
A Transcriptomic Atlas of the Ectomycorrhizal Fungus Laccaria bicolor

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Fungal Isolate and Maintenance of Cultures
2.2. Growth in Different Nutritional Conditions
2.3. Plant Material and Fungal–Plant Co-Cultures
2.4. Basidiocarp Production and Sampling
2.5. P Determinations
2.6. Microscopy
2.7. RNA Extractions and RNAseq
3. Results
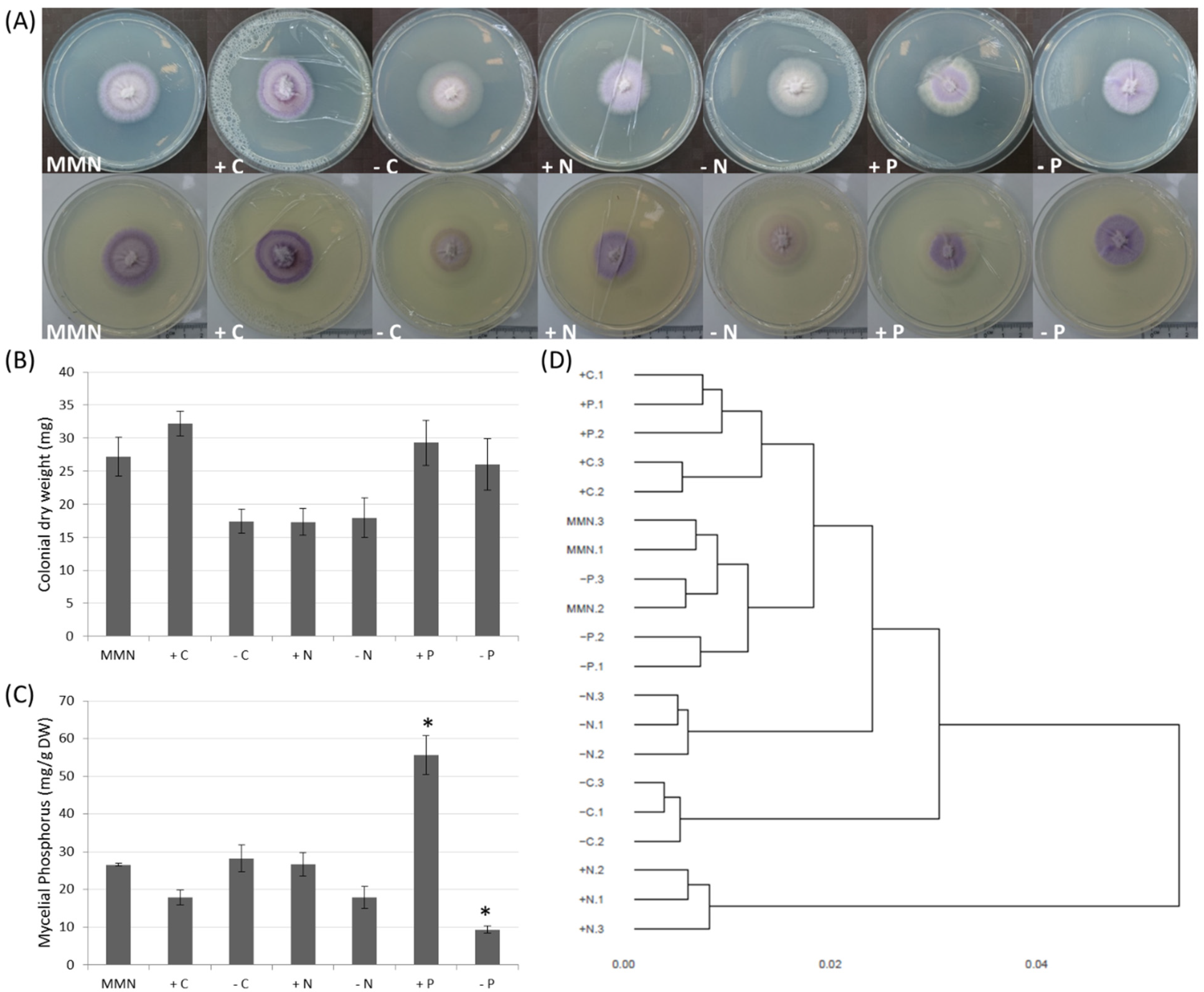
3.1. Effect of Nutrition on Growth and Mycelial P Accumulation
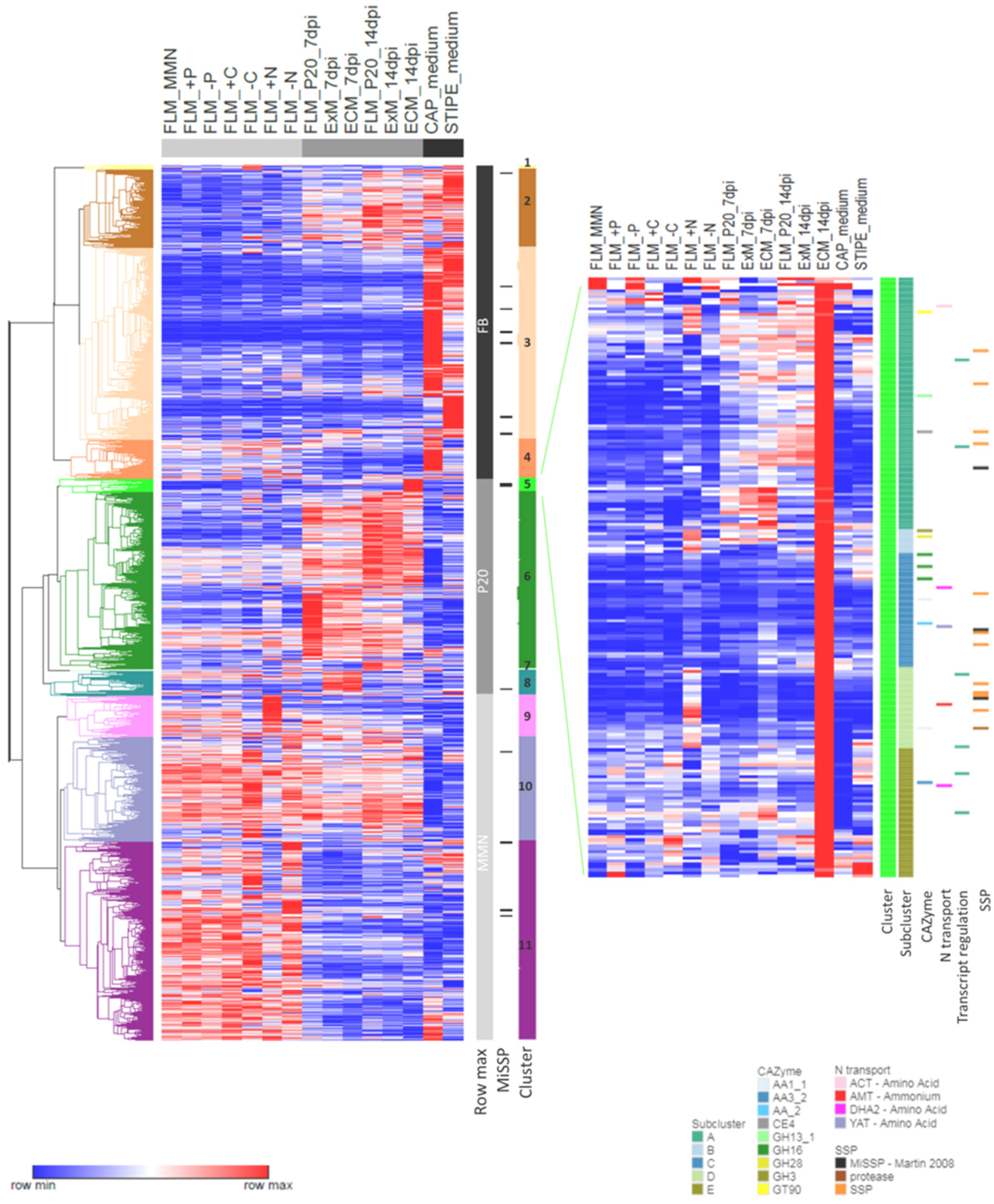
3.2. Nutrition-Regulated Transcriptome
3.3. Time Course of Ectomycorrhizal Development
3.4. Symbiosis-Regulated Transcriptome
3.5. Transcriptomics of the Developing Basidiocarp
3.6. Similarities and Dissimilarities between Transcriptomes of Different Morphological and Functional L. bicolor Structures
4. Discussion
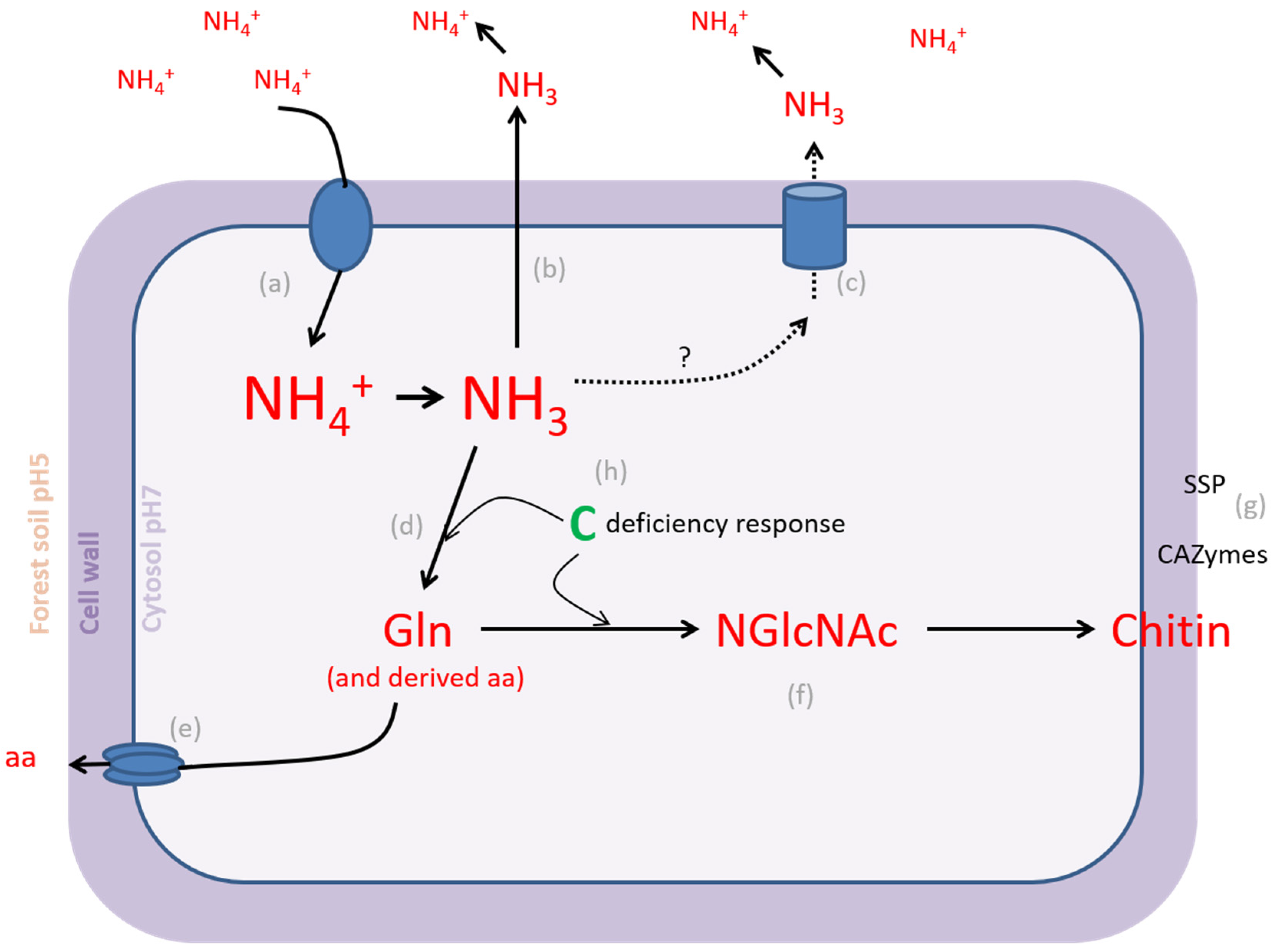
4.1. L. bicolor PO4− Homeostatic Range Is Larger Than NH4+ Homeostatic Range
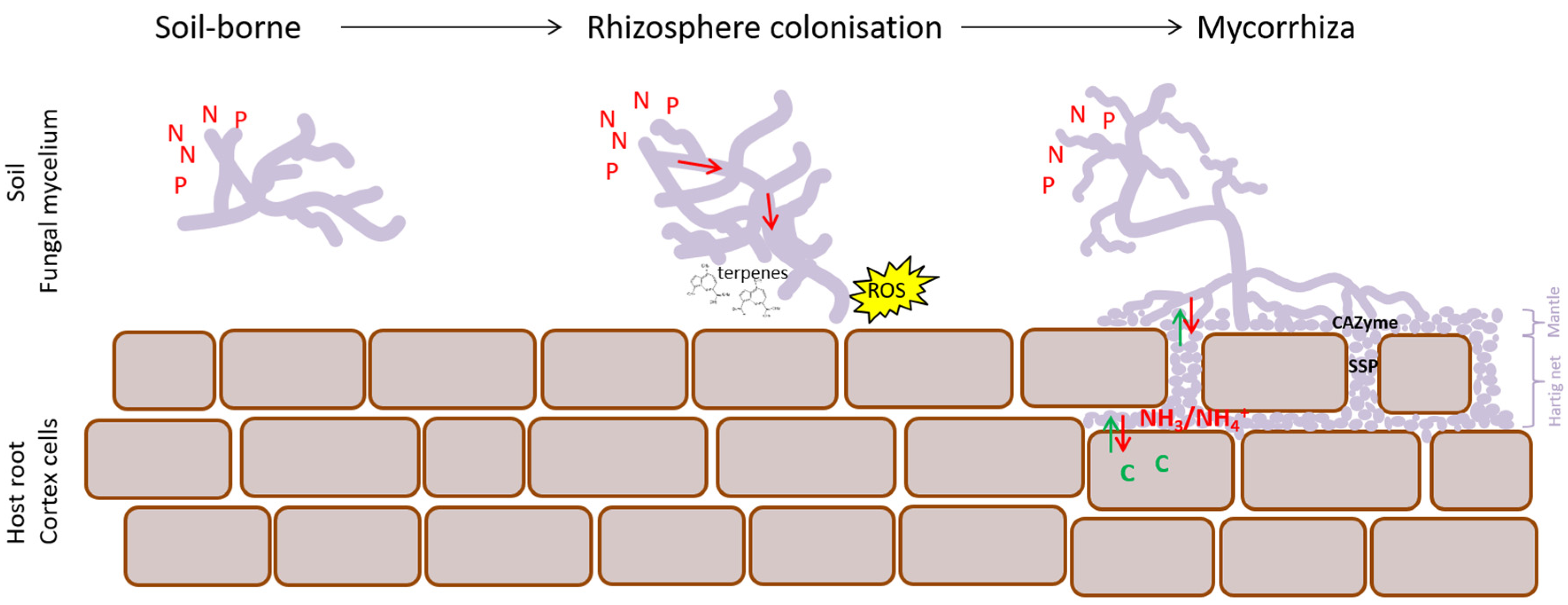
4.2. EcM Development Is a Two-Step Process
4.3. L. bicolor Mushroom Development Shares Transcriptomic Signatures with Other Agaricomyctes
4.4. Nutrients Are Key in Functional Differentiation of EcM Fungi
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Felipe-Lucia, M.R.; Soliveres, S.; Penone, C.; Manning, P.; van der Plas, F.; Boch, S.; Prati, D.; Ammer, C.; Schall, P.; Gossner, M.M.; et al. Multiple forest attributes underpin the supply of multiple ecosystem services. Nat. Commun. 2018, 9, 4839. [Google Scholar] [CrossRef]
- Baldrian, P. Microbial activity and the dynamics of ecosystem processes in forest soils. Curr. Opin. Microbiol. 2017, 37, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Policelli, N.; Bruns, T.D.; Vilgalys, R.; Nuñez, M.A. Suilloid fungi as global drivers of pine invasions. New Phytol. 2019, 222, 714–725. [Google Scholar] [CrossRef]
- Tedersoo, L.; May, T.W.; Smith, M.E. Ectomycorrhizal lifestyle in fungi: Global diversity, distribution, and evolution of phylogenetic lineages. Mycorrhiza 2010, 20, 217–263. [Google Scholar] [CrossRef]
- Tedersoo, L.; Bahram, M.; Zobel, M. How mycorrhizal associations drive plant population and community biology. Science 2020, 367, 6480. [Google Scholar] [CrossRef]
- Kohler, A.; Kuo, A.; Nagy, L.G.; Morin, E.; Barry, K.W.; Buscot, F.; Canbäck, B.; Choi, C.; Cichocki, N.; Clum, A.; et al. Convergent losses of decay mechanisms and rapid turnover of symbiosis genes in mycorrhizal mutualists. Nat. Genet. 2015, 47, 410–415. [Google Scholar] [CrossRef]
- Martin, F.; Kohler, A.; Murat, C.; Veneault-Fourrey, C.; Hibbett, D.S. Unearthing the roots of ectomycorrhizal symbioses. Nat. Rev. Microbiol. 2016, 14, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Genre, A.; Lanfranco, L.; Perotto, S.; Bonfante, P. Unique and common traits in mycorrhizal symbioses. Nat. Rev. Microbiol. 2020, 18, 649–660. [Google Scholar] [CrossRef]
- Miyauchi, S.; Kiss, E.; Kuo, A.; Drula, E.; Kohler, A.; Sánchez-García, M.; Morin, E.; Andreopoulos, B.; Barry, K.W.; Bonito, G.; et al. Large-scale genome sequencing of mycorrhizal fungi provides insights into the early evolution of symbiotic traits. Nat. Commun. 2020, 11, 5125. [Google Scholar] [CrossRef]
