, Z. Post-transcriptional gene regulation by an Hfq-independent small RNA in Caulobacter crescentus. Nucleic Acids Res. 2018, 46, 10969–10982. [Google Scholar] [CrossRef]
- Van Acker, H.; Coenye, T. The Role of Reactive Oxygen Species in Antibiotic-Mediated Killing of Bacteria. Trends Microbiol. 2017, 25, 456–466. [Google Scholar] [CrossRef]
- Dibrov, P.; Dzioba, J.; Gosink, K.K.; Häse, C.C. Chemiosmotic Mechanism of Antimicrobial Activity of Ag+ in Vibrio cholerae. Antimicrob. Agents Chemother. 2002, 46, 2668–2670. [Google Scholar] [CrossRef] [PubMed]
- Balhesteros, H.; Shipelskiy, Y.; Long, N.J.; Majumdar, A.; Katz, B.B.; Santos, N.M.; Leaden, L.; Newton, S.M.; Marques, M.V.; Klebba, P.E. TonB-Dependent Heme/Hemoglobin Utilization by Caulobacter crescentus HutA. J. Bacteriol. 2017, 199. [Google Scholar] [CrossRef] [PubMed]
- Mazzon, R.R.; Braz, V.S.; da Silva Neto, J.F.; do Valle Marques, M. Analysis of the Caulobacter crescentus Zur regulon reveals novel insights in zinc acquisition by TonB-dependent outer membrane proteins. BMC Genom. 2014, 15, 734. [Google Scholar] [CrossRef] [PubMed]
- García-Bayona, L.; Guo, M.S.; Laub, M.T. Contact-dependent killing by Caulobacter crescentus via cell surface-associated, glycine zipper proteins. eLife 2017, 6, e24869. [Google Scholar] [CrossRef]
- Davis, A.V.; O’Halloran, T.V. A place for thioether chemistry in cellular copper ion recognition and trafficking. Nat. Chem. Biol. 2008, 4, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; van Tonder, A.J.; Vilchèze, C.; Mendes, V.; Thomas, S.E.; Malek, A.; Chen, B.; Chen, M.; Kim, J.; Blundell, T.L.; et al. Arginine-deprivation–induced oxidative damage sterilizes Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2018, 115, 9779–9784. [Google Scholar] [CrossRef] [PubMed]
- Barrientos-Moreno, L.; Molina-Henares, M.A.; Pastor-García, M.; Ramos-González, M.I.; Espinosa-Urgel, M. Arginine Biosynthesis Modulates Pyoverdine Production and Release in Pseudomonas putida as Part of the Mechanism of Adaptation to Oxidative Stress. J. Bacteriol. 2019, 201. [Google Scholar] [CrossRef]
- Chakraborty, B.; Burne, R.A. Effects of Arginine on Streptococcus mutans Growth, Virulence Gene Expression, and Stress Tolerance. Appl. Environ. Microbiol. 2017, 83, e00496-17. [Google Scholar] [CrossRef]
- Xue, Y.; Davis, A.V.; Balakrishnan, G.; Stasser, J.P.; Staehlin, B.M.; Focia, P.; Spiro, T.G.; Penner-Hahn, J.E.; O’Halloran, T.V. Cu(I) recognition via cation-pi and methionine interactions in CusF. Nat. Chem. Biol. 2008, 4, 107–109. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Lei, Y.; Patel, J. Bioremediation of soluble heavy metals with recombinant Caulobacter crescentus. Bioeng. Bugs 2010, 1, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Dietl, A.-M.; Amich, J.; Leal, S.; Beckmann, N.; Binder, U.; Beilhack, A.; Pearlman, E.; Haas, H. Histidine biosynthesis plays a crucial role in metal homeostasis and virulence of Aspergillus fumigatus. Virulence 2016, 7, 465–476. [Google Scholar] [CrossRef]
- Hör, J.; Gorski, S.A.; Vogel, J. Bacterial RNA Biology on a Genome Scale. Mol. Cell 2018, 70, 785–799. [Google Scholar] [CrossRef] [PubMed]
- Assis, N.G.; Ribeiro, R.A.; Da Silva, L.G.; Vicente, A.M.; Hug, I.; Marques, M.V. Identification of Hfq-binding RNAs in Caulobacter crescentus. RNA Biol. 2019, 16, 719–726. [Google Scholar] [CrossRef]
