
Combination Regimens of Favipiravir Plus Interferon Alpha Inhibit Chikungunya Virus Replication in Clinically Relevant Human Cell Lines


 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Virus
2.3. Antivirals
2.4. Drug Assays
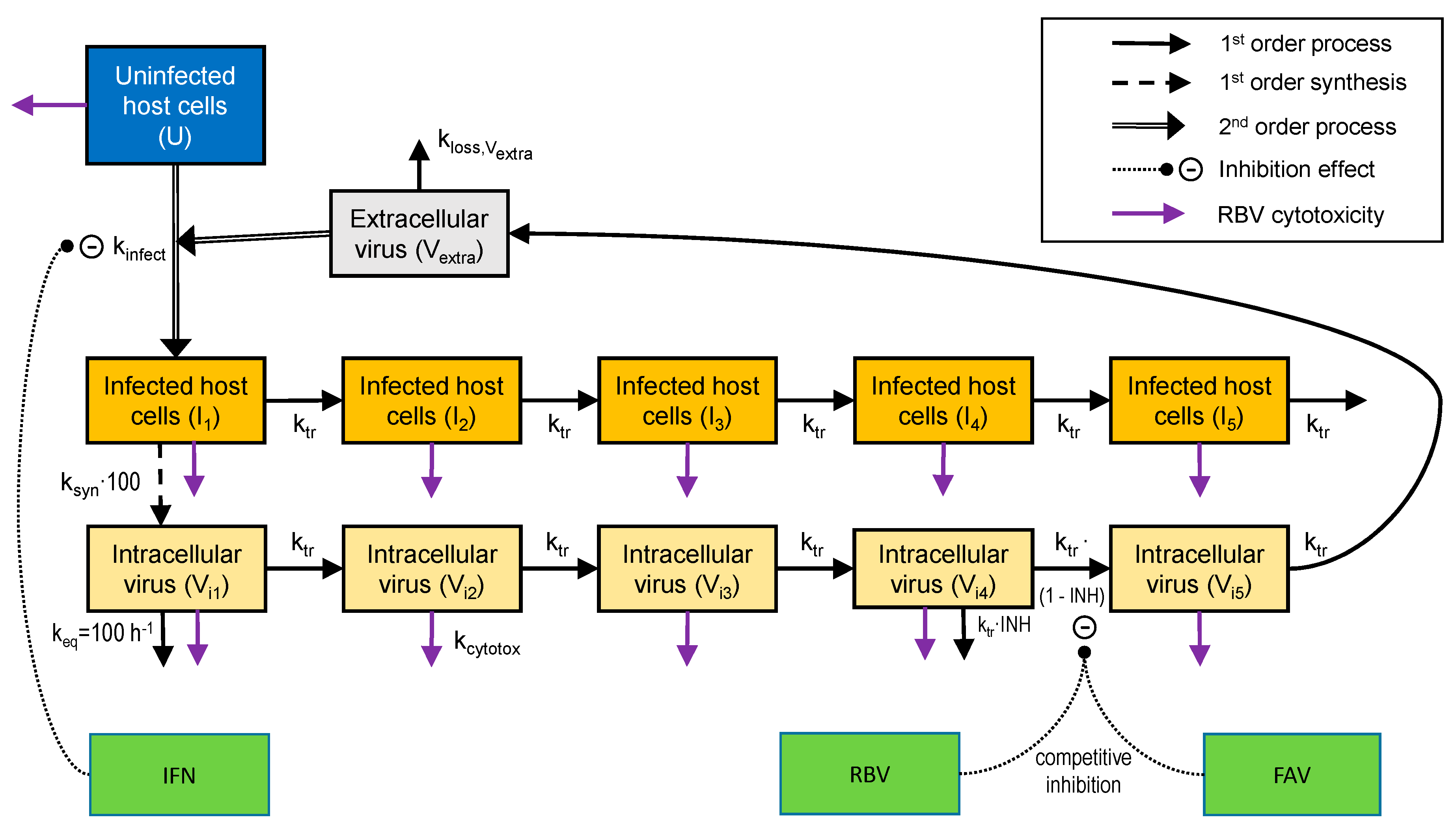
2.5. Mathematical Modeling and Simulation
3. Results
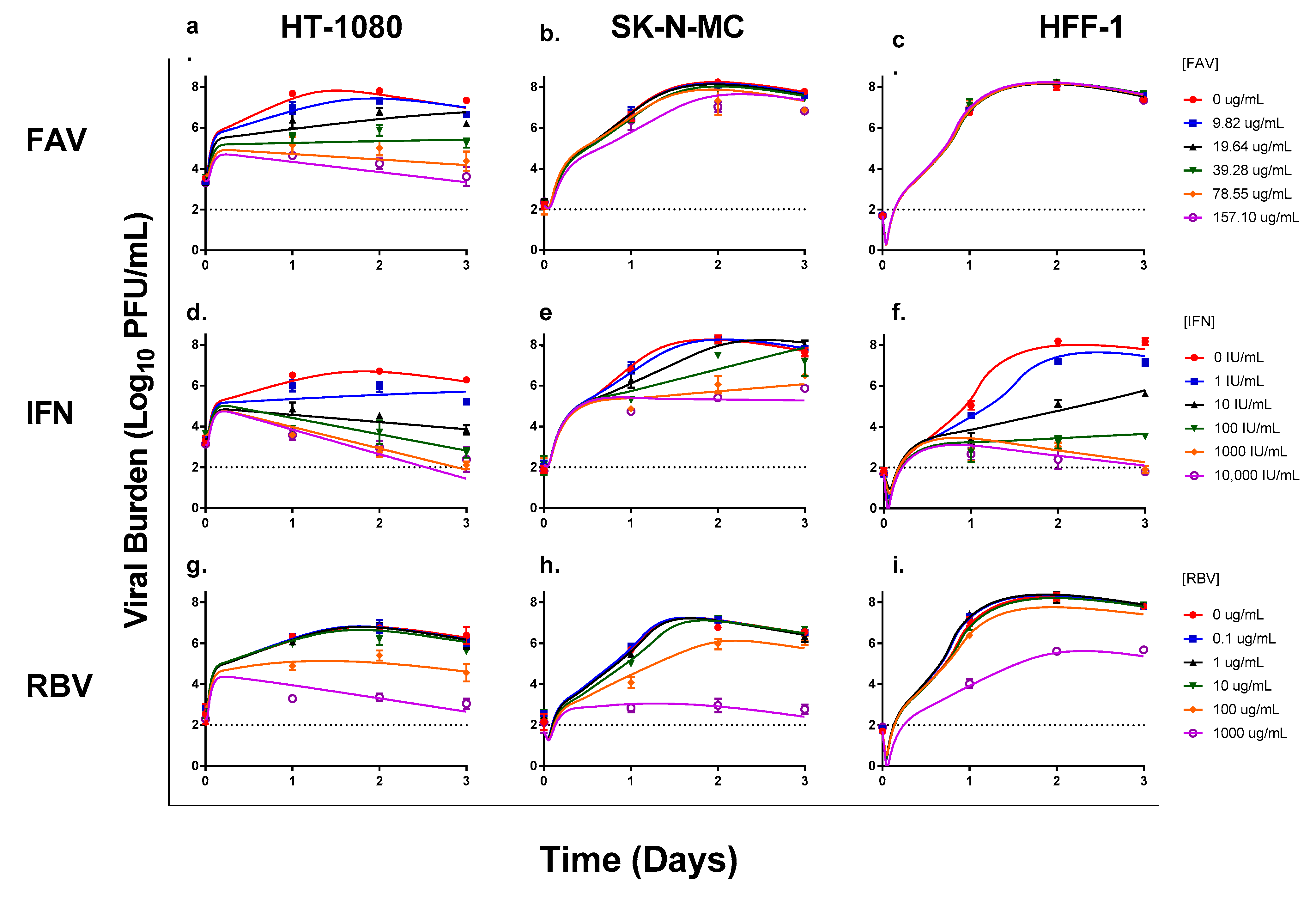
3.1. Antiviral Evaluations of Broad-Spectrum Agents as Monotherapy