- Martin, F.; Aerts, A.; Ahrén, D.; Brun, A.; Danchin, E.; Duchaussoy, F.; Gibon, J.; Kohler, A.; Lindquist, E.; Pereda, V.; et al. The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis. Nature 2008, 452, 88–92. [Google Scholar] [CrossRef]
- Peter, M.; Kohler, A.; Ohm, R.A.; Kuo, A.; Kruetzmann, J.; Morin, E.; Arend, M.; Barry, K.; Binder, M.; Choi, C.; et al. Ectomycorrhizal ecology is imprinted in the genome of the dominant symbiotic fungus Cenococcum geophilum. Nat. Commun. 2016, 7, 12662. [Google Scholar] [CrossRef]
- Murat, C.; Payen, T.; Noel, B.; Kuo, A.; Morin, E.; Chen, J.; Kohler, A.; Krizsán, K.; Balestrini, R.; Da Silva, C.; et al. Pezizomycetes genomes reveal the molecular basis of ectomycorrhizal truffle lifestyle. Nat. Ecol. Evol. 2018, 2, 1956–1965. [Google Scholar] [CrossRef] [PubMed]
- Plett, J.M.; Daguerre, Y.; Wittulsky, S.; Vayssières, A.; Deveau, A.; Melton, S.J.; Kohler, A.; Morrell-Falvey, J.L.; Brun, A.; Veneault-Fourrey, C.; et al. Effector MiSSP7 of the mutualistic fungus Laccaria bicolor stabilizes the Populus JAZ6 protein and represses jasmonic acid (JA) responsive genes. Proc. Natl. Acad. Sci. USA 2014, 111, 8299–8304. [Google Scholar] [CrossRef]
- Plett, J.M.; Plett, K.L.; Wong-Bajracharya, J.; de Freitas Pereira, M.; Costa, M.D.; Kohler, A.; Martin, F.; Anderson, I.C. Mycorrhizal effector PaMiSSP10b alters polyamine biosynthesis in Eucalyptus root cells and promotes root colonization. New Phytol. 2020, 228, 718–727. [Google Scholar] [CrossRef]
- Kang, H.; Chen, X.; Kemppainen, M.; Pardo, A.G.; Veneault-Fourrey, C.; Kohler, A.; Martin, F.M. The small secreted effector protein MiSSP7.6 of Laccaria bicolor is required for the establishment of ectomycorrhizal symbiosis. Environ. Microbiol. 2020, 22, 1435–1446. [Google Scholar] [CrossRef]
- Pellegrin, C.; Daguerre, Y.; Ruytinx, J.; Guinet, F.; Kemppainen, M.; Frey, N.F.D.; Puech-Pagès, V.; Hecker, A.; Pardo, A.G.; Martin, F.M.; et al. Laccaria bicolor MiSSP8 is a small-secreted protein decisive for the establishment of the ectomycorrhizal symbiosis. Environ. Microbiol. 2019, 21, 3765–3779. [Google Scholar] [CrossRef]
- Veneault-Fourrey, C.; Commun, C.; Kohler, A.; Morin, E.; Balestrini, R.; Plett, J.; Danchin, E.; Coutinho, P.; Wiebenga, A.; de Vries, R.P.; et al. Genomic and transcriptomic analysis of Laccaria bicolor CAZome reveals insights into polysaccharides remodelling during symbiosis establishment. Fungal Genet. Biol. 2014, 72, 168–181. [Google Scholar] [CrossRef]
- Zhang, F.; Anasontzis, G.E.; Labourel, A.; Champion, C.; Haon, M.; Kemppainen, M.; Commun, C.; Deveau, A.; Pardo, A.; Veneault-Fourrey, C.; et al. The ectomycorrhizal basidiomycete Laccaria bicolor releases a secreted β-1, 4 endoglucanase that plays a key role in symbiosis development. New Phytol. 