- Georg, J.; Lalaouna, D.; Hou, S.; Lott, S.C.; Caldelari, I.; Marzi, S.; Hess, W.R.; Romby, P. The power of cooperation: Experimental and computational approaches in the functional characterization of bacterial sRNAs. Mol. Microbiol. 2019, 113, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.L.; Markich, S.J. Evaluation of the free ion activity model of metal-organism interaction: Extension of the conceptual model. Aquat. Toxicol. 2000, 51, 177–194. [Google Scholar] [CrossRef]
- Hoffman, D.R.; Okon, J.L.; Sandrin, T.R. Medium composition affects the degree and pattern of cadmium inhibition of naphthalene biodegradation. Chemosphere 2005, 59, 919–927. [Google Scholar] [CrossRef]
- Rathnayake, I.; Megharaj, M.; Krishnamurti, G.; Bolan, N.S.; Naidu, R. Heavy metal toxicity to bacteria—Are the existing growth media accurate enough to determine heavy metal toxicity? Chemosphere 2013, 90, 1195–1200. [Google Scholar] [CrossRef]
- Nies, D.H. The biological chemistry of the transition metal “transportome” of Cupriavidus metallidurans. Metallomics 2016, 8, 481–507. [Google Scholar] [CrossRef]
- Smith, R.M.; Martell, A.E. Critical Stability Constants; Plenum Press: New York, NY, USA, 1989; pp. 173–174. [Google Scholar]
- Hiroshi, O. The Stability Constants of Ethylenediaminetetraacetato, Trimethylenediaminetetraacetato and Propylenediaminetetraacetato Complexes of Some Divalent Metal Ions. BCSJ 1965, 38, 771–777. [Google Scholar] [CrossRef]
- Sezonov, G.; Joseleau-Petit, D.; D’Ari, R. Escherichia coli Physiology in Luria-Bertani Broth. J. Bacteriol. 2007, 189, 8746–8749. [Google Scholar] [CrossRef]
- De Spiegeleer, P.; Sermon, J.; Lietaert, A.; Aertsen, A.; Michiels, C. Source of tryptone in growth medium affects oxidative stress resistance in Escherichia coli. J. Appl. Microbiol. 2004, 97, 124–133. [Google Scholar] [CrossRef]
- Mosser, M.; Kapel, R.; Chevalot, I.; Olmos, E.; Marc, I.; Marc, A.; Oriol, E. Fractionation of yeast extract by nanofiltration process to assess key compounds involved in CHO cell culture improvement. Biotechnol. Prog. 2015, 31, 875–882. [Google Scholar] [CrossRef]
- El-Helow, E.; Sabry, S.; Amer, R. Cadmium biosorption by a cadmium resistant strain of Bacillus thuringiensis: Regulation and optimization of cell surface affinity for metal cations. BioMetals 2000, 13, 273–280. [Google Scholar] [CrossRef]
- Hottes, A.K.; Meewan, M.; Yang, D.; Arana, N.; Romero, P.; McAdams, H.H.; Stephens, C. Transcriptional Profiling of Caulobacter crescentus during Growth on Complex and Minimal Media. J. Bacteriol. 2004, 186, 1448–1461. [Google Scholar] [CrossRef] [PubMed]
- Laub, M.T.; Shapiro, L.; McAdams, H.H. Systems biology of Caulobacter. Annu. Rev. Genet. 2007, 41, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Sun, L.; Ding, X.; Sun, D.; Liu, J.; Wang, W. Complete genome sequence of Caulobacter flavus RHGG3T, a type species of the genus Caulobacter with plant growth-promoting traits and heavy metal resistance. 3 Biotech 2019, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.; Janakiraman, B.; Kumar, L.; Radhakrishnan, S.K. A cell cycle-controlled redox switch regulates the topoisomerase IV activity. Genes Dev. 2015, 29, 1175–1187. [Google Scholar] [CrossRef]
