
Comparative Genomic Analysis Determines the Functional Genes Related to Bile Salt Resistance in Lactobacillus salivarius

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation and Genome Sequencing of L. salivarius
2.2. Determination of the Tolerance of L. salivarius Strains in Bile Salt Solutions
2.3. Cluster Analysis of Three Subtypes of BSHs
2.4. Comparative Genomic Analysis
2.5. Quantitative RT-PCR
2.6. Gene Knockout of L. salivarius FWXBH36M1
2.7. Statistical Analysis
3. Results
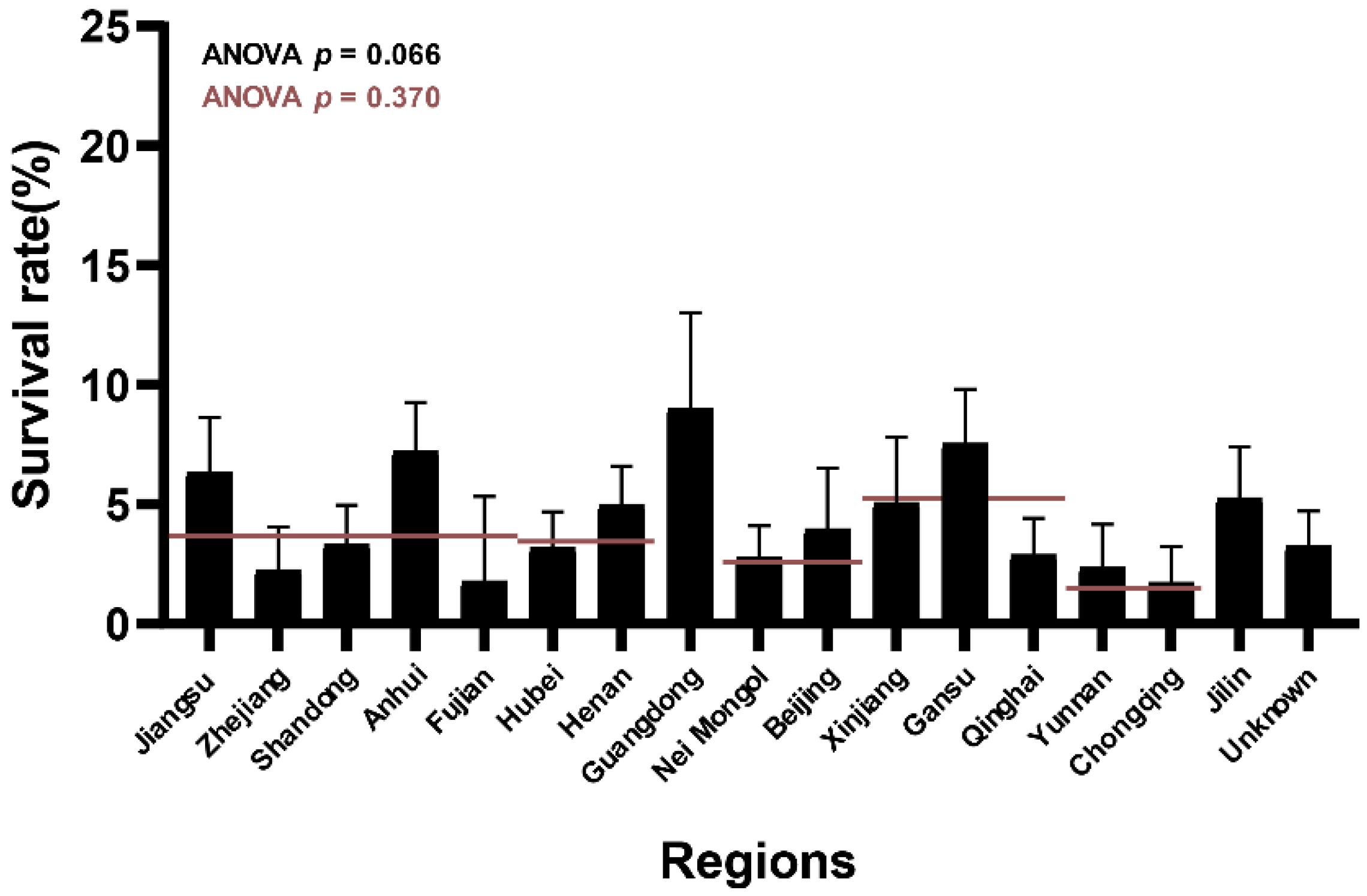
3.1. Different L. salivarius Strains Showed Diverse Survival Rates in Bile Salt Solutions
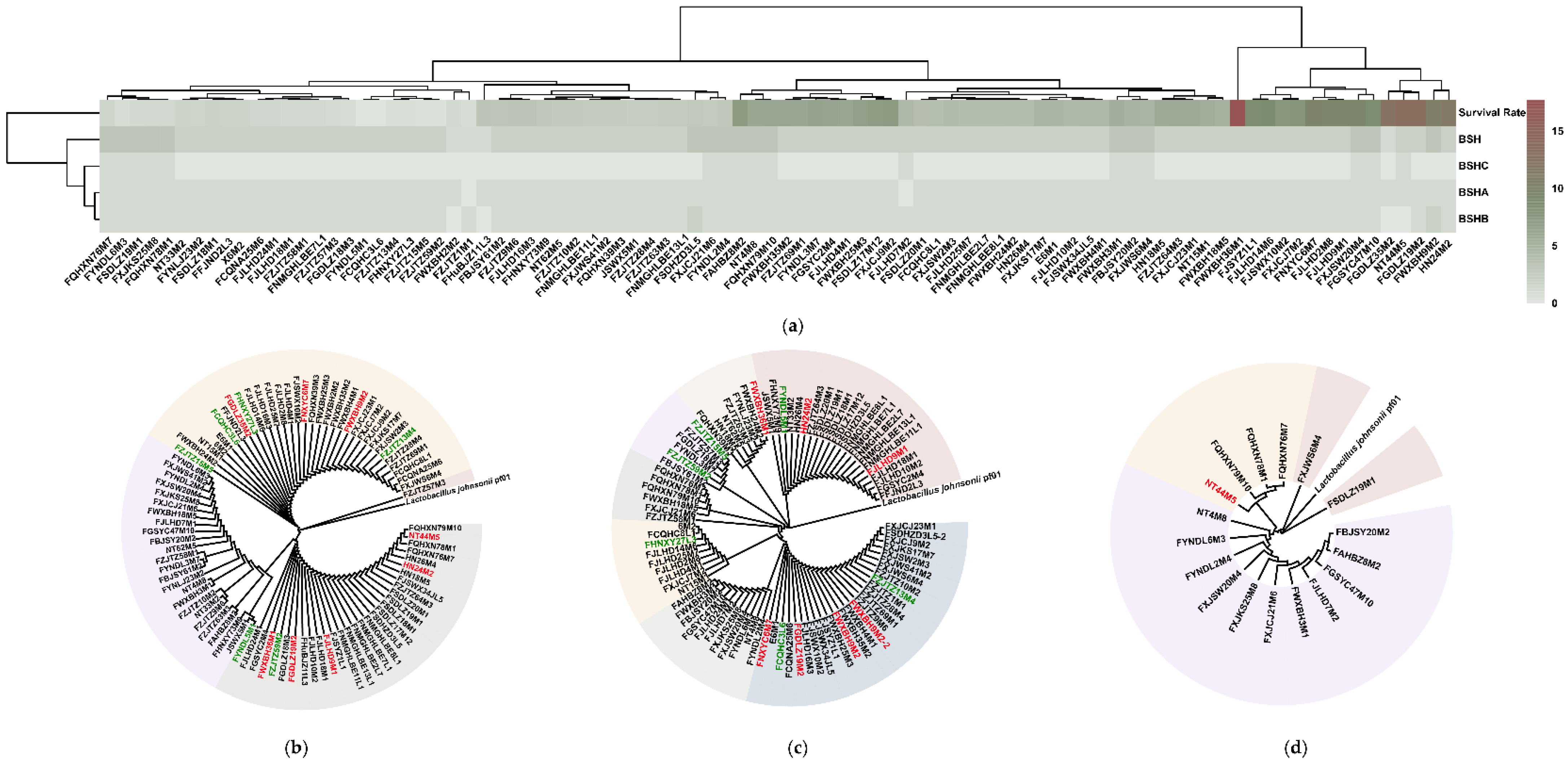
3.2. Strain Tolerance in Bile Salt Solutions Was Not Associated with BSH Variations
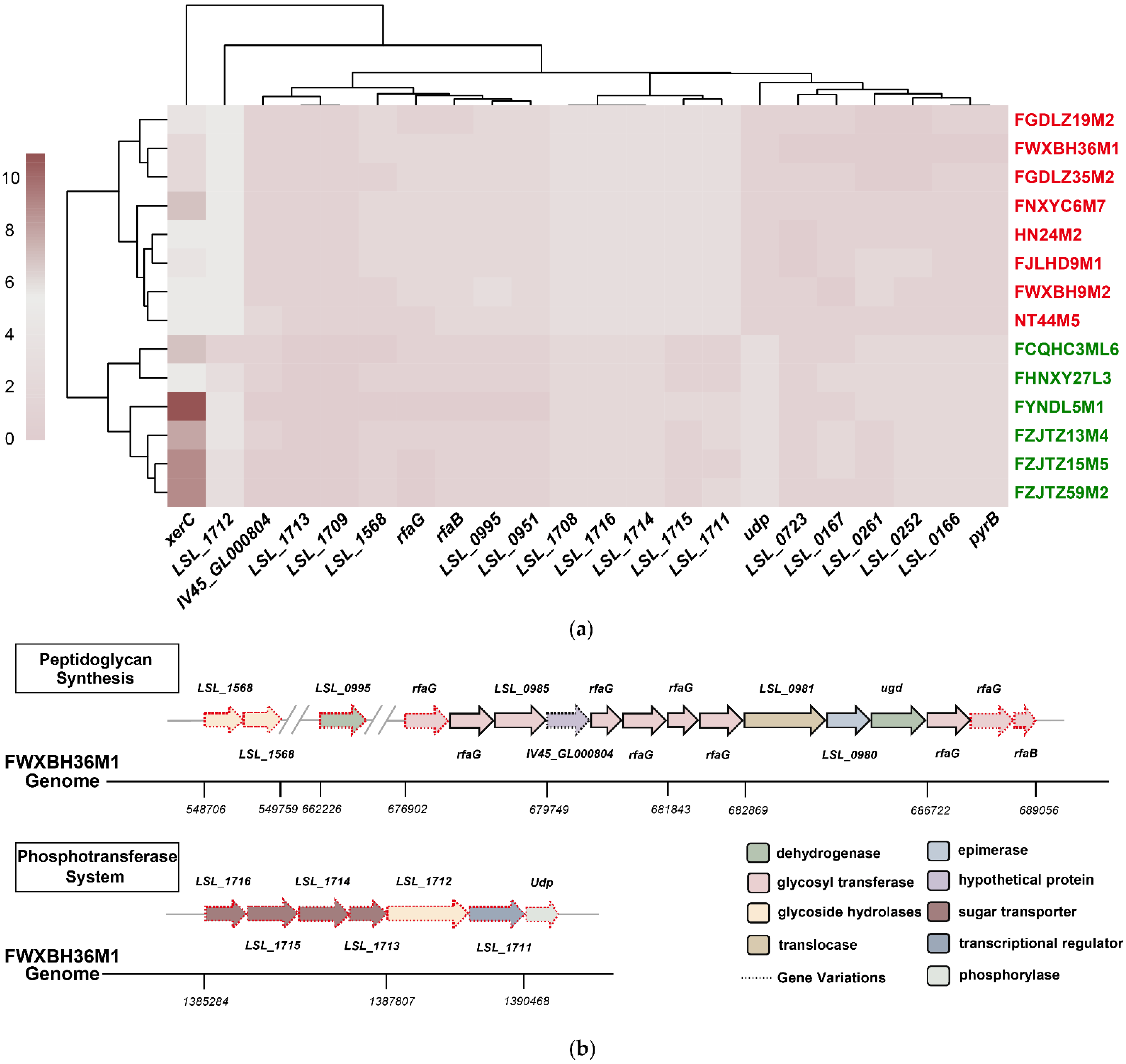
3.3. Comparative Genomic Analysis Determined the Potential L. salivarius Genes Responsible for Bile Salt Tolerance
3.4. Possible Redundant L. salivarius Genes for Bile Salt Tolerance Were Identified via Comparative Genomic Analysis
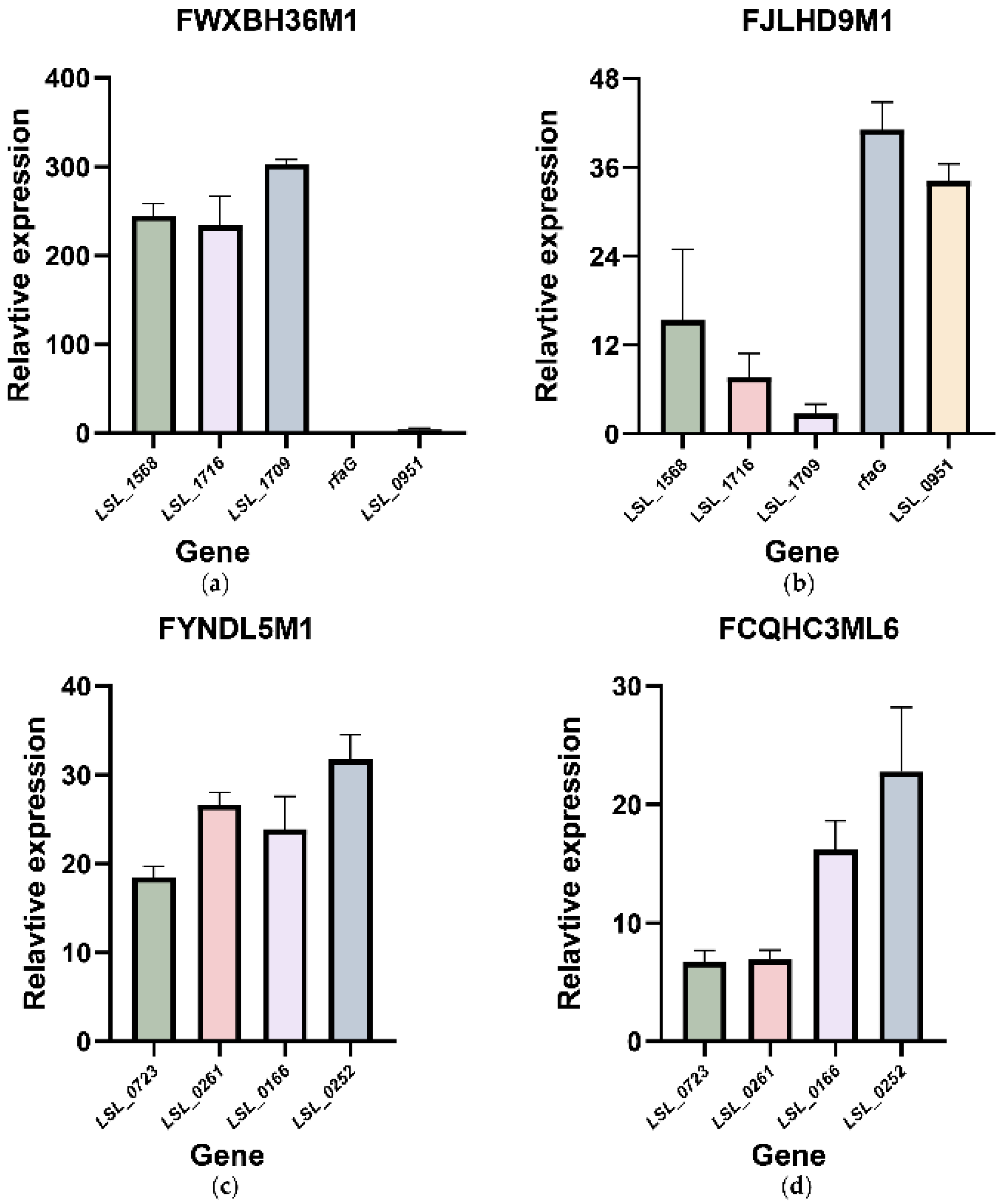
3.5. qRT-PCR Showed the Transcriptional Changes of the Possible Functional and Redundant Genes Related to Tolerance in Bile Salt Solutions
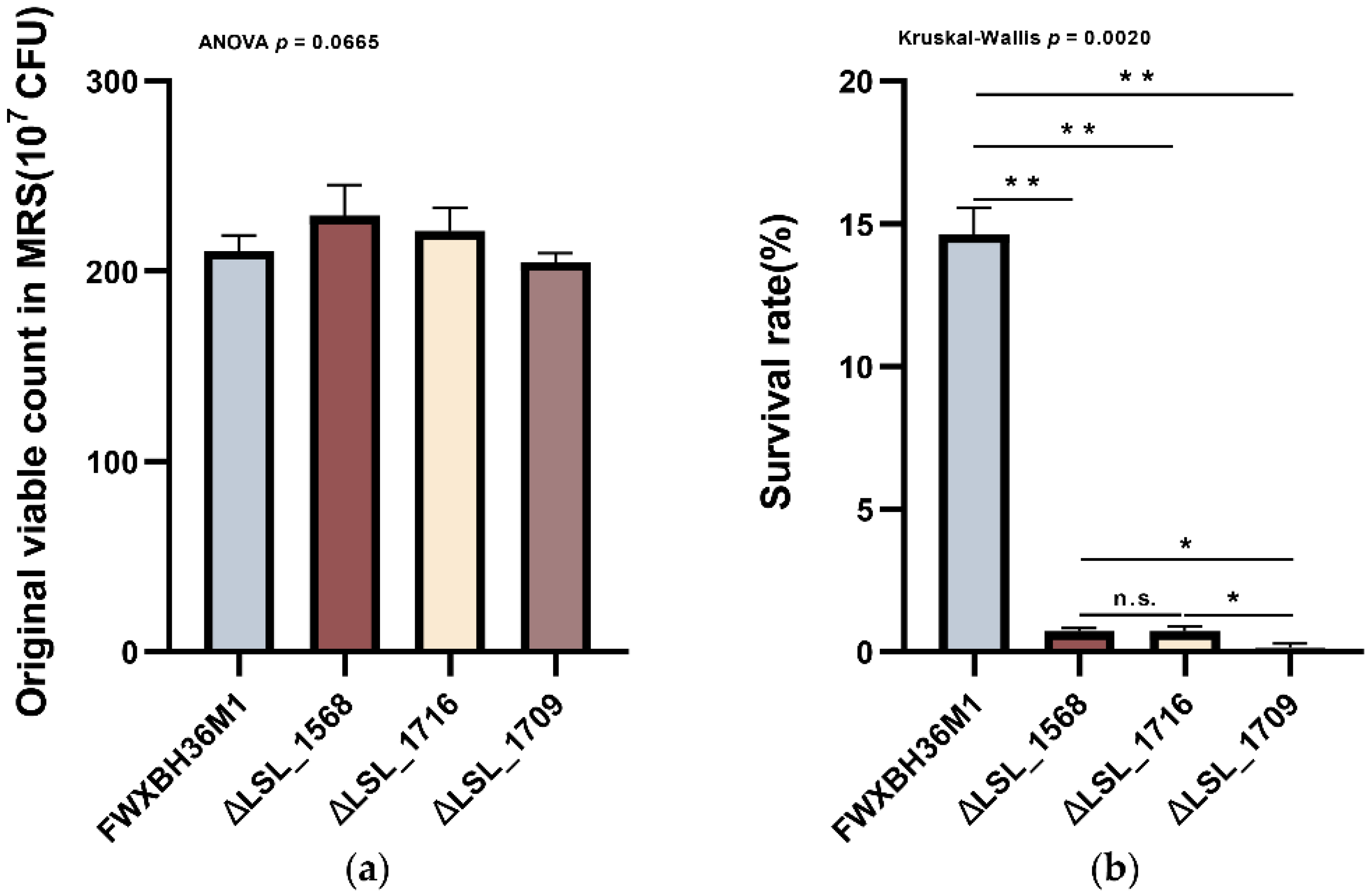
3.6. Knockout of the Functional Genes in L. salivarius FWXBH36M1 Verified Their Contributions to the Bile Salt Tolerance
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rogosa, M.; Wiseman, R.F.; Mitchell, J.A.; Disraely, M.N.; Beaman, A.J. Species differentiation of oral lactobacilli from man including description of Lactobacillus salivarius nov spec and Lactobacillus cellobiosus nov spec. J. Bacteriol. 1953, 65, 681–699. [Google Scholar] [CrossRef]
- Guo, X.-H.; Kim, J.-M.; Nam, H.-M.; Park, S.-Y.; Kim, J.-M. Screening lactic acid bacteria from swine origins for multistrain probiotics based on in vitro functional properties. Anaerobe 2010, 16, 321–326. [Google Scholar] [CrossRef]