2018, 220, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- De Beeck, M.O.; Troein, C.; Siregar, S.; Gentile, L.; Abbondanza, G.; Peterson, C.; Persson, P.; Tunlid, A. Regulation of fungal decomposition at single-cell level. ISME J. 2020, 14, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Labourel, A.; Frandsen, K.E.; Zhang, F.; Brouilly, N.; Grisel, S.; Haon, M.; Ciano, L.; Ropartz, D.; Fanuel, M.; Martin, F.; et al. A fungal family of lytic polysaccharide monooxygenase-like copper proteins. Nat. Chem. Biol. 2020, 16, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Deveau, A.; Palin, B.; Delaruelle, C.; Peter, M.; Kohler, A.; Pierrat, J.C.; Sarniguet, A.; Garbaye, J.; Martin, F.; Frey-Klett, P. The mycorrhiza helper Pseudomonas fluorescens BBc6R8 has a specific priming effect on the growth, morphology and gene expression of the ectomycorrhizal fungus Laccaria bicolor S238N. New Phytol. 2007, 175, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Kottke, I.; Guttenberger, M.; Hampp, R.; Oberwinkler, F. An in vitro method for establishing mycorrhizae on coniferous tree seedling. Trees—Struct. Funct. 1987, 1, 191–194. [Google Scholar] [CrossRef]
- Felten, J.; Kohler, A.; Morin, E.; Bhalerao, R.P.; Palme, K.; Martin, F.; Ditengou, F.A.; Legué, V. The ectomycorrhizal fungus Laccaria bicolor stimulates lateral root formation in poplar and Arabidopsis through auxin transport and signaling. Plant Physiol. 2009, 151, 1991–2005. [Google Scholar] [CrossRef] [PubMed]
- Le Tacon, F.; Jung, G.; Mugnier, J.; Michelot, P.; Mauperin, C. Efficiency in a forest nursery of an ectomycorrhizal fungus inoculum produced in a fermentor and entrapped in polymeric gels. Can. J. Bot. 1985, 63, 1664–1668. [Google Scholar] [CrossRef]
- Chang, S.; Puryear, J.; Cairney, J. A simple and efficient method for isolating RNA from pine trees. Plant Mol. Biol. Rep. 1993, 11, 113–116. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dis- persion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Miyauchi, S.; Navarro, D.; Grigoriev, I.; Lipzen, A.; Riley, R.; Chevret, D.; Grisel, S.; Berrin, J.-G.; Henrissat, B.; Rosso, M.-N. Visual comparative omics of fungi for plant biomass deconstruction. Front. Microbiol. 2016, 7, 1335. [Google Scholar] [CrossRef]
- Miyauchi, S.; Navarro, D.; Grisel, S.; Chevret, D.; Berrin, J.G.; Rosso, M.N. The integrative omics of white-rot fungus Pycnoporus coccineus reveals co-regulated CAZymes for orchestrated lignocellulose breakdown. PLoS ONE 2017, 12, e0175528. [Google Scholar] [CrossRef]
- Miyauchi, S.; Rancon, A.; Drula, E.; Hage, H.; Chaduli, D.; Favel, A.; Grisel, S.; Henrissat, B.; Herpoël-Gimbert, I.; Ruiz-Dueñas, F.J.; et al. Integrative visual omics of the white-rot fungus Polyporus brumalis exposes the biotechnological potential of its oxidative enzymes for delignifying raw plant biomass. Biotechnol. Biofuels 2018, 11, 201. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology. R Package Version 2.44.0. Available online: https://bioconductor.org/packages/release/bioc/html/topGO.html (accessed on 21 October 2021).
- Dietz, S.; von Bülow, J.; Beitz, E.; Nehls, U. The aquaporin gene family of the ectomycorrhizal fungus Laccaria bicolor: Lessons for symbiotic functions. New Phytol. 2011, 190, 927–940. [Google Scholar] [CrossRef]