3.2. Antiviral Evaluations of Combination Therapy
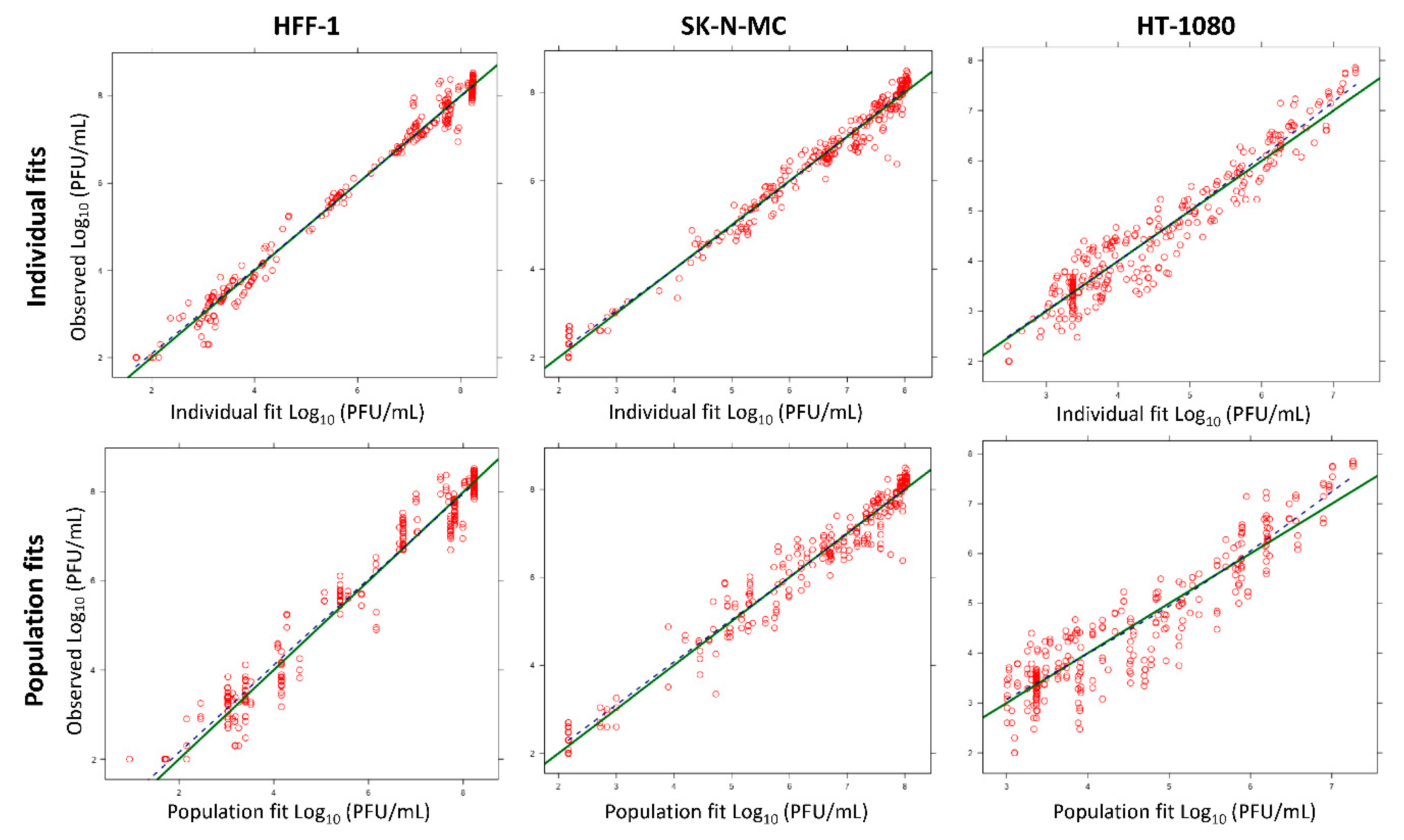
3.3. Mathematical Modeling
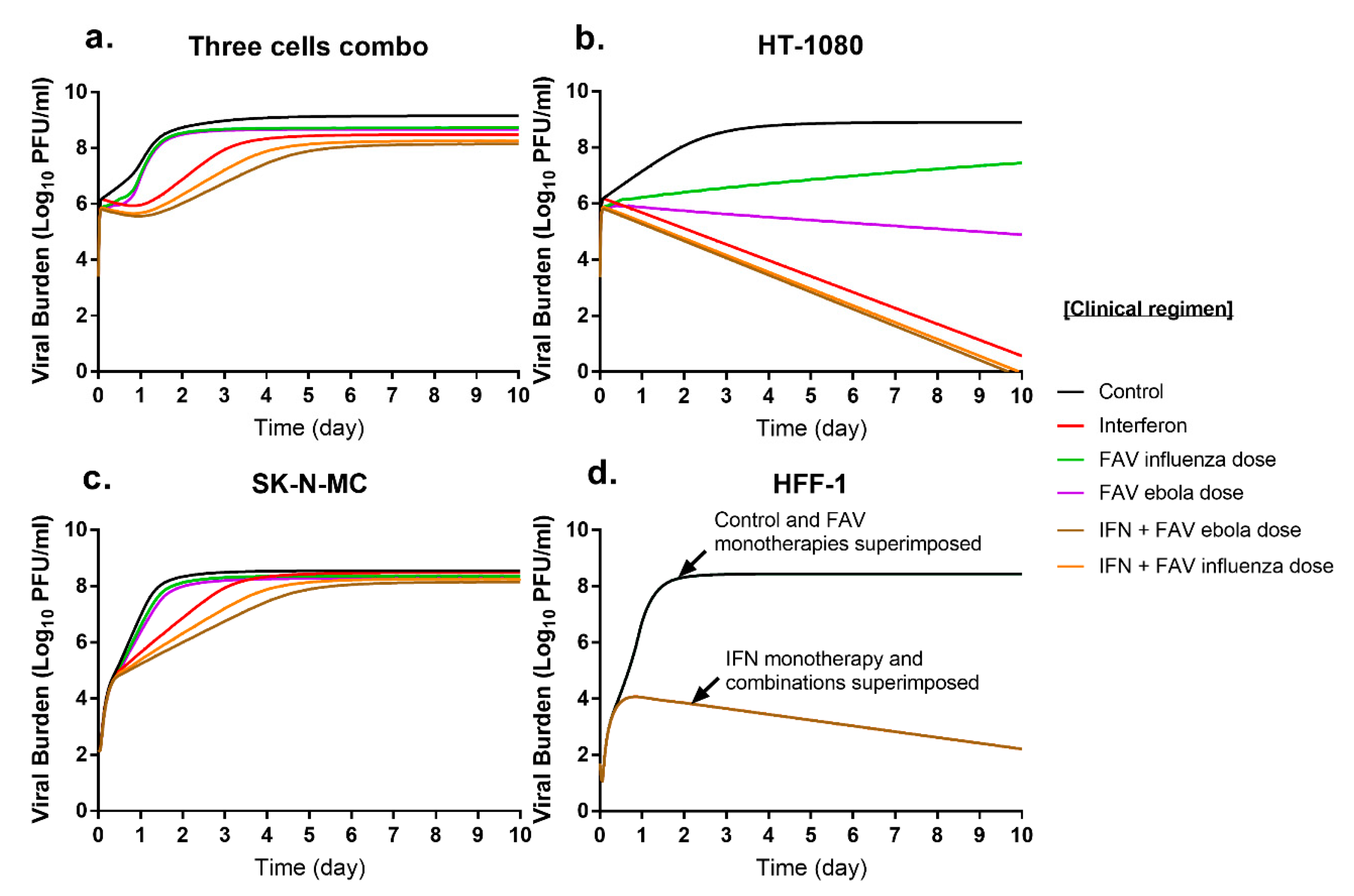
3.4. Simulated Antiviral Activity of Favipiravir (FAV) and Interferon-Alpha (IFN) Combination Regimens against Chikungunya Virus (CHIKV)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nasci, R.S. Movement of chikungunya virus into the Western hemisphere. Emerg. Infect. Dis. 2014, 20, 1394–1395. [Google Scholar] [CrossRef] [PubMed]
- Zeller, H.; Van Bortel, W.; Sudre, B. Chikungunya: Its History in Africa and Asia and Its Spread to New Regions in 2013–2014. J. Infect. Dis. 2016, 214, S436–S440. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.A.; Dermody, T.S. Chikungunya virus: Epidemiology, replication, disease mechanisms, and prospective intervention strategies. J. Clin. Investig. 2017, 127, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Pan American Health Organization. Geographic Spread of Chikungunya in the Americas December 2013–December 2017. Available online: http://ais.paho.org/phip/viz/ed_chikungunya_amro.asp (accessed on 23 September 2019).
- Centers for Disease Control and Prevention. Chikungunya Virus. Available online: https://www.cdc.gov/chikungunya/symptoms/index.html (accessed on 23 September 2019).