- Soro-Yao, A.A.; Schumann, P.; Thonart, P.; Djè, K.M.; Pukall, R. The use of MALDI-TOF mass spectrometry, ribotyping and phenotypic tests to identify lactic acid bacteria from fermented cereal foods in Abidjan (Côte d’Ivoire). Open Microbiol. J. 2014, 8, 78–86. [Google Scholar] [CrossRef]
- Kang, C.-H.; Han, S.H.; Kim, Y.; Paek, N.-S.; So, J.-S. In vitro probiotic properties of Lactobacillus salivarius MG242 isolated from human vagina. Probiotics Antimicrob. Proteins 2018, 10, 343–349. [Google Scholar] [CrossRef]
- Neville, B.A.; O’Toole, P.W. Probiotic properties of Lactobacillus salivarius and closely related Lactobacillus species. Future Microbiol. 2010, 5, 759–774. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, L.; Zheng, X.; Fu, T.; Guo, H.; Ren, F. Lactobacillus salivarius strain FDB89 induced longevity in Caenorhabditis elegans by dietary restriction. J. Microbiol. 2013, 51, 183–188. [Google Scholar] [CrossRef]
- Zhai, Q.; Shen, X.; Cen, S.; Zhang, C.; Tian, F.; Zhao, J.; Zhang, H.; Xue, Y.; Chen, W. Screening of Lactobacillus salivarius strains from the feces of Chinese populations and the evaluation of their effects against intestinal inflammation in mice. Food Funct. 2020, 11, 221–235. [Google Scholar] [CrossRef]
- Kang, M.-S.; Lim, H.-S.; Oh, J.-S.; Lim, Y.-j.; Wuertz-Kozak, K.; Harro, J.M.; Shirtliff, M.E.; Achermann, Y. Antimicrobial activity of Lactobacillus salivarius and Lactobacillus fermentum against Staphylococcus aureus. Pathog. Dis. 2017, 75, ftx009. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Adams, M.C. In vitro assessment of the upper gastrointestinal tolerance of potential probiotic dairy propionibacteria. Int. J. Food Microbiol. 2004, 91, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, L.; Margolles, A.; Sánchez, B. Bile resistance mechanisms in Lactobacillus and Bifidobacterium. Front Microbiol. 2013, 4, 396. [Google Scholar] [CrossRef] [PubMed]