- Xu, H.; Navarro-Ro’denas, A.; Cooke, J.E.; Zwiazek, J.J. Transcript profiling of aquaporins during basidiocarp development in Laccaria bicolor ectomycorrhizal with Picea glauca. Mycorrhiza 2016, 26, 19–31. [Google Scholar] [CrossRef]
- Ezawa, T.; Saito, K. How do arbuscular mycorrhizal fungi handle phosphate? New insight into fine-tuning of phosphate metabolism. New Phytol. 2018, 220, 1116–1121. [Google Scholar] [CrossRef]
- Nehls, U.; Plassard, C. Nitrogen and phosphate metabolism in ectomycorrhizas. New Phytol. 2018, 220, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Plassard, C.; Becquer, A.; Garcia, K. Phosphorus transport in mycorrhiza: How far are we? Trends Plant Sci. 2019, 24, 794–801. [Google Scholar] [CrossRef]
- Ashford, A.E.; Ryde, S.; Barrow, K.D. Demonstration of a short chain polyphosphate in Pisolithus tinctorius and the implications for phosphorus transport. New Phytol. 1994, 126, 239–247. [Google Scholar] [CrossRef]
- Bücking, H.; Heyser, W. Elemental composition and function of polyphosphates in ectomycorrhizal fungi–an X-ray microanalytical study. Mycol. Res. 1999, 103, 31–39. [Google Scholar] [CrossRef]
- Casieri, L.; Lahmidi, N.A.; Doidy, J.; Veneault-Fourrey, C.; Migeon, A.; Bonneau, L.; Courty, P.-E.; Garcia, K.; Charbonnier, M.; Delteil, A.; et al. Biotrophic transportome in mutualistic plant–fungal interactions. Mycorrhiza 2013, 23, 597–625. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Marchal, J.P.; Timinska, A.; Canet, D. The metabolism and physical state of polyphosphates in ectomycorrhizal fungi. A31p nuclear magnetic resonance study. New Phytol. 1985, 101, 275–290. [Google Scholar] [CrossRef]
- Tatry, M.V.; El Kassis, E.; Lambilliotte, R.; Corratgé, C.; Van Aarle, I.; Amenc, L.K.; Alary, R.; Zimmermann, S.; Sentenac, H.; Plassard, C. Two differentially regulated phosphate transporters from the symbiotic fungus Hebeloma cylindrosporum and phosphorus acquisition by ectomycorrhizal Pinus pinaster. Plant J. 2009, 57, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Torres-Aquino, M.; Becquer, A.; Le Guernevé, C.; Louche, J.; Amenc, L.K.; Staunton, S.; Quiquampoix, H.; Plassard, C. The host plant Pinus pinaster exerts specific effects on phosphate efflux and polyphosphate metabolism of the ectomycorrhizal fungus Hebeloma cylindrosporum: A radiotracer, cytological staining and 31P NMR spectroscopy study. Plant Cell Environ. 2017, 40, 190–202. [Google Scholar] [CrossRef]
- Secco, D.; Wang, C.; Arpat, B.; Wang, Z.; Poirier, Y.; Tyerman, S.; Wu, P.; Shou, H.; Whelan, J. The emerging importance of the SPX domain-containing proteins in phosphate homeostasis. New Phytol. 2012, 193, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Colpaert, J.V.; Van Tichelen, K.K.; Van Assche, J.A.; Van Laere, A. Short-term phosphorus uptake rates in mycorrhizal and non-mycorrhizal roots of intact Pinus sylvestris seedlings. New Phytol. 1999, 143, 589–597. [Google Scholar] [CrossRef]
