
The Characterization of a Novel Phage, pPa_SNUABM_DT01, Infecting Pseudomonas aeruginosa

 ,
,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strain and Growth Conditions
2.2. Isolation and Characterization of Bacteriophage
2.3. Transmission Electron Microscopy (TEM) of pPa_SNUABM_DT01
2.4. In Vitro Planktonic Bacterial Lysis Assay
2.5. Complete Genome Sequencing and Annotation
2.6. Comparative Genome Analysis
3. Results
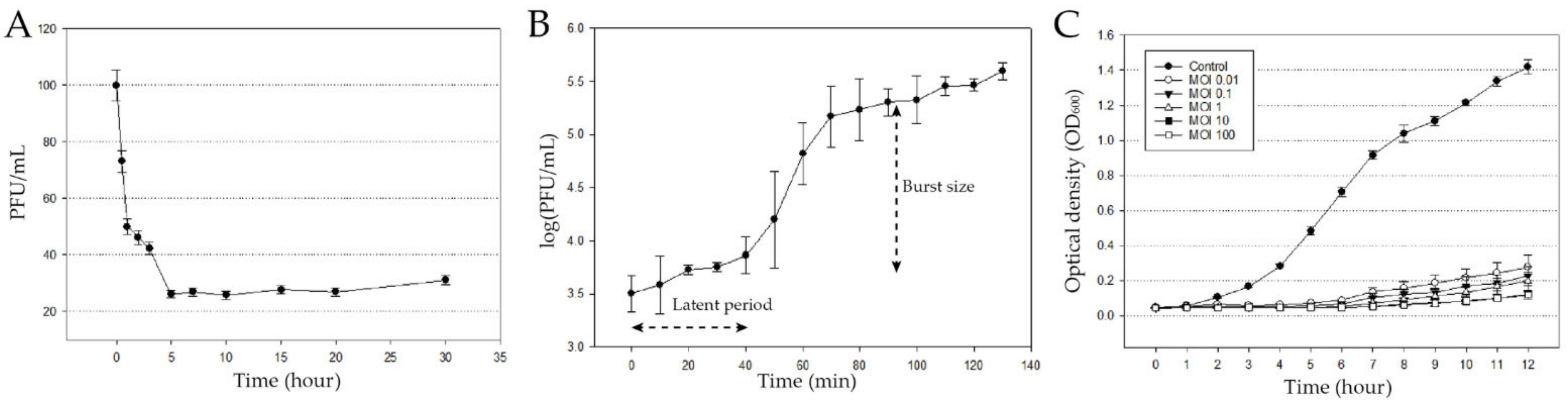
3.1. Isolation and Biological Characterization of pPa_SNUABM_DT01
3.2. Genome Analysis of pPa_SNUABM_DT01
3.2.1. General Features of the Phage Genome
3.2.2. Specific Features of the Phage Genome
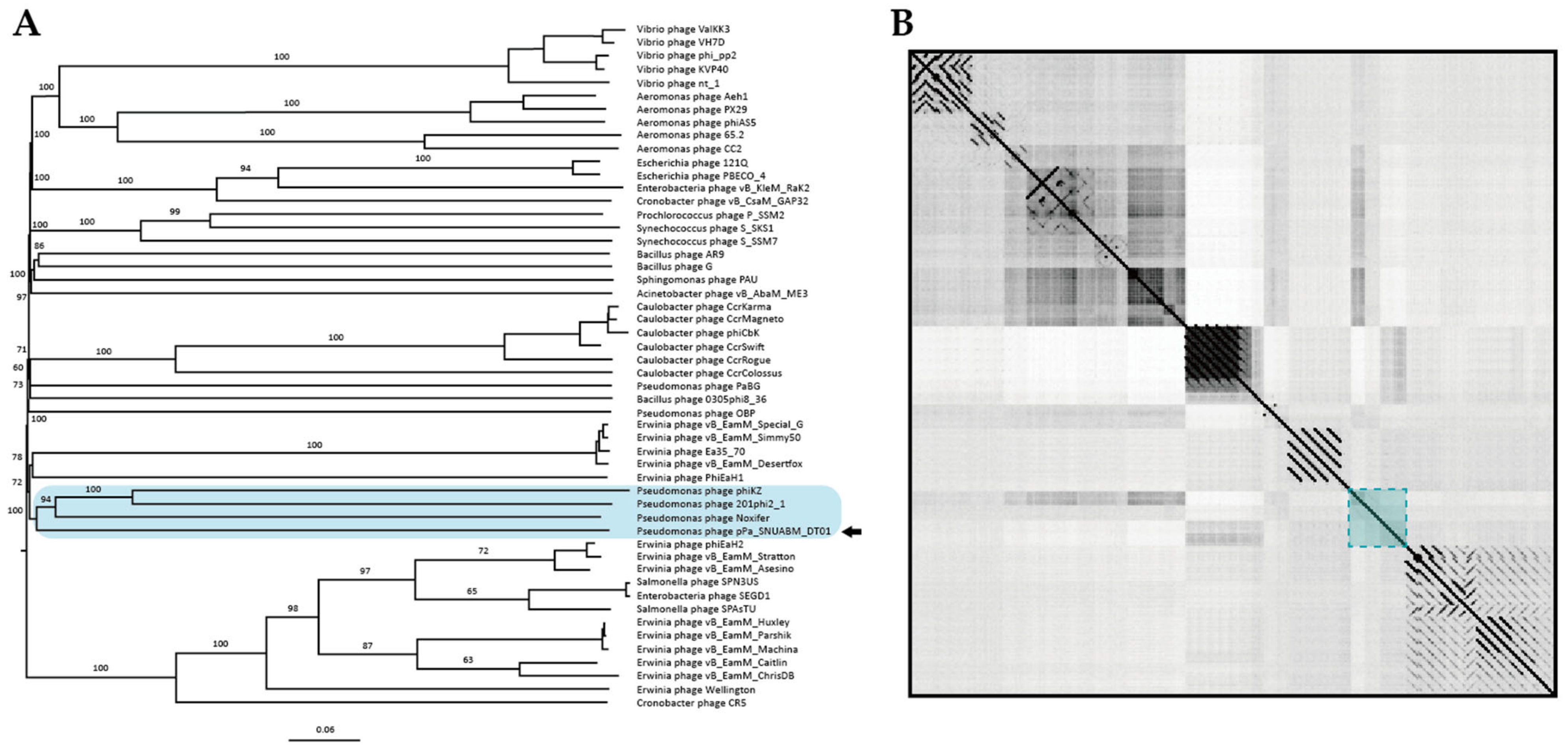
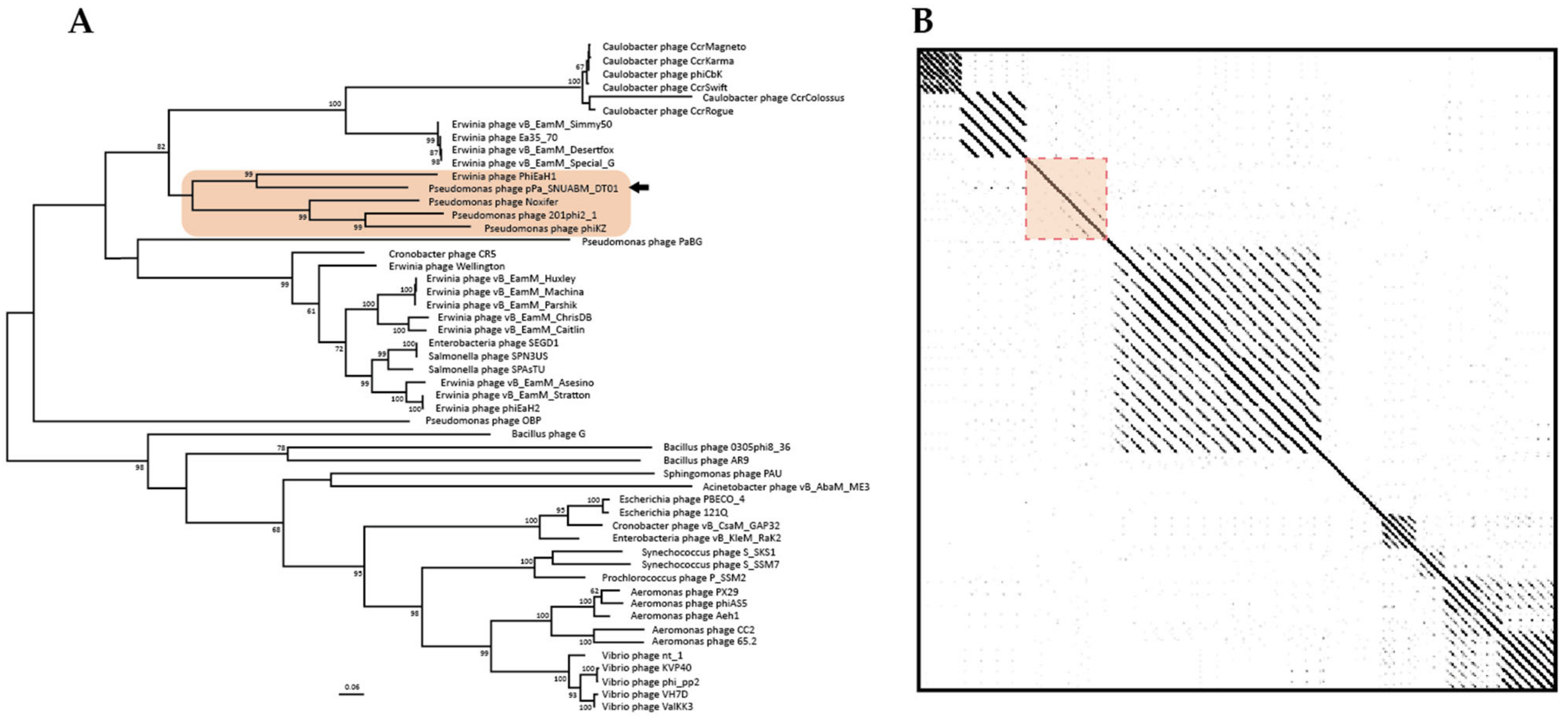
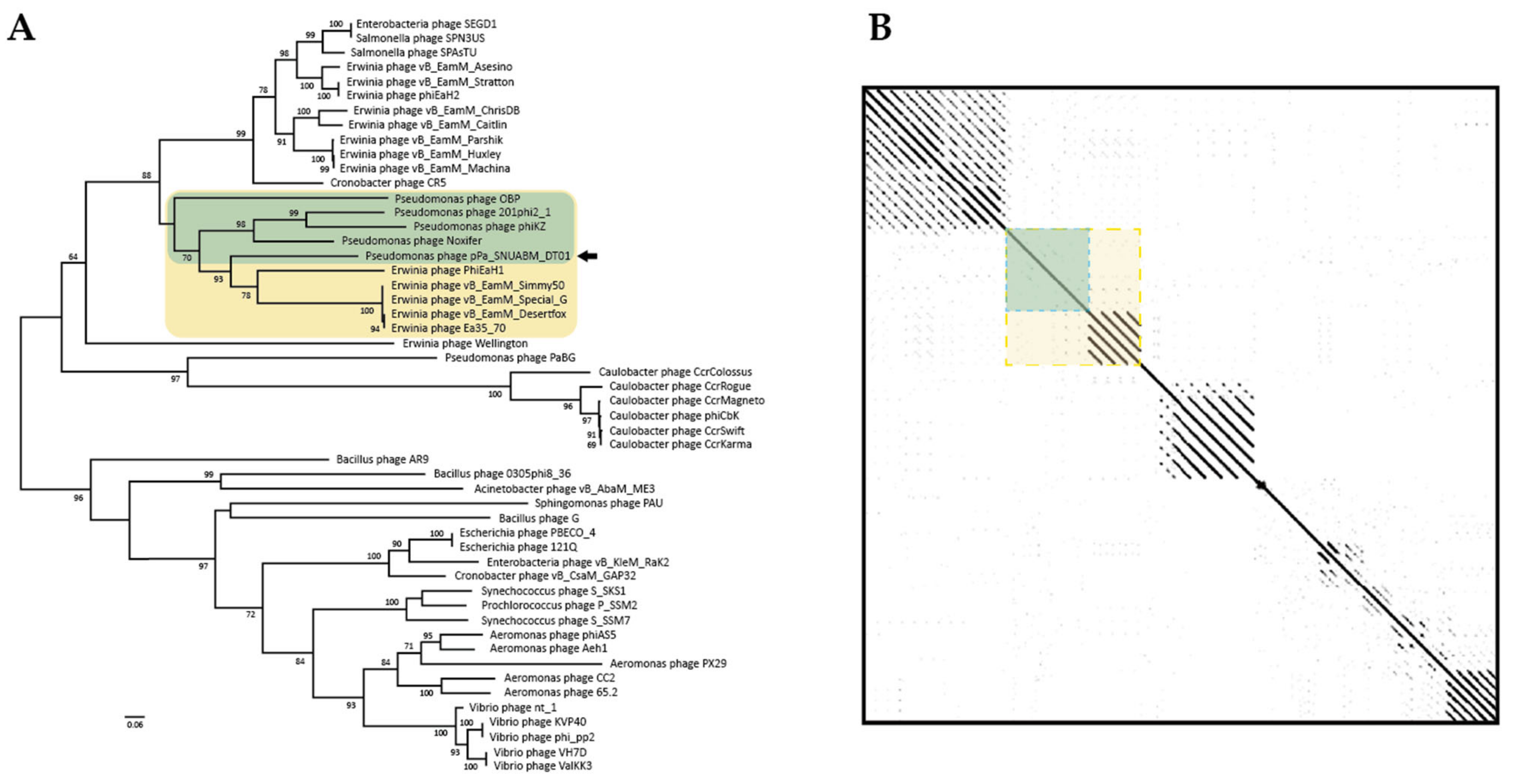
3.3. Comparative Analysis of the Phage Genome