- Bron, P.A.; Marco, M.; Hoffer, S.M.; Van Mullekom, E.; de Vos, W.M.; Kleerebezem, M. Genetic characterization of the bile salt response in Lactobacillus plantarum and analysis of responsive promoters in vitro and in situ in the gastrointestinal tract. J. Bacteriol 2004, 186, 7829–7835. [Google Scholar] [CrossRef]
- Leverrier, P.; Dimova, D.; Pichereau, V.; Auffray, Y.; Boyaval, P.; Jan, G. Susceptibility and adaptive response to bile salts in Propionibacterium freudenreichii: Physiological and proteomic analysis. Appl. Environ. Microbiol. 2003, 69, 3809–3818. [Google Scholar] [CrossRef]
- Noh, D.O.; Gilliland, S.E. Influence of bile on cellular integrity and β-galactosidase activity of Lactobacillus acidophilus. J. Dairy Sci. 1993, 76, 1253–1259. [Google Scholar] [CrossRef]
- Wu, M.-H.; Pan, T.-M.; Wu, Y.-J.; Chang, S.-J.; Chang, M.-S.; Hu, C.-Y. Exopolysaccharide activities from probiotic bifidobacterium: Immunomodulatory effects (on J774A.1 macrophages) and antimicrobial properties. Int. J. Food Microbiol. 2010, 144, 104–110. [Google Scholar] [CrossRef]
- Pfeiler, E.A.; Klaenhammer, T.R. Role of transporter proteins in bile tolerance of Lactobacillus acidophilus. Appl. Environ. Microbiol. 2009, 75, 6013–6016. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Lee, B.H. Bile salt hydrolases: Structure and function, substrate preference, and inhibitor development. Protein Sci. 2018, 27, 1742–1754. [Google Scholar] [CrossRef] [PubMed]
- Hamon, E.; Horvatovich, P.; Izquierdo, E.; Bringel, F.; Marchioni, E.; Aoudé-Werner, D.; Ennahar, S. Comparative proteomic analysis of Lactobacillus plantarum for the identification of key proteins in bile tolerance. BMC Microbiol. 2011, 11, 63. [Google Scholar] [CrossRef]
- Lee, J.Y.; Pajarillo, E.A.B.; Kim, M.J.; Chae, J.P.; Kang, D.-K. Proteomic and transcriptional analysis of Lactobacillus johnsonii PF01 during bile salt exposure by iTRAQ shotgun proteomics and quantitative RT-PCR. J. Proteome Res. 2013, 12, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Ali, S.A.; Kumar, S.; Mohanty, A.K.; Behare, P. Label-free quantitative proteomic analysis of Lactobacillus fermentum NCDC 400 during bile salt exposure. J. Proteom. 