- Garcia, K.; Haider, M.Z.; Delteil, A.; Corratgé-Faillie, C.; Conéjero, G.; Tatry, M.V.; Becquer, A.; Amenc, L.; Sentenac, H.; Plassard, C.; et al. Promoter-dependent expression of the fungal transporter HcPT1. 1 under Pi shortage and its spatial localization in ectomycorrhiza. Fungal Genet. Biol. 2013, 58, 53–61. [Google Scholar] [CrossRef]
- Brun, A.; Chalot, M.; Botton, B.; Martin, F. Purification and characterization of glutamine synthetase and NADP-glutamate dehydrogenase from the ectomycorrhizal fungus Laccaria laccata. Plant Physiol. 1992, 99, 938–944. [Google Scholar] [CrossRef]
- Montanini, B.; Viscomi, A.R.; Bolchi, A.; Martin, Y.; Siverio, J.M.; Balestrini, R.; Bonfante, P.; Ottonello, S. Functional properties and differential mode of regulation of the nitrate transporter from a plant symbiotic ascomycete. Biochem. J. 2006, 394, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, B.D.; Tunlid, A. Ectomycorrhizal fungi–potential organic matter decomposers, yet not saprotrophs. New Phytol. 2015, 205, 1443–1447. [Google Scholar] [CrossRef]
- Martinell, K.; Westlund, A.; Häggström, L. Ammonium ion transport—A cause of cell death. Cytotechnology 1996, 22, 251–254. [Google Scholar] [CrossRef]
- Rutherford, J.C.; Lin, X.; Nielsen, K.; Heitman, J. Amt2 permease is required to induce ammonium-responsive invasive growth and mating in Cryptococcus neoformans. Eukaryot. Cell 2008, 7, 237–246. [Google Scholar] [CrossRef]
- Hess, D.C.; Lu, W.; Rabinowitz, J.D.; Botstein, D. Ammonium toxicity and potassium limitation in yeast. PLoS Biol. 2006, 4, e351. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Carleton, T.J.; Malloch, D.W.; Hellebust, J.A. Nitrogen metabolism in the ectomycorrhizal fungus Laccaria bicolor (R. Mre.) Orton. New Phytol. 1990, 116, 431–441. [Google Scholar] [CrossRef]
- Javelle, A.; Rodríguez-Pastrana, B.-R.; Jacob, C.; Botton, B.; Brun, A.; André, B.; Marini, A.-M.; Chalot, M. Molecular characterization of two ammonium transporters from the ectomycorrhizal fungus Hebeloma cylindrosporum. FEBS Lett. 2001, 505, 393–398. [Google Scholar] [CrossRef]
- Lucic, E.; Fourrey, C.; Kohler, A.; Martin, F.; Chalot, M.; Brun-Jacob, A. A gene repertoire for nitrogen transporters in Laccaria bicolor. New Phytol. 2008, 180, 343–364. [Google Scholar] [CrossRef]
- Adlimoghaddam, A.; Boeckstaens, M.; Marini, A.M.; Treberg, J.R.; Brassinga, A.K.C.; Weihrauch, D. Ammonia excretion in Caenorhabditis elegans: Mechanism and evidence of ammonia transport of the Rhesus protein CeRhr-1. J. Exp. Biol. 2015, 218, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Boo, M.V.; Hiong, K.C.; Goh, E.J.; Choo, C.Y.; Wong, W.P.; Chew, S.F.; Ip, Y.K. The ctenidium of the giant clam, Tridacna squamosa, expresses an ammonium transporter 1 that displays light-suppressed gene and protein expression and may be involved in ammonia excretion. J. Comp. Physiol. B 2018, 188, 765–777. [Google Scholar] [CrossRef]
- Guaragnella, N.; Butow, R.A. ATO3 encoding a putative outward ammonium transporter is an RTG-independent retrograde responsive gene regulated by GCN4 and the Ssy1-Ptr3-Ssy5 amino acid sensor system. J. Biol. Chem. 2003, 278, 45882–45887. [Google Scholar] [CrossRef]