- Landt, S.G.; Abeliuk, E.; McGrath, P.T.; Lesley, J.A.; McAdams, H.H.; Shapiro, L. Small non-coding RNAs in Caulobacter crescentus. Mol. Microbiol. 2008, 68, 600–614. [Google Scholar] [CrossRef] [PubMed]
- McGrath, P.T.; Lee, H.; Zhang, L.; Iniesta, A.; Hottes, A.K.; Tan, M.H.; Hillson, N.J.; Hu, P.; Shapiro, L.; McAdams, H.H. High-throughput identification of transcription start sites, conserved promoter motifs and predicted regulons. Nat. Biotechnol. 2007, 25, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Schrader, J.M.; Zhou, B.; Li, G.-W.; Lasker, K.; Childers, W.S.; Williams, B.; Long, T.; Crosson, S.; McAdams, H.H.; Weissman, J.S.; et al. The Coding and Noncoding Architecture of the Caulobacter crescentus Genome. PLoS Genet. 2014, 10, e1004463. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Schrader, J.M.; Kalogeraki, V.S.; Abeliuk, E.; Dinh, C.B.; Pham, J.Q.; Cui, Z.Z.; Dill, D.L.; McAdams, H.H.; Shapiro, L. The Global Regulatory Architecture of Transcription during the Caulobacter Cell Cycle. PLoS Genet. 2015, 11, e1004831. [Google Scholar] [CrossRef] [PubMed]
- Romilly, C.; Caldelari, I.; Parmentier, D.; Lioliou, E.; Romby, P.; Fechter, P. Current knowledge on regulatory RNAs and their machineries inStaphylococcus aureus. RNA Biol. 2012, 9, 402–413. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Makarova, A.A.; Grachova, E.V.; Neudachina, V.S.; Yashina, L.; Blüher, A.; Molodtsov, S.L.; Mertig, M.; Ehrlich, H.; Adamchuk, V.K.; Laubschat, C.; et al. Insight into Bio-metal Interface Formation in vacuo: Interplay of S-layer Protein with Copper and Iron. Sci. Rep. 2015, 5, srep08710. [Google Scholar] [CrossRef]
- Kawasaki, Y.; Kurosaki, K.; Kan, D.; Borges, I.K.; Atagui, A.S.; Sato, M.; Kondo, K.; Katahira, M.; Suzuki, I.; Takeda, M. Identification and characterization of the S-layer formed on the sheath of Thiothrix nivea. Arch. Microbiol. 2018, 200, 1257–1265. [Google Scholar] [CrossRef]
- Saberi, F.; Kamali, M.; Najafi, A.; Yazdanparast, A.; Moghaddam, M.M. Natural antisense RNAs as mRNA regulatory elements in bacteria: A review on function and applications. Cell. Mol. Biol. Lett. 2016, 21, 6. [Google Scholar] [CrossRef]
- Del Medico, L.; Cerletti, D.; Schächle, P.; Christen, M.; Christen, B. The type IV pilin PilA couples surface attachment and cell-cycle initiation in Caulobacter crescentus. Proc. Natl. Acad. Sci. USA 2020, 117, 9546–9553. [Google Scholar] [CrossRef]
- Skerker, J.M.; Shapiro, L. Identification and cell cycle control of a novel pilus system in Caulobacter crescentus. EMBO J. 2000, 19, 3223–3234. [Google Scholar] [CrossRef]
- Fang, G.; Passalacqua, K.D.; Hocking, J.; Llopis, P.M.; Gerstein, M.; Bergman, N.H.; Jacobs-Wagner, C. Transcriptomic and phylogenetic analysis of a bacterial cell cycle reveals strong associations between gene co-expression and evolution. BMC Genom. 2013, 14, 450. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway or Functional Relation | Specific Pathway | M2G ST | M2G SW | PYE ST | PYE SW | ||||
|---|---|---|---|---|---|---|---|---|---|
| Up | Down | Up | Down | Up | Down | Up | Down | ||
| Metal resistance mechanisms | 19 | 0 | 5 | 0 | 6 | 1 | 0 | 0 | |
| Proteases and peptidases | 32 | 9 | 13 | 6 | 4 | 0 | 0 | 0 | |
| Ribosome synthesis | 2 | 78 | 0 | 24 | 0 | 0 | 0 | 0 | |
| ATP synthesis | 0 | 10 | 0 | 6 | 0 | 0 | 0 | 0 | |
