
Dynamics of a Dual SARS-CoV-2 Lineage Co-Infection on a Prolonged Viral Shedding COVID-19 Case: Insights into Clinical Severity and Disease Duration

 , , ,
, , ,  ,
,  , , , , , , and
, , , , , , and 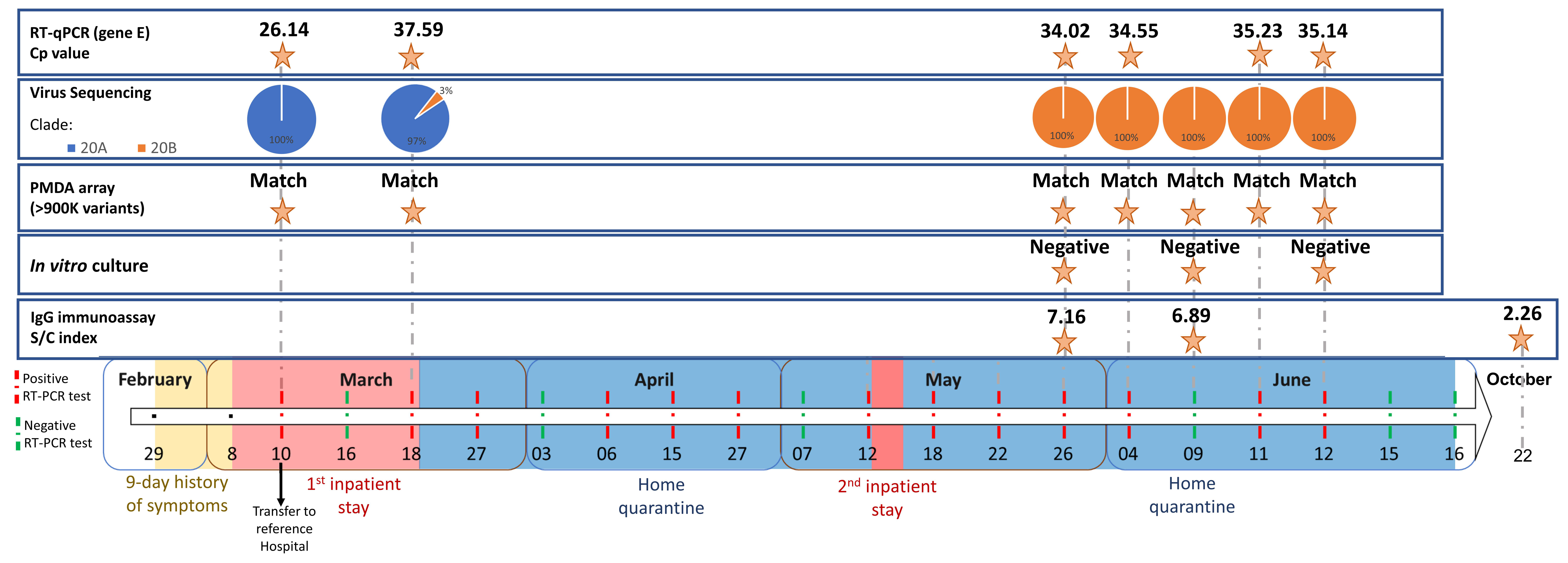{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. RT-PCR and Antibody Testing
2.2. Viral Whole Genome-Sequencing and Phylogenetic Analysis
2.3. Genome-Wide Array Screening and Calculation of the Polygenic Risk Score
2.4. In Vitro Culture of the Virus
3. Results
3.1. Clinical Features
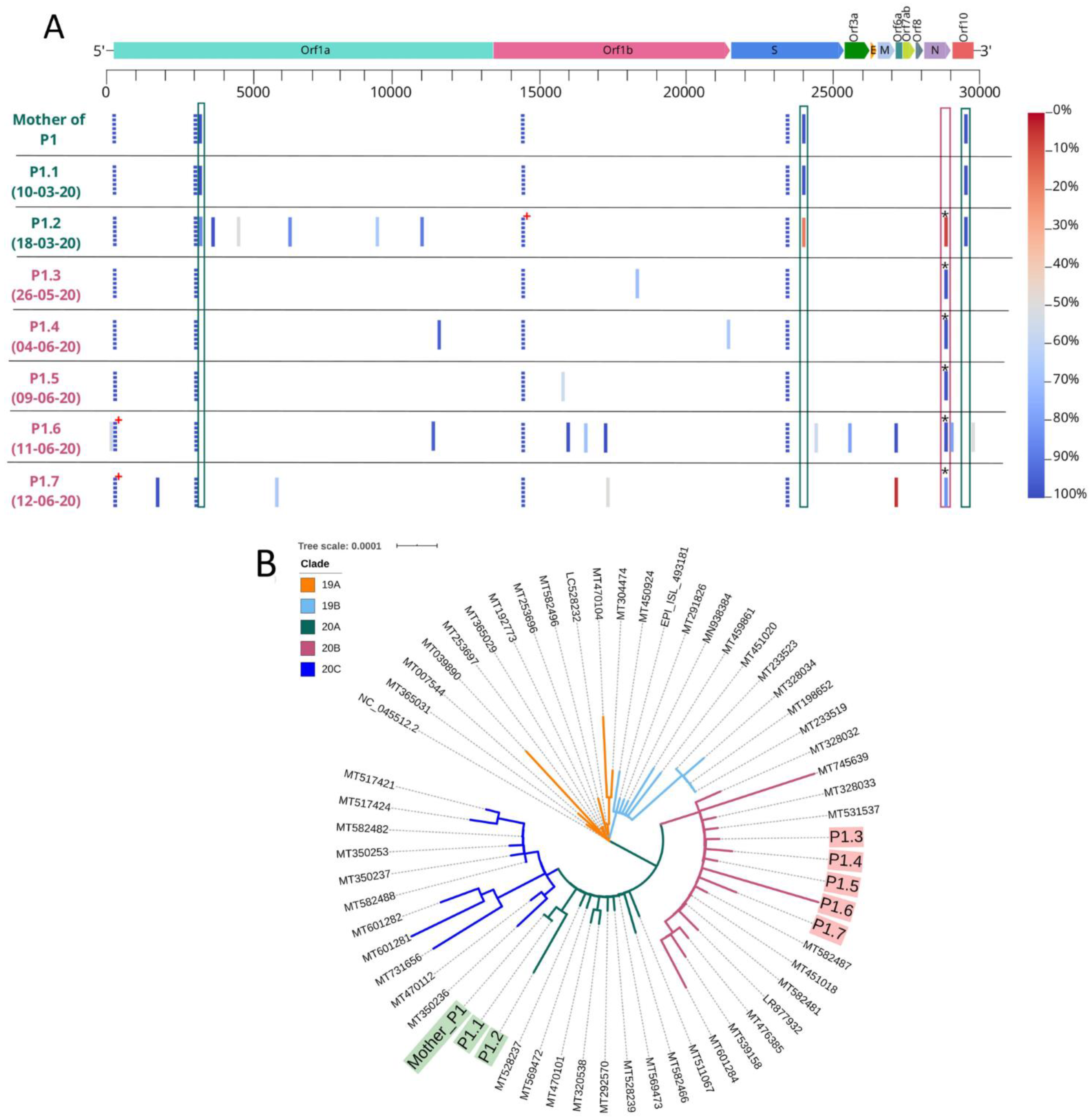
3.2. Viral Viability and Genome Analysis
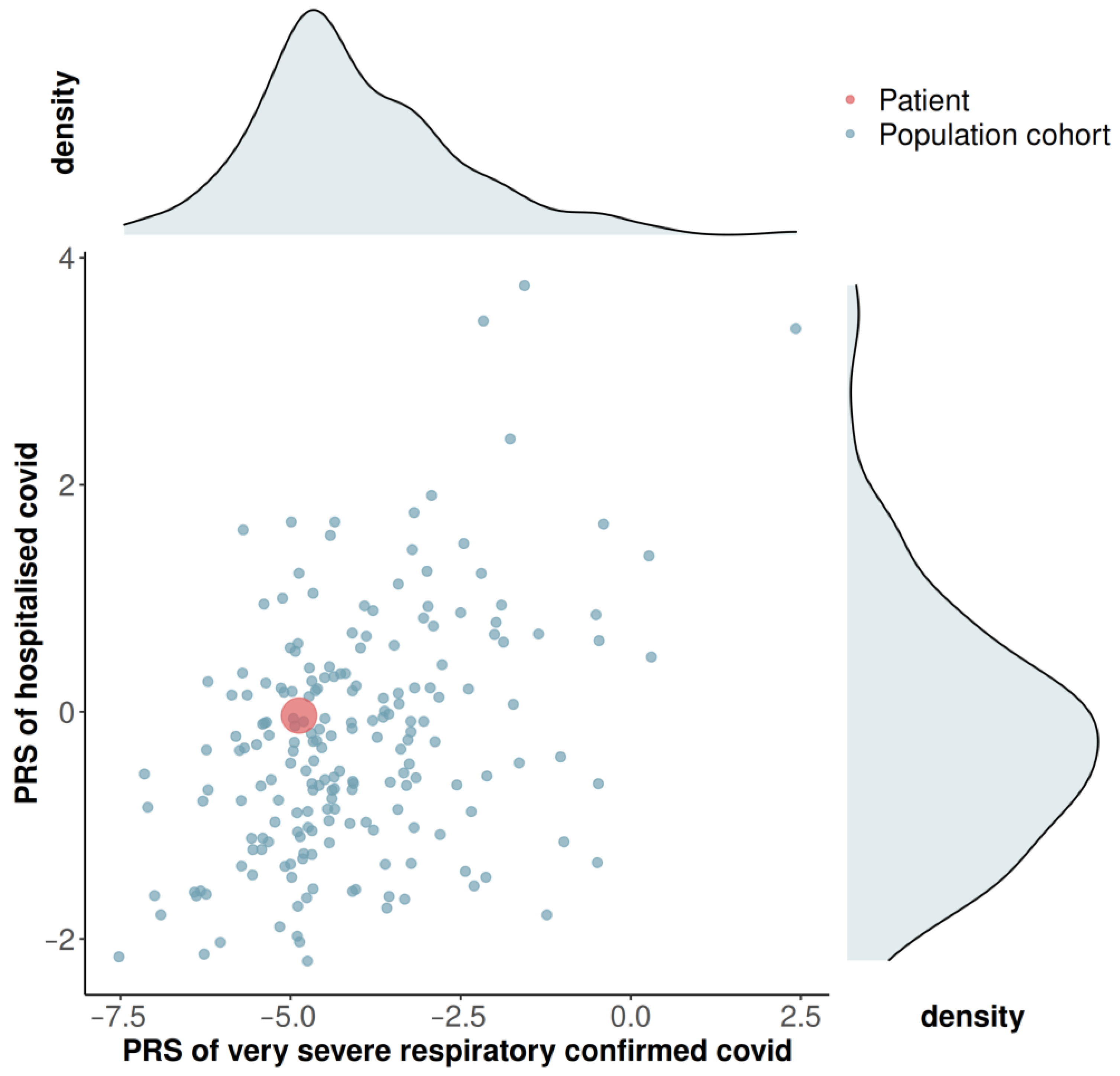
3.3. Genome-Wide Array Screening and Polygenic Risk Score
4. Discussions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, W.G. Impact of virus genetic variability and host immunity for the success of COVID-19 vaccines. Biomed. Pharmacother. 2021, 136, 111272. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Liu, H.Q.; Yang, Z.R.; Chen, Y.X.; Liu, Z.Y.; Zhang, K.; Wang, C.; Li, W.X.; An, Y.W.; Wang, J.C.; et al. Recurrence of positive SARS-CoV-2 viral RNA in recovered COVID-19 patients during medical isolation observation. Sci. Rep. 2020, 10, 11887. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, X.; Lv, T. Prolonged SARS-CoV-2 RNA shedding: Not a rare phenomenon. J. Med. Virol. 2020, 92, 2286–2287. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.Y.; Gandhi, R.T. Re-infection with SARS-CoV-2: What Goes Around May Come Back Around. Clin. Infect. Dis. 2020, ciaa1541. [Google Scholar] [CrossRef]