- Maillard, F.; Didion, M.; Fauchery, L.; Bach, C.; Buée, M. N-Acetylglucosaminidase activity, a functional trait of chitin degradation, is regulated differentially within two orders of ectomycorrhizal fungi: Boletales and Agaricales. Mycorrhiza 2018, 28, 391–397. [Google Scholar] [CrossRef]
- Gow, N.A.; Latge, J.P.; Munro, C.A. The fungal cell wall: Structure, biosynthesis, and function. Microbiol. Spectr. 2017, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Paul, J.A.; Wallen, R.M.; Zhao, C.; Shi, T.; Perlin, M.H. Coordinate regulation of Ustilago maydis ammonium transporters and genes involved in mating and pathogenicity. Fungal Biol. 2018, 122, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Doré, J.; Kohler, A.; Dubost, A.; Hundley, H.; Singan, V.; Peng, Y.; Kuo, A.; Grigoriev, I.V.; Martin, F.; Marmeisse, R.; et al. The ectomycorrhizal basidiomycete Hebeloma cylindrosporum undergoes early waves of transcriptional reprogramming prior to symbiotic structures differentiation. Environ. Microbiol. 2017, 19, 1338–1354. [Google Scholar] [CrossRef]
- Tang, N.; Lebreton, A.; Xu, W.; Dai, Y.; Yu, F.; Martin, F.M. Transcriptome profiling reveals differential gene expression of secteted proteases and highly specific gene repertoires involved in Lactarius-Pinus symbiosis. Front. Plant Sci. 2021, 12, 714393. [Google Scholar] [CrossRef]
- Casarrubia, S.; Sapienza, S.; Fritz, H.; Daghino, S.; Rosenkranz, M.; Schnitzler, J.-P.; Martin, F.; Perotto, S.; Martino, E. Ecologically different fungi affect Arabidopsis development: Contribution of soluble and volatile compounds. PLoS ONE 2016, 11, e0168236. [Google Scholar] [CrossRef]
- Duplessis, S.; Courty, P.E.; Tagu, D.; Martin, F. Transcript patterns associated with ectomycorrhiza development in Eucalyptus globulus and Pisolithus microcarpus. New Phytol. 2005, 165, 599–611. [Google Scholar] [CrossRef]
- Morel, M.; Jacob, C.; Kohler, A.; Johansson, T.; Martin, F.; Chalot, M.; Brun, A. Identification of genes differentially expressed in extraradical mycelium and ectomycorrhizal roots during Paxillus involutus-Betula pendula ectomycorrhizal symbiosis. Appl. Environ. Microbiol. 2005, 71, 382–391. [Google Scholar] [CrossRef]
- Lofgren, L.A.; Nguyen, N.H.; Vilgalys, R.; Ruytinx, J.; Liao, H.L.; Branco, S.; Kuo, A.; Lipzen, A.; Adreopoulos, W.; Pangilinan, J.; et al. Comparative genomics reveals dynamic genome evolution in host specialist ectomycorrhizal fungi. New Phytol. 2021, 230, 774–792. [Google Scholar] [CrossRef]
- Wright, D.P.; Johansson, T.; Le Quéré, A.; Söderström, B.; Tunlid, A. Spatial patterns of gene expression in the extramatrical mycelium and mycorrhizal root tips formed by the ectomycorrhizal fungus Paxillus involutus in association with birch (Betula pendula) seedlings in soil microcosms. New Phytol. 2005, 167, 579–596. [Google Scholar] [CrossRef]
- Ditengou, F.A.; Müller, A.; Rosenkranz, M.; Felten, J.; Lasok, H.; Van Doorn, M.M.; Legué, V.; Palme, K.; Schnitzler, J.P.; Polle, A. Volatile signalling by sesquiterpenes from ectomycorrhizal fungi reprogrammes root architecture. Nat. Commun. 2015, 6, 6279. [Google Scholar] [CrossRef]
