Sulfur and Methane-Oxidizing Microbial Community in a Terrestrial Mud Volcano Revealed by Metagenomics



Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling, Field Measurements, Chemical Analyses, and DNA Isolation
2.2. 16S rRNA Gene Sequencing and Analysis
2.3. Sequencing of Metagenomic DNA, Assembly, and Annotation of MAGs
2.4. Genome -to-Genome Distance Estimation and Phylogenetic Analysis
2.5. Nucleotide Sequence Accession Numbers
3. Results and Discussion
3.1. Study Site and Fluid Chemistry
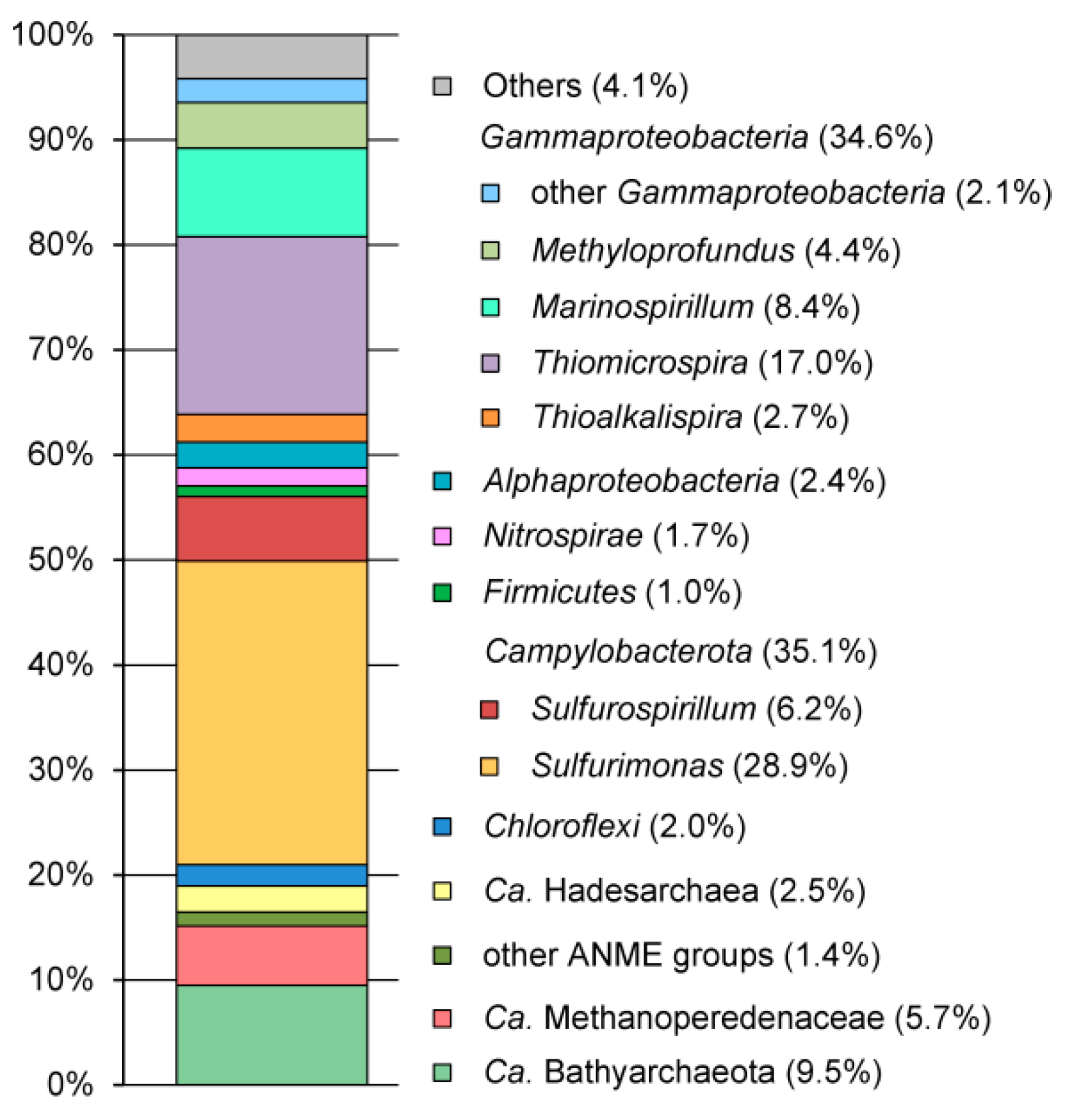
3.2. Microbial Community Structure Revealed by 16S rRNA Profiling
3.3. Metagenome Sequencing and Assembly of MAGs
3.4. Sulfur-Oxidizing Bacteria
3.5. Sulfate-Reducing Bacteria