- Gidari, A.; Nofri, M.; Saccarelli, L.; Bastianelli, S.; Sabbatini, S.; Bozza, S.; Camilloni, B.; Fusco-Moffa, I.; Monari, C.; De Robertis, E.; et al. Is recurrence possible in coronavirus disease 2019 (COVID-19)? Case series and systematic review of literature. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 1–12. [Google Scholar] [CrossRef]
- Cevik, M.; Tate, M.; Lloyd, O.; Maraolo, A.E.; Schafers, J.; Ho, A. SARS-CoV-2, SARS-CoV, and MERS-CoV viral load dynamics, duration of viral shedding, and infectiousness: A systematic review and meta-analysis. Lancet Microbe 2021, 2, e13–e22. [Google Scholar] [CrossRef]
- Iwasaki, A. What reinfections mean for COVID-19. Lancet Infect. Dis. 2021, 21, 3–5. [Google Scholar] [CrossRef]
- To, K.K.; Hung, I.F.; Ip, J.D.; Chu, A.W.; Chan, W.M.; Tam, A.R.; Fong, C.H.; Yuan, S.; Tsoi, H.W.; Ng, A.C.; et al. COVID-19 re-infection by a phylogenetically distinct SARS-coronavirus-2 strain confirmed by whole genome sequencing. Clin. Infect. Dis. 2020, ciaa1275, Online ahead of print. [Google Scholar] [CrossRef]
- Van Elslande, J.; Vermeersch, P.; Vandervoort, K.; Wawina-Bokalanga, T.; Vanmechelen, B.; Wollants, E.; Laenen, L.; André, E.; Van Ranst, M.; Lagrou, K.; et al. Symptomatic SARS-CoV-2 reinfection by a phylogenetically distinct strain. Clin. Infect. Dis. 2020, ciaa1330, Online ahead of print. [Google Scholar] [CrossRef]
- Prado-Vivar, B.; Becerra-Wong, M.; Guadalupe, J.; Marquez, S.; Gutierrez, B.; Rojas-Silva, P.; Grunauer, M.; Trueba, G.; Barragan, V.; Cardenas, P. COVID-19 re-infection by a phylogenetically distinct SARS-CoV-2 variant, first confirmed event in South America. SSRN 2020. [Google Scholar] [CrossRef]