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carlton, R.M. Phage therapy: Past history and future prospects. Arch. Immunol. Ther. Exp. 1999, 47, 267–274. [Google Scholar]
- Kwon, J.; Kim, S.G.; Kim, H.J.; Giri, S.S.; Kim, S.W.; Lee, S.B.; Park, S.C. Isolation and characterization of Salmonella jumbo-phage pSal-SNUABM-04. Viruses 2021, 13, 27. [Google Scholar] [CrossRef]
- Golkar, Z.; Bagasra, O.; Pace, D.G. Bacteriophage therapy: A potential solution for the antibiotic resistance crisis. J. Infect. Dev. Ctries. 2014, 8, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Sulakvelidze, A.; Alavidze, Z.; Morris, J.G., Jr. Bacteriophage therapy. Antimicrob. Agents Chemother. 2001, 45, 649–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakasis, A.; Panitsa, G. Bacteriophage therapy as an alternative treatment for human infections. A comprehensive review. Int. J. Antimicrob. Agents 2019, 53, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Weber-Dąbrowska, B.; Jończyk-Matysiak, E.; Żaczek, M.; Łobocka, M.; Łusiak-Szelachowska, M.; Górski, A. Bacteriophage procurement for therapeutic purposes. Front. Microbiol. 2016, 7, 1177. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Gao, M. Jumbo bacteriophages: An overview. Front. Microbiol. 2017, 8, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham-Mize, C.A.; Rosser, E.J., Jr. Comparison of microbial isolates and susceptibility patterns from the external ear canal of dogs with otitis externa. J. Am. Anim. Hosp. Assoc. 2004, 40, 102–108. [Google Scholar] [CrossRef]
- Bajwa, J. Canine otitis externa—Treatment and complications. Can. Vet. J. 2019, 60, 97. [Google Scholar]
- Korbelik, J.; Singh, A.; Rousseau, J.; Weese, J.S. Analysis of the otic mycobiota in dogs with otitis externa compared to healthy individuals. Vet. Dermatol. 2018, 29, 417-e138. [Google Scholar] [CrossRef]
- Poole, K. Pseudomonas aeruginosa: Resistance to the max. Front. Microbiol. 2011, 2, 65. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kim, S.G.; Jun, J.W.; Giri, S.S.; Yun, S.; Kim, H.J.; Kang, J.W.; Han, S.J.; Jeong, D.; Park, S.C. Isolation and characterisation of pVa-21, a giant bacteriophage with anti-biofilm potential against Vibrio alginolyticus. Sci. Rep. 2019, 9, 6284. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farris, J.S. Estimating phylogenetic trees from distance matrices. Am. Nat. 1972, 106, 645–668. [Google Scholar] [CrossRef]
- FigTree 1.4.3—A Graphical Viewer of Phylogenetic Trees and a Program for Producing Publication-Ready Figures. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 11 November 2020).