2017, 167, 36–45. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Douillard, F.P.; Wang, G.; Zhai, Z.; Yang, J.; Song, S.; Cui, J.; Ren, F.; Luo, Y.; Zhang, B.; et al. Integrated transcriptomic and proteomic analysis of the bile stress response in a centenarian-originated probiotic Bifidobacterium longum BBMN68. Mol. Cell Proteom. 2014, 13, 2558–2572. [Google Scholar] [CrossRef]
- Kapse, N.G.; Engineer, A.S.; Gowdaman, V.; Wagh, S.; Dhakephalkar, P.K. Functional annotation of the genome unravels probiotic potential of Bacillus coagulans HS243. Genomics 2019, 111, 921–929. [Google Scholar] [CrossRef]
- Lv, L.-X.; Yan, R.; Shi, H.-Y.; Shi, D.; Fang, D.-Q.; Jiang, H.-Y.; Wu, W.-R.; Guo, F.-F.; Jiang, X.-W.; Gu, S.-L.; et al. Integrated transcriptomic and proteomic analysis of the bile stress response in probiotic Lactobacillus salivarius LI01. J. Proteom. 2017, 150, 216–229. [Google Scholar] [CrossRef]
- Wang, G.; Zhai, Z.; Ren, F.; Li, Z.; Zhang, B.; Hao, Y. Combined transcriptomic and proteomic analysis of the response to bile stress in a centenarian-originated probiotic Lactobacillus salivarius Ren. Food Res. Int. 2020, 137, 109331. [Google Scholar] [CrossRef]
- Jiang, H.; Dong, H.; Zhang, G.; Yu, B.; Chapman, L.R.; Fields, M.W. Microbial diversity in water and sediment of Lake Chaka, an Athalassohaline Lake in Northwestern China. Appl. Environ. Microbiol. 2006, 72, 7430. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Guo, L.; Gu, S.; Wang, O.; Zhang, R.; Peters, B.A.; Fan, G.; Liu, X.; Xu, X.; Deng, L.; et al. TGS-GapCloser: A fast and accurate gap closer for large genomes with low coverage of error-prone long reads. GigaScience 2020, 9, giaa094. [Google Scholar] [CrossRef]
- Borodovsky, M.; McIninch, J. GENMARK: Parallel gene recognition for both DNA strands. Comput. Chem. 1993, 17, 123–133. [Google Scholar] [CrossRef]