- Tillett, R.; Sevinsky, J.; Hartley, P.; Kerwin, H.; Crawford, N.; Gorzalski, A.; Laverdure, C.; Verma, S.; Rossetto, C.; Jackson, D.; et al. Genomic evidence for reinfection with SARS-CoV-2: A case study. Lancet Infect. Dis. 2021, 21, 52–58. [Google Scholar] [CrossRef]
- Arvin, A.M.; Fink, K.; Schmid, M.A.; Cathcart, A.; Spreafico, R.; Havenar-Daughton, C.; Lanzavecchia, A.; Corti, D.; Virgin, H.W. A perspective on potential antibody-dependent enhancement of SARS-CoV-2. Nature 2020, 584, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Bhoyar, R.C.; Jain, A.; Srivastava, S.; Upadhayay, R.; Imran, M.; Jolly, B.; Divakar, M.K.; Sharma, D.; Sehgal, P.; et al. Asymptomatic reinfection in two healthcare workers from India with genetically distinct SARS-CoV-2. Clin. Infect. Dis. 2020, ciaa1451, Online ahead of print. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B.; Hunt, V.; Liu, M.; Kumar, S.; Zaremba, S.; Gu, Z.; et al. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012, 40, D593–D598. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Staples, J.; Witherspoon, D.J.; Jorde, L.B.; Nickerson, D.A.; Below, J.E.; Huff, C.D. PADRE: Pedigree-Aware Distant-Relationship Estimation. Am. J. Hum. Genet. 2016, 99, 154–162. [Google Scholar] [CrossRef]
- Initiative, C.-H.G. The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur. J. Hum. Genet. 2020, 28, 715–718. [Google Scholar] [CrossRef] [PubMed]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Borges, V.; Isidro, J.; Cortes-Martins, H.; Duarte, S.; Vieira, L.; Leite, R.; Gordo, I.; Caetano, C.P.; Nunes, B.; Sá, R.; et al. Massive dissemination of a SARS-CoV-2 Spike Y839 variant in Portugal. Emerg. Microbes Infect. 2020, 9, 2488–2496. [Google Scholar] [CrossRef]