- Burt, F.J.; Chen, W.; Miner, J.J.; Lenschow, D.J.; Merits, A.; Schnettler, E.; Kohl, A.; Rudd, P.A.; Taylor, A.; Herrero, L.J.; et al. Chikungunya virus: An update on the biology and pathogenesis of this emerging pathogen. Lancet Infect. Dis. 2017, 17, e107–e117. [Google Scholar] [CrossRef]
- Schwartz, O.; Albert, M.L. Biology and pathogenesis of chikungunya virus. Nat. Rev. Microbiol. 2010, 8, 491–500. [Google Scholar] [CrossRef]
- Suhrbier, A.; Jaffar-Bandjee, M.C.; Gasque, P. Arthritogenic alphaviruses—An overview. Nat. Rev. Rheumatol. 2012, 8, 420–429. [Google Scholar] [CrossRef]
- Agarwal, A.; Vibha, D.; Srivastava, A.K.; Shukla, G.; Prasad, K. Guillain-Barre syndrome complicating chikungunya virus infection. J. Neurovirol. 2017, 23, 504–507. [Google Scholar] [CrossRef]
- McFadden, G.; Mohamed, M.R.; Rahman, M.M.; Bartee, E. Cytokine determinants of viral tropism. Nat. Rev. Immunol. 2009, 9, 645–655. [Google Scholar] [CrossRef]
- Matusali, G.; Colavita, F.; Bordi, L.; Lalle, E.; Ippolito, G.; Capobianchi, M.R.; Castilletti, C. Tropism of the Chikungunya Virus. Viruses 2019, 11, 175. [Google Scholar] [CrossRef]
- Roberts, G.C.; Zothner, C.; Remenyi, R.; Merits, A.; Stonehouse, N.J.; Harris, M. Evaluation of a range of mammalian and mosquito cell lines for use in Chikungunya virus research. Sci. Rep. 2017, 7, 14641. [Google Scholar] [CrossRef]
- Franco, E.J.; Rodriquez, J.L.; Pomeroy, J.J.; Hanrahan, K.C.; Brown, A.N. The effectiveness of antiviral agents with broad-spectrum activity against chikungunya virus varies between host cell lines. Antivir. Chem. Chemother. 2018, 26, 2040206618807580. [Google Scholar] [CrossRef] [PubMed]
- Gallegos, K.M.; Drusano, G.L.; DZ, D.A.; Brown, A.N. Chikungunya Virus: In Vitro Response to Combination Therapy with Ribavirin and Interferon Alfa 2a. J. Infect. Dis. 2016, 214, 1192–1197. [Google Scholar] [CrossRef] [PubMed]
- Vanderlinden, E.; Vrancken, B.; Van Houdt, J.; Rajwanshi, V.K.; Gillemot, S.; Andrei, G.; Lemey, P.; Naesens, L. Distinct Effects of T-705 (Favipiravir) and Ribavirin on Influenza Virus Replication and Viral RNA Synthesis. Antimicrob. Agents Chemother. 2016, 60, 6679–6691. [Google Scholar] [CrossRef] [PubMed]
- Sissoko, D.; Laouenan, C.; Folkesson, E.; M’Lebing, A.B.; Beavogui, A.H.; Baize, S.; Camara, A.M.; Maes, P.; Shepherd, S.; Danel, C.; et al. Experimental Treatment with Favipiravir for Ebola Virus Disease (the JIKI Trial): A Historically Controlled, Single-Arm Proof-of-Concept Trial in Guinea. PLoS Med. 2016, 13, e1001967. [Google Scholar] [CrossRef]
- Hong, Z.; Cameron, C.E. Pleiotropic mechanisms of ribavirin antiviral activities. Prog. Drug. Res. 2002, 59, 41–69. [Google Scholar] [CrossRef]
- Te, H.S.; Randall, G.; Jensen, D.M. Mechanism of action of ribavirin in the treatment of chronic hepatitis C. Gastroenterol. Hepatol. 2007, 3, 218–225. [Google Scholar]
- Furuta, Y.; Gowen, B.B.; Takahashi, K.; Shiraki, K.; Smee, D.F.; Barnard, D.L. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antivir. Res. 2013, 100, 446–454. [Google Scholar] [CrossRef]
- Furuta, Y.; Komeno, T.; Nakamura, T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 449–463. [Google Scholar] [CrossRef]
- Feld, J.J.; Hoofnagle, J.H. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 2005, 436, 967–972. [Google Scholar] [CrossRef]