- Haghshenas, B.; Haghshenas, M.; Nami, Y.; Khosroushahi, A.Y.; Abdullah, N.; Barzegari, A.; Rosli, R.; Hejazi, M.S. Probiotic assessment of Lactobacillus plantarum 15HN and Enterococcus mundtii 50H isolated from traditional dairies microbiota. Adv. Pharm. Bull. 2016, 6, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Chae, J.P.; Valeriano, V.D.; Kim, G.B.; Kang, D.K. Molecular cloning, characterization and comparison of bile salt hydrolases from Lactobacillus johnsonii PF01. J. Appl. Microbiol. 2013, 114, 121–133. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Brunk, B.P.; Chen, F.; Gao, X.; Harb, O.S.; Iodice, J.B.; Shanmugam, D.; Roos, D.S.; Stoeckert, C.J., Jr. Using OrthoMCL to assign proteins to OrthoMCL-DB groups or to cluster proteomes into new ortholog groups. Curr. Protoc. Bioinform. 2011, 35, 6–12. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Li, T.T.; Jalbani, Y.M.; Zhang, G.L.; Zhao, Z.Y.; Wang, Z.Y.; Zhao, X.Y.; Chen, A.L. Detection of goat meat adulteration by real-time PCR based on a reference primer. Food Chem. 2019, 277, 554–557. [Google Scholar] [CrossRef]
- Jernberg, C.; Löfmark, S.; Edlund, C.; Jansson, J.K. Long-term ecological impacts of antibiotic administration on the human intestinal microbiota. ISME J. 2007, 1, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Bongers, R.S.; Kleerebezem, M. Cre-lox-based system for multiple gene deletions and selectable-marker removal in Lactobacillus plantarum. Appl. Environ. Microbiol. 2007, 73, 1126–1135. [Google Scholar] [CrossRef] [PubMed]
- van Pijkeren, J.-P.; Canchaya, C.; Ryan, K.A.; Li, Y.; Claesson, M.J.; Sheil, B.; Steidler, L.; Mahony, L.; Fitzgerald, G.F.; van Sinderen, D.; et al. Comparative and functional analysis of sortase-dependent proteins in the predicted secretome of Lactobacillus salivarius UCC118. Appl. Environ. Microbiol. 2006, 72, 4143–4153. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Skoneczna, A.; Miciałkiewicz, A.; Skoneczny, M. Saccharomyces cerevisiae Hsp31p, a stress response protein conferring protection against reactive oxygen species. Free Radic. Biol. Med. 2007, 42, 1409–1420. [Google Scholar] [CrossRef]