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Krumsiek, J.; Arnold, R.; Rattei, T. Gepard: A rapid and sensitive tool for creating dot plots on genome scale. Bioinformatics 2007, 23, 1026–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darling, A.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple Alignment of Conserved Genomic Sequence with Rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackermann, H.-W. 5500 Phages examined in the electron microscope. Arch. Virol. 2006, 152, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Hyman, P.; Abedon, S.T. Practical Methods for Determining Phage Growth Parameters. In Bioinformatics in MicroRNA Research; Springer Science and Business Media LLC.: New York, NY, USA, 2009; Volume 501, pp. 175–202. [Google Scholar]
- Oechslin, F. Resistance development to bacteriophages occurring during bacteriophage therapy. Viruses 2018, 10, 351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heilmann, S.; Sneppen, K.; Krishna, S. Coexistence of phage and bacteria on the boundary of self-organized refuges. Proc. Natl. Acad. Sci. USA 2012, 109, 12828–12833. [Google Scholar] [CrossRef] [Green Version]
- Blazanin, M.; Turner, P.E. Community context matters for bacteria-phage ecology and evolution. ISME J. 2021. [Google Scholar] [CrossRef]
- Gu, J.; Liu, X.; Li, Y.; Han, W.; Lei, L.; Yang, Y.; Zhao, H.; Gao, Y.; Song, J.; Lu, R.; et al. A method for generation phage cocktail with great therapeutic potential. PLoS ONE 2012, 7, e31698. [Google Scholar] [CrossRef] [Green Version]
- Kelly, D.; McAuliffe, O.; O’Mahony, J.; Coffey, A. Development of a broad-host-range phage cocktail for biocontrol. Bioeng. Bugs 2011, 2, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Jun, J.W.; Giri, S.S.; Kim, S.G.; Kim, S.W.; Kwon, J.; Lee, S.B.; Chi, C.; Park, S.C. Bacteriophage cocktail for the prevention of multiple-antibiotic-resistant and mono-phage-resistant Vibrio coralliilyticus infection in pacific oyster (Crassostrea gigas) larvae. Pathogens 2020, 9, 831. [Google Scholar] [CrossRef]
- Kim, S.G.; Giri, S.S.; Yun, S.; Kim, H.J.; Kim, S.W.; Kang, J.W.; Han, S.J.; Kwon, J.; Oh, W.T.; Jun, J.W.; et al. Synergistic phage–surfactant combination clears IgE-promoted Staphylococcus aureus aggregation in vitro and enhances the effect in vivo. Int. J. Antimicrob. Agents 2020, 56, 105997. [Google Scholar] [CrossRef]
- Segall, A.M.; Roach, D.R.; Strathdee, S.A. Stronger together? Perspectives on phage-antibiotic synergy in clinical applications of phage therapy. Curr. Opin. Microbiol. 2019, 51, 46–50. [Google Scholar] [CrossRef]
- Sokolova, M.L.; Misovetc, I.; Severinov, K.V. Multisubunit RNA polymerases of jumbo bacteriophages. Viruses 2020, 12, 1064. [Google Scholar] [CrossRef]
- Thomas, J.A.; Quintana, A.D.B.; Bosch, M.A.; De Peña, A.C.; Aguilera, E.; Coulibaly, A.; Wu, W.; Osier, M.V.; Hudson, A.O.; Weintraub, S.T.; et al. Identification of Essential Genes in the Salmonella Phage SPN3US Reveals Novel Insights into Giant Phage Head Structure and Assembly. J. Virol. 2016, 90, 10284–10298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burroughs, A.M.; Zhang, D.; Schäffer, D.E.; Iyer, L.M.; Aravind, L. Comparative genomic analyses reveal a vast, novel network of nucleotide-centric systems in biological conflicts, immunity and signaling. Nucleic Acids Res. 2015, 43, 10633–10654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Souza, R.F.; Aravind, L. Identification of novel components of NAD-utilizing metabolic pathways and prediction of their biochemical functions. Mol. Biosyst. 