- Rito, T.; Richards, M.B.; Pala, M.; Correia-Neves, M.; Soares, P.A. Phylogeography of 27,000 SARS-CoV-2 Genomes: Europe as the Major Source of the COVID-19 Pandemic. Microorganisms 2020, 8, 1678. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Carballa, A.; Bello, X.; Pardo-Seco, J.; Martinón-Torres, F.; Salas, A. Mapping genome variation of SARS-CoV-2 worldwide highlights the impact of COVID-19 super-spreaders. Genome Res. 2020, 30, 1434–1448. [Google Scholar] [CrossRef]
- Karamitros, T.; Papadopoulou, G.; Bousali, M.; Mexias, A.; Tsiodras, S.; Mentis, A. SARS-CoV-2 exhibits intra-host genomic plasticity and low-frequency polymorphic quasispecies. J. Clin. Virol. 2020, 131, 104585. [Google Scholar] [CrossRef]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Li, W.; Su, Y.Y.; Zhi, S.S.; Huang, J.; Zhuang, C.L.; Bai, W.Z.; Wan, Y.; Meng, X.R.; Zhang, L.; Zhou, Y.B.; et al. Virus shedding dynamics in asymptomatic and mildly symptomatic patients infected with SARS-CoV-2. Clin. Microbiol. Infect. 2020, 26, 1556.e1–1556.e6. [Google Scholar] [CrossRef]
- Wölfel, R.; Corman, V.M.; Guggemos, W.; Seilmaier, M.; Zange, S.; Müller, M.A.; Niemeyer, D.; Jones, T.C.; Vollmar, P.; Rothe, C.; et al. Virological assessment of hospitalized patients with COVID-2019. Nature 2020, 581, 465–469. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pedro, N.; Silva, C.N.; Magalhães, A.C.; Cavadas, B.; Rocha, A.M.; Moreira, A.C.; Gomes, M.S.; Silva, D.; Sobrinho-Simões, J.; Ramos, A.; et al. Dynamics of a Dual SARS-CoV-2 Lineage Co-Infection on a Prolonged Viral Shedding COVID-19 Case: Insights into Clinical Severity and Disease Duration. Microorganisms 2021, 9, 300. https://doi.org/10.3390/microorganisms9020300
Pedro N, Silva CN, Magalhães AC, Cavadas B, Rocha AM, Moreira AC, Gomes MS, Silva D, Sobrinho-Simões J, Ramos A, et al. Dynamics of a Dual SARS-CoV-2 Lineage Co-Infection on a Prolonged Viral Shedding COVID-19 Case: Insights into Clinical Severity and Disease Duration. Microorganisms. 2021; 9(2):300. https://doi.org/10.3390/microorganisms9020300
Chicago/Turabian StylePedro, Nicole, Cláudio N. Silva, Ana C. Magalhães, Bruno Cavadas, Ana M. Rocha, Ana C. Moreira, Maria Salomé Gomes, Diogo Silva, Joana Sobrinho-Simões, Angélica Ramos, and et al. 2021. "Dynamics of a Dual SARS-CoV-2 Lineage Co-Infection on a Prolonged Viral Shedding COVID-19 Case: Insights into Clinical Severity and Disease Duration" Microorganisms 9, no. 2: 300. https://doi.org/10.3390/microorganisms9020300
APA StylePedro, N., Silva, C. N., Magalhães, A. C., Cavadas, B., Rocha, A. M., Moreira, A. C., Gomes, M. S., Silva, D., Sobrinho-Simões, J., Ramos, A., Cardoso, M. J., Filipe, R., Palma, P., Ceia, F., Silva, S., Guimarães, J. T., Sarmento, A., Fernandes, V., Pereira, L., & Tavares, M. (2021). Dynamics of a Dual SARS-CoV-2 Lineage Co-Infection on a Prolonged Viral Shedding COVID-19 Case: Insights into Clinical Severity and Disease Duration. Microorganisms, 9(2), 300. https://doi.org/10.3390/microorganisms9020300