- Veneault-Fourrey, C.; Martin, F. Mutualistic interactions on a knife-edge between saprotrophy and pathogenesis. Curr. Opin. Plant Biol. 2011, 14, 444–450. [Google Scholar] [CrossRef]
- Becquer, A.; Garcia, K.; Amenc, L.; Rivard, C.; Doré, J.; Trives-Segura, C.; Szponarski, W.; Russet, S.; Baeza, Y.; Lassalle-Kaiser, B.; et al. The Hebeloma cylindrosporum HcPT2 Pi transporter plays a key role in ectomycorrhizal symbiosis. New Phytol. 2018, 220, 1185–1199. [Google Scholar] [CrossRef]
- Guerrero-Galán, C.; Garcia, K.; Houdinet, G.; Zimmermann, S.D. Hc TOK1 participates in the maintenance of K+ homeostasis in the ectomycorrhizal fungus Hebeloma cylindrosporum, which is essential for the symbiotic K+ nutrition of Pinus pinaster. Plant Signal. Behav. 2018, 13, e1480845. [Google Scholar] [CrossRef]
- Liao, H.L.; Chen, Y.; Vilgalys, R. Metatranscriptomic study of common and host-specific patterns of gene expression between pines and their symbiotic ectomycorrhizal fungi in the genus Suillus. PLoS Genet. 2016, 12, e1006348. [Google Scholar] [CrossRef]
- Kües, U. Life history and developmental processes in the basidiomycete Coprinus cinereus. Microbiol. Mol. Biol. Rev. 2000, 64, 316–353. [Google Scholar] [CrossRef]
- Ohm, R.A.; De Jong, J.F.; Lugones, L.G.; Aerts, A.; Kothe, E.; Stajich, J.E.; de Vries, R.P.; Record, E.; Levasseur, A.; Baker, S.E.; et al. Genome sequence of the model mushroom Schizophyllum commune. Nat. Biotechnol. 2010, 9, 957–963. [Google Scholar] [CrossRef]
- Pelkmans, J.F.; Vos, A.M.; Scholtmeijer, K.; Hendrix, E.; Baars, J.J.P.; Gehrmann, T.; Reinders, M.J.T.; Lugones, L.G.; Wösten, H.A.B. The transcriptional regulator c2h2 accelerates mushroom formation in Agaricus bisporus. Appl. Microbiol. Biotechnol. 2016, 100, 7151–7159. [Google Scholar] [CrossRef]
- Sipos, G.; Prasanna, A.N.; Walter, M.C.; O’Connor, E.; Bálint, B.; Krizsán, K.; Kiss, B.; Hess, J.; Varga, T.; Slot, J.; et al. Genome expansion and lineage-specific genetic innovations in the forest pathogenic fungi Armillaria. Nat. Ecol. Evol. 2017, 1, 1931. [Google Scholar] [CrossRef]
- Gupta, D.K.; Rühl, M.; Mishra, B.; Kleofas, V.; Hofrichter, M.; Herzog, R.; Pecyna, M.J.; Sharma, R.; Kellner, H.; Hennicke, F.; et al. The genome sequence of the commercially cultivated mushroom Agrocybe aegerita reveals a conserved repertoire of fruiting-related genes and a versatile suite of biopolymer-degrading enzymes. BMC Genom. 2018, 19, 48. [Google Scholar] [CrossRef]
- Orban, A.; Weber, A.; Herzog, R.; Hennicke, F.; Rühl, M. Transcriptome of different fruiting stages in the cultivated mushroom Cyclocybe aegerita suggests a complex regulation of fruiting and reveals enzymes putatively involved in fungal oxylipin biosynthesis. BMC Genom. 2021, 22, 324. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Xu, Z.; Knudson, A.; Carlson, A.; Chen, N.; Kovaka, S.; LaButti, K.; Lipzen, A.; Pennachio, C.; Riley, R.; et al. Genomics and development of Lentinus tigrinus: A white-rot wood-decaying mushroom with dimorphic fruiting bodies. Genome Biol. Evol. 2018, 10, 3250–3261. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.B.; Xia, E.H.; Li, M.; Cui, Y.Y.; Wang, P.M.; Zhang, J.-X.; Xie, B.G.; Xu, J.P.; Yan, J.J.; Li, J.; et al. Transcriptome data reveal conserved patterns of fruiting body development and response to heat stress in the mushroom-forming fungus Flammulina filiformis. PLoS ONE 2020, 15, e0239890. [Google Scholar]