2012, 8, 1661–1677. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.Q.; Lee, S.J.; Richardson, C.C.; Tabor, S. A novel nucleotide kinase encoded by gene 1.7 of bacteriophage T7. Mol. Microbiol. 2010, 77, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Pielstick, B.A.; Bell, K.A.; Nieman, T.B.; Stubbs, O.A.; Yeates, E.L.; Baltrus, D.A.; Grose, J.H. A novel, highly related jumbo family of bacteriophages that were isolated against Erwinia. Front. Microbiol. 2019, 10, 1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, R.L.; Morrical, S.W. Coordinated binding of single-stranded and double-stranded DNA by UvsX recombinase. PLoS ONE 2013, 8, e66654. [Google Scholar]
- Timms, A.R.; Cambray-Young, J.; Scott, A.E.; Petty, N.K.; Connerton, P.L.; Clarke, L.; Seeger, K.; Quail, M.; Cummings, N.; Maskell, D.J.; et al. Evidence for a lineage of virulent bacteriophages that target Campylobacter. BMC Genom. 2010, 11, 214. [Google Scholar] [CrossRef] [Green Version]
- Danis-Wlodarczyk, K.; Vandenheuvel, D.; Jang, H.B.; Briers, Y.; Olszak, T.; Arabski, M.; Wasik, S.; Drabik, M.; Higgins, G.; Tyrrel, J.; et al. A proposed integrated approach for the preclinical evaluation of phage therapy in Pseudomonas infections. Sci. Rep. 2016, 6, 28115. [Google Scholar] [CrossRef] [Green Version]
- Struthers-Schlinke, J.S.; Robins, W.P.; Kemp, P.; Molineux, I.J. The internal head protein Gp16 controls DNA ejection from the bacteriophage T7 virion. J. Mol. Biol. 2000, 301, 35–45. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, J.G.; Lin, A.; Dennis, J.J. Isolation and Characterization of the Novel Bacteriophage AXL3 against Stenotrophomonas maltophilia. Int. J. Mol. Sci. 2020, 21, 6338. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.S.; Yu, Z.; Maxwell, K.L.; Davidson, A.R. Ig-like domains on bacteriophages: A tale of promiscuity and deceit. J. Mol. Biol. 2006, 359, 496–507. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, J.; Kim, S.W.; Kim, S.G.; Kang, J.W.; Jung, W.J.; Lee, S.B.; Lee, Y.M.; Giri, S.S.; Chi, C.; Park, S.C. The Characterization of a Novel Phage, pPa_SNUABM_DT01, Infecting Pseudomonas aeruginosa. Microorganisms 2021, 9, 2040. https://doi.org/10.3390/microorganisms9102040
Kwon J, Kim SW, Kim SG, Kang JW, Jung WJ, Lee SB, Lee YM, Giri SS, Chi C, Park SC. The Characterization of a Novel Phage, pPa_SNUABM_DT01, Infecting Pseudomonas aeruginosa. Microorganisms. 2021; 9(10):2040. https://doi.org/10.3390/microorganisms9102040
Chicago/Turabian StyleKwon, Jun, Sang Wha Kim, Sang Guen Kim, Jeong Woo Kang, Won Joon Jung, Sung Bin Lee, Young Min Lee, Sib Sankar Giri, Cheng Chi, and Se Chang Park. 2021. "The Characterization of a Novel Phage, pPa_SNUABM_DT01, Infecting Pseudomonas aeruginosa" Microorganisms 9, no. 10: 2040. https://doi.org/10.3390/microorganisms9102040
APA StyleKwon, J., Kim, S. W., Kim, S. G., Kang, J. W., Jung, W. J., Lee, S. B., Lee, Y. M., Giri, S. S., Chi, C., & Park, S. C. (2021). The Characterization of a Novel Phage, pPa_SNUABM_DT01, Infecting Pseudomonas aeruginosa. Microorganisms, 9(10), 2040. https://doi.org/10.3390/microorganisms9102040