| sRNAs | 16 | 0 | 3 | 1 | 5 | 1 | 0 | 0 | |
| Antibiotic resistance | 12 | 3 | 5 | 1 | 0 | 0 | 0 | 0 | |
| Chaperons | 13 | 2 | 5 | 2 | 0 | 0 | 0 | 0 | |
| Transporters | ABC transport | 18 | 4 | 9 | 3 | 1 | 0 | 0 | 0 |
| TonB-dependent receptors | 6 | 10 | 2 | 4 | 4 | 2 | 1 | 0 | |
| Other | 19 | 19 | 2 | 9 | 4 | 0 | 3 | 0 | |
| Regulators | Known function | 19 | 19 | 6 | 13 | 6 | 2 | 1 | 0 |
| Other | 33 | 16 | 11 | 3 | 4 | 0 | 1 | 0 | |
| Amino acid metabolism | Methionine biosynthesis | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 |
| Cysteine biosynthesis & S assimilation | 8 | 2 | 0 | 0 | 1 | 3 | 0 | 0 | |
| Arginine biosynthesis | 1 | 7 | 1 | 5 | 0 | 0 | 0 | 0 | |
| Serine biosynthesis | 2 | 7 | 1 | 1 | 0 | 0 | 0 | 0 | |
| Histidine | 2 | 1 | 0 | 0 | 4 | 0 | 1 | 0 | |
| Redox and oxidative stress-related | Radical removal & antioxidants | 16 | 2 | 11 | 0 | 4 | 0 | 2 | 0 |
| Glutathione cycle | 7 | 2 | 2 | 1 | 0 | 0 | 0 | 0 | |
| Other | 37 | 39 | 14 | 5 | 7 | 0 | 2 | 0 | |
| Hypothetical proteins | 73 | 33 | 48 | 23 | 22 | 2 | 2 | 0 | |
| Locus Tag 1 | Gene Product | Log2 Fold Change | |||
|---|---|---|---|---|---|
| M2G | PYE | ||||
| ST | SW | ST | SW | ||
| 00028 $ | TonB-dependent receptor protein | 1.85 | 1.87 | 4.64 | 2.26 |
| 02833 | Sulfoxide reductase heme-binding subunit | 5.32 | 4.18 | 3.13 | 1.84 |
| 02834 | Sulfoxide reductase catalytic subunit | 5.84 | 5.73 | 3.34 | 2.60 |
| 02999 | DNA-binding domain-containing protein | 5.03 | 4.32 | 3.24 | 2.04 |
| 03000 | DUF692 domain-containing protein | 5.94 | 5.77 | 3.63 | 2.80 |
| 03001 | Hypothetical protein | 5.67 | 6.21 | 3.17 | 2.00 |
| 03273 | Anti-sigma factor NrsF | 4.15 | 4.27 | 2.88 | 1.84 * |
| 03362 | ECF-family sigma factor SigF | 4.44 | 4.70 | 2.78 | 2.17 |
| 03363 | DUF2282 domain-containing protein | 5.79 | 6.07 | 3.46 | 2.01 |
| 03364 | DUF692 domain-containing protein | 6.16 | 6.31 | 4.22 | 3.53 |
| 03365 | Hypothetical protein | 5.86 | 5.06 | 4.15 | 2.74 |
| 03366 | DoxX-family protein | 5.50 | 4.35 | 4.27 | 1.40 * |
| 03372 $ | Bacterioferritin-associated ferredoxin | 4.50 | 5.59 | 2.97 | 5.33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maertens, L.; Cherry, P.; Tilquin, F.; Van Houdt, R.; Matroule, J.-Y. Environmental Conditions Modulate the Transcriptomic Response of Both Caulobacter crescentus Morphotypes to Cu Stress. Microorganisms 2021, 9, 1116. https://doi.org/10.3390/microorganisms9061116
Maertens L, Cherry P, Tilquin F, Van Houdt R, Matroule J-Y. Environmental Conditions Modulate the Transcriptomic Response of Both Caulobacter crescentus Morphotypes to Cu Stress. Microorganisms. 2021; 9(6):1116. https://doi.org/10.3390/microorganisms9061116
Chicago/Turabian StyleMaertens, Laurens, Pauline Cherry, Françoise Tilquin, Rob Van Houdt, and Jean-Yves Matroule. 2021. "Environmental Conditions Modulate the Transcriptomic Response of Both Caulobacter crescentus Morphotypes to Cu Stress" Microorganisms 9, no. 6: 1116. https://doi.org/10.3390/microorganisms9061116
APA StyleMaertens, L., Cherry, P., Tilquin, F., Van Houdt, R., & Matroule, J.-Y. (2021). Environmental Conditions Modulate the Transcriptomic Response of Both Caulobacter crescentus Morphotypes to Cu Stress. Microorganisms, 9(6), 1116. https://doi.org/10.3390/microorganisms9061116