- Samuel, C.E. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001, 14, 778–809. [Google Scholar] [CrossRef] [PubMed]
- Pires de Mello, C.P.; Tao, X.; Kim, T.H.; Bulitta, J.B.; Rodriquez, J.L.; Pomeroy, J.J.; Brown, A.N. Zika Virus Replication Is Substantially Inhibited by Novel Favipiravir and Interferon Alpha Combination Regimens. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.N.; Bulitta, J.B.; McSharry, J.J.; Weng, Q.; Adams, J.R.; Kulawy, R.; Drusano, G.L. Effect of half-life on the pharmacodynamic index of zanamivir against influenza virus delineated by a mathematical model. Antimicrob. Agents Chemother. 2011, 55, 1747–1753. [Google Scholar] [CrossRef]
- Beal, S.L. Ways to fit a PK model with some data below the quantification limit. J. Pharmacokinet. Pharmacodyn. 2001, 28, 481–504. [Google Scholar] [CrossRef] [PubMed]
- Bulitta, J.B.; Bingölbali, A.; Shin, B.S.; Landersdorfer, C.B. Development of a new pre-and post-processing tool (SADAPT-TRAN) for nonlinear mixed-effects modeling in S-ADAPT. AAPS J. 2011, 13, 201–211. [Google Scholar] [PubMed]
- Mdvi, L. Phase 3 Efficacy and Safety Study of Favipiravir for Treatment of Uncomplicated Influenza in Adults; ClinicalTrials. gov [Internet]; National Library of Medicine (US): Bethesda, MD, USA, 2015. [Google Scholar]
- Mentré, F.; Taburet, A.-M.; Guedj, J.; Anglaret, X.; Keïta, S.; de Lamballerie, X.; Malvy, D. Dose regimen of favipiravir for Ebola virus disease. Lancet Infect. Dis. 2015, 15, 150–151. [Google Scholar] [CrossRef]
- Bauer, R.J.; Guzy, S. Monte Carlo parametric expectation maximization (MC-PEM) method for analyzing population pharmacokinetic/pharmacodynamic data. In Advanced Methods of Pharmacokinetic and Pharmacodynamic Systems Analysis Volume 3; Springer: Berlin, Germany, 2004; pp. 135–163. [Google Scholar]
- Bulitta, J.B.; Landersdorfer, C.B. Performance and robustness of the Monte Carlo importance sampling algorithm using parallelized S-ADAPT for basic and complex mechanistic models. AAPS J. 2011, 13, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Krause, A.; Lowe, P. Visualization and communication of pharmacometric models with Berkeley Madonna. CPT Pharmacomet. Syst. Pharmacol. 2014, 3, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Murrell, P. R Graphics; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Gutterman, J.U.; Fine, S.; Quesada, J.; Horning, S.J.; Levine, J.F.; Alexanian, R.; Bernhardt, L.; Kramer, M.; Spiegel, H.; Colburn, W.; et al. Recombinant leukocyte A interferon: Pharmacokinetics, single-dose tolerance, and biologic effects in cancer patients. Ann. Intern. Med. 1982, 96, 549–556. [Google Scholar] [CrossRef]
- Nagata, T.; Lefor, A.K.; Hasegawa, M.; Ishii, M. Favipiravir: A new medication for the Ebola virus disease pandemic. Disaster Med. Public Health Prep. 2015, 9, 79–81. [Google Scholar] [CrossRef]
- Detje, C.N.; Meyer, T.; Schmidt, H.; Kreuz, D.; Rose, J.K.; Bechmann, I.; Prinz, M.; Kalinke, U. Local type I IFN receptor signaling protects against virus spread within the central nervous system. J. Immunol. 2009, 182, 2297–2304. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter Name | Symbol | Unit | HFF-1 | SK-N-MC | HT-1080 |
|---|---|---|---|---|---|
| Log10 of 2nd order infection rate constant for batch with fast growth | Log10 (kinfect) fast | - | −6.17 (2.4%) | −7.41 (2.0%) | −8.53 (2.6%) |