- Krizsán, K.; Almási, É.; Merényi, Z.; Sahu, N.; Virágh, M.; Kószó, T.; Mondo, S.; Kiss, B.; Bálint, B.; Kües, U.; et al. Transcriptomic atlas of mushroom development reveals conserved genes behind complex multicellularity in fungi. Proc. Natl. Acad. Sci. USA 2019, 116, 7409–7418. [Google Scholar] [CrossRef]
- Zhang, T.Q.; Xu, Z.G.; Shang, G.D.; Wang, J.W. A single-cell RNA sequencing profiles the developmental landscape of Arabidopsis root. Mol. Plant 2019, 12, 648–660. [Google Scholar] [CrossRef]
- Cha, J.; Lee, I. Single-cell network biology for resolving cellular heterogeneity in human disease. Exp. Mol. Med. 2020, 52, 1798–1808. [Google Scholar] [CrossRef]
- Paparokidou, C.; Leake, J.R.; Beerling, D.J.; Rolfe, S.A. Phosphate availability and ectomycorrhizal symbiosis with Pinus sylvestris have independent effects on the Paxillus involutus transcriptome. Mycorrhiza 2021, 31, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Garcia, K.; Doidy, J.; Zimmermann, S.; Wipf, D.; Courty, P.E. Take a trip through the plant and fungal transportome of mycorrhiza. Trends Plant Sci. 2016, 21, 937–950. [Google Scholar] [CrossRef] [PubMed]
- Hortal, S.; Plett, K.L.; Plett, J.M.; Cresswell, T.; Johansen, M.; Pendall, E.; Anderson, I.C. Role of plant-fungal nutrient trading and host control in determining the competitive success of ectomycorrhizal fungi. ISME J. 2017, 11, 2666–2676. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruytinx, J.; Miyauchi, S.; Hartmann-Wittulsky, S.; de Freitas Pereira, M.; Guinet, F.; Churin, J.-L.; Put, C.; Le Tacon, F.; Veneault-Fourrey, C.; Martin, F.; et al. A Transcriptomic Atlas of the Ectomycorrhizal Fungus Laccaria bicolor. Microorganisms 2021, 9, 2612. https://doi.org/10.3390/microorganisms9122612
Ruytinx J, Miyauchi S, Hartmann-Wittulsky S, de Freitas Pereira M, Guinet F, Churin J-L, Put C, Le Tacon F, Veneault-Fourrey C, Martin F, et al. A Transcriptomic Atlas of the Ectomycorrhizal Fungus Laccaria bicolor. Microorganisms. 2021; 9(12):2612. https://doi.org/10.3390/microorganisms9122612
Chicago/Turabian StyleRuytinx, Joske, Shingo Miyauchi, Sebastian Hartmann-Wittulsky, Maíra de Freitas Pereira, Frédéric Guinet, Jean-Louis Churin, Carine Put, François Le Tacon, Claire Veneault-Fourrey, Francis Martin, and et al. 2021. "A Transcriptomic Atlas of the Ectomycorrhizal Fungus Laccaria bicolor" Microorganisms 9, no. 12: 2612. https://doi.org/10.3390/microorganisms9122612
APA StyleRuytinx, J., Miyauchi, S., Hartmann-Wittulsky, S., de Freitas Pereira, M., Guinet, F., Churin, J.-L., Put, C., Le Tacon, F., Veneault-Fourrey, C., Martin, F., & Kohler, A. (2021). A Transcriptomic Atlas of the Ectomycorrhizal Fungus Laccaria bicolor. Microorganisms, 9(12), 2612. https://doi.org/10.3390/microorganisms9122612