- Imane, H.A.; Amel, D. Characterization and screening of the potential probiotic lactic acid bacteria and Bifidobacterium strains isolated of different biotopes. Mediterr. J. Nutr. Metab. 2018, 11, 145–173. [Google Scholar] [CrossRef]
- Horackova, S.; Vesela, K.; Klojdova, I.; Bercikova, M.; Plockova, M. Bile salt hydrolase activity, growth characteristics and surface properties in Lactobacillus acidophilus. Eur. Food Res. Technol. 2020, 246, 1627–1636. [Google Scholar] [CrossRef]
- Harris, H.M.B.; Bourin, M.J.B.; Claesson, M.J.; O’Toole, P.W. Phylogenomics and comparative genomics of Lactobacillus salivarius, a mammalian gut commensal. Microb. Genom. 2017, 3, e000115. [Google Scholar] [CrossRef]
- Martino, M.E.; Bayjanov, J.R.; Caffrey, B.E.; Wels, M.; Joncour, P.; Hughes, S.; Gillet, B.; Kleerebezem, M.; van Hijum, S.A.F.T.; Leulier, F. Nomadic lifestyle of Lactobacillus plantarum revealed by comparative genomics of 54 strains isolated from different habitats. Environ. Microbiol. 2016, 18, 4974–4989. [Google Scholar] [CrossRef]
- Choi, S.; Jin, G.-D.; Park, J.; You, I.; Kim, E.B. Pan-Genomics of Lactobacillus plantarum revealed group-specific genomic profiles without habitat association. J. Microbiol. Biotechnol. 2018, 28, 1352–1359. [Google Scholar] [CrossRef] [PubMed]
- Egan, A.J.F.; Errington, J.; Vollmer, W. Regulation of peptidoglycan synthesis and remodelling. Nat. Rev. Microbiol. 2020, 18, 446–460. [Google Scholar] [CrossRef] [PubMed]
- Saier, M.H., Jr. The bacterial phosphotransferase system: New frontiers 50 years after its discovery. J. Mol. Microbiol. Biotechnol. 2015, 25, 73–78. [Google Scholar] [CrossRef]
- Sharma, V.; Smolin, J.; Nayak, J.; Ayala, J.E.; Scott, D.A.; Peterson, S.N.; Freeze, H.H. Mannose alters gut microbiome, prevents diet-induced obesity, and improves host metabolism. Cell Rep. 2018, 24, 3087–3098. [Google Scholar] [CrossRef] [PubMed]