| Log10 of 2nd order infection rate constant for batch with slow growth | Log10 (kinfect) slow | - | −5.56 (1.5%) | −6.09 (13.7%) | −7.91 (1.3%) |
| Synthesis rate constant for intracellular virus (batch with fast growth) | ksyn fast | 1/h | 221 (9.0%) | 100 (10.7%) | 31.3 (20.3%) |
| Synthesis rate constant for intracellular virus (batch with slow growth) | ksyn slow | 1/h | 137 (11.6%) | 14.5 (13.5%) | 5.83 (14.3%) |
| Mean delay time until release of intracellular virus in absence of drug; equivalent to the mean survival time of infected host cells | TDelay = 5/ktr | h | 22.4 (4.0%) | 11.7 (9.5%) | 0.965 (55.7%) |
| Mean survival time for extracellular virus | MSTVirus = 1/kloss,virus | h | 14.4 (8.3%) | 21.1 (10.8%) | 18.7 (5.9%) |
| IFN concentration causing 50% of Imax | IC50,IFN | IU/mL | 0.0841 (21.5%) | 5.44 (36.9%) | 4.86 (20.3%) |
| FAV concentration causing 50% of Imax | IC50,FAV | ug/mL | n/a | 79.9 (11.3%) | 24.6 (16.3%) |
| RBV concentration causing 50% of Imax | IC50,RBV | ug/mL | 664 (5.8%) | 196 (8.7%) | 294 (14.4%) |
| Hill function for IFN | HillIFN | - | 0.725 (4.1%) | 0.558 (4.4%) | 0.902 (61.2%) |
| Hill function for FAV | HillFAV | - | n/a | 0.784 (18.9%) | 1.04 (12.8%) |
| Hill function for RBV | HillRBV | - | 1.53 (12.8%) | 0.917 (14.5%) | 1.08 (30.6%) |
| Maximum cytotoxicity rate constant by RBV | kmaxRBV | 1/h | 0.1 (fixed) | 0.1 (fixed) | 0.1 (fixed) |
| RBV concentration causing 50% of RBV related cytotoxicity | SC50,RBV | ug/mL | 150 (fixed) | 150 (fixed) | 150 (fixed) |
| Log10 of initial concentration of uninfected cells (cells/mL) | Log_U | - | 6.30 (fixed) | 6.30 (fixed) | 6.30 (fixed) |
| Log10 of initial concentration of infected cells (ICI1 in cells/mL) | Log_I | - | 3.22 (2.3%) | 3.37 (4.8%) | 4.73 (1.8%) |
| Initial condition for extracellular virus | ICVextra | PFU/mL | 50 (fixed) | 148 (9%) | 2,330 (9.5%) |
| SD of additive error for viral load on log10 scale | SDin | - | 0.307 (5.6%) | 0.318 (6.7%) | 0.420 (5.4%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco, E.J.; Tao, X.; Hanrahan, K.C.; Zhou, J.; Bulitta, J.B.; Brown, A.N. Combination Regimens of Favipiravir Plus Interferon Alpha Inhibit Chikungunya Virus Replication in Clinically Relevant Human Cell Lines. Microorganisms 2021, 9, 307. https://doi.org/10.3390/microorganisms9020307
Franco EJ, Tao X, Hanrahan KC, Zhou J, Bulitta JB, Brown AN. Combination Regimens of Favipiravir Plus Interferon Alpha Inhibit Chikungunya Virus Replication in Clinically Relevant Human Cell Lines. Microorganisms. 2021; 9(2):307. https://doi.org/10.3390/microorganisms9020307
Chicago/Turabian StyleFranco, Evelyn J., Xun Tao, Kaley C. Hanrahan, Jieqiang Zhou, Jürgen B. Bulitta, and Ashley N. Brown. 2021. "Combination Regimens of Favipiravir Plus Interferon Alpha Inhibit Chikungunya Virus Replication in Clinically Relevant Human Cell Lines" Microorganisms 9, no. 2: 307. https://doi.org/10.3390/microorganisms9020307
APA StyleFranco, E. J., Tao, X., Hanrahan, K. C., Zhou, J., Bulitta, J. B., & Brown, A. N. (2021). Combination Regimens of Favipiravir Plus Interferon Alpha Inhibit Chikungunya Virus Replication in Clinically Relevant Human Cell Lines. Microorganisms, 9(2), 307. https://doi.org/10.3390/microorganisms9020307