- Quigley, P.M.; Korotkov, K.; Baneyx, F.; Hol, W.G.J. A new native EcHsp31 structure suggests a key role of structural flexibility for chaperone function. Protein Sci. 2004, 13, 269–277. [Google Scholar] [CrossRef]
- Zheng, Z.Y.; Bernstein, C. Bile salt/acid induction of DNA damage in bacterial cells: Effect of taurine conjugation. Nutr. Cancer 1992, 18, 157–164. [Google Scholar] [CrossRef]
- Prieto, A.I.; Ramos-Morales, F.; Casadesús, J. Repair of DNA damage induced by bile salts in Salmonella enterica. Genetics 2006, 174, 575–584. [Google Scholar] [CrossRef]
- Garay-Arroyo, A.; Covarrubias, A.A.; Clark, I.; Niño, I.; Gosset, G.; Martinez, A. Response to different environmental stress conditions of industrial and laboratory Saccharomyces cerevisiae strains. Appl. Microbiol. Biotechnol. 2004, 63, 734–741. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; de Jong, A.; Frenzel, E.; Kuipers, O.P. Comparative transcriptomics of Bacillus mycoides strains in response to potato-root exudates reveals different genetic adaptation of endophytic and soil isolates. Front. Microbiol. 2017, 8, 1487. [Google Scholar] [CrossRef] [PubMed]
- Jan, G.; Leverrier, P.; Proudy, I.; Lait, N.R.J. Survival and beneficial effects of propionibacteria in the human gut: In vivo and in vitro investigations. Lait 2002, 82, 131–144. [Google Scholar] [CrossRef][Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subtypes | Function | Accession No. |
|---|---|---|
| BSHA | Taurine-conjugated bile salt hydrolase | EGP12224 |
| BSHB | Taurine-conjugated bile salt hydrolase | EGP13287 |
| BSHC | Glycosyl-conjugated bile salt hydrolase | EGP12391 |
| Group | Gene | Function |
|---|---|---|
| Variable Genes | LSL_1568 | beta-N-acetylhexosaminidase |
| LSL_0995 | UDP-D-quinovosamine 4-dehydrogenase | |
| rfaG | glycosyl transferase | |
| IV45_GL000804 | hypothetical protein | |
| rfaB | glycosyl transferase | |
| LSL_0951 | hypothetical protein | |
| LSL_1716 | PTS mannose transporter subunit IIB | |
| LSL_1715 | PTS sugar transporter subunit IIC | |
| LSL_1714 | PTS N-acetylglucosamine transporter subunit IIABC | |
| LSL_1713 | PTS N-acetylglucosamine transporter subunit IIBC | |
| LSL_1712 | alpha-glucosidase | |
| LSL_1711 | LacI family transcriptional regulator | |
| LSL_1709 | type 1 glutamine amido transferase | |
| LSL_1708 | ArsR family transcriptional regulator | |
| Udp | phosphorylase | |
| Redundant Genes | LSL_0723 | transcriptional regulator |
| LSL_0261 | hypothetical protein | |
| LSL_0166 | hypothetical protein | |
| LSL_0167 | hypothetical protein | |
| pyrB | aspartate carbamoyltransferase catalytic subunit | |
| xerC | site-specific integrase | |
| LSL_0252 | hypothetical protein |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, Q.; Shen, X.; Yu, L.; Tian, F.; Zhao, J.; Zhang, H.; Chen, W.; Zhai, Q. Comparative Genomic Analysis Determines the Functional Genes Related to Bile Salt Resistance in Lactobacillus salivarius. Microorganisms 2021, 9, 2038. https://doi.org/10.3390/microorganisms9102038
Pan Q, Shen X, Yu L, Tian F, Zhao J, Zhang H, Chen W, Zhai Q. Comparative Genomic Analysis Determines the Functional Genes Related to Bile Salt Resistance in Lactobacillus salivarius. Microorganisms. 2021; 9(10):2038. https://doi.org/10.3390/microorganisms9102038
Chicago/Turabian StylePan, Qiqi, Xudan Shen, Leilei Yu, Fengwei Tian, Jianxin Zhao, Hao Zhang, Wei Chen, and Qixiao Zhai. 2021. "Comparative Genomic Analysis Determines the Functional Genes Related to Bile Salt Resistance in Lactobacillus salivarius" Microorganisms 9, no. 10: 2038. https://doi.org/10.3390/microorganisms9102038
APA StylePan, Q., Shen, X., Yu, L., Tian, F., Zhao, J., Zhang, H., Chen, W., & Zhai, Q. (2021). Comparative Genomic Analysis Determines the Functional Genes Related to Bile Salt Resistance in Lactobacillus salivarius. Microorganisms, 9(10), 2038. https://doi.org/10.3390/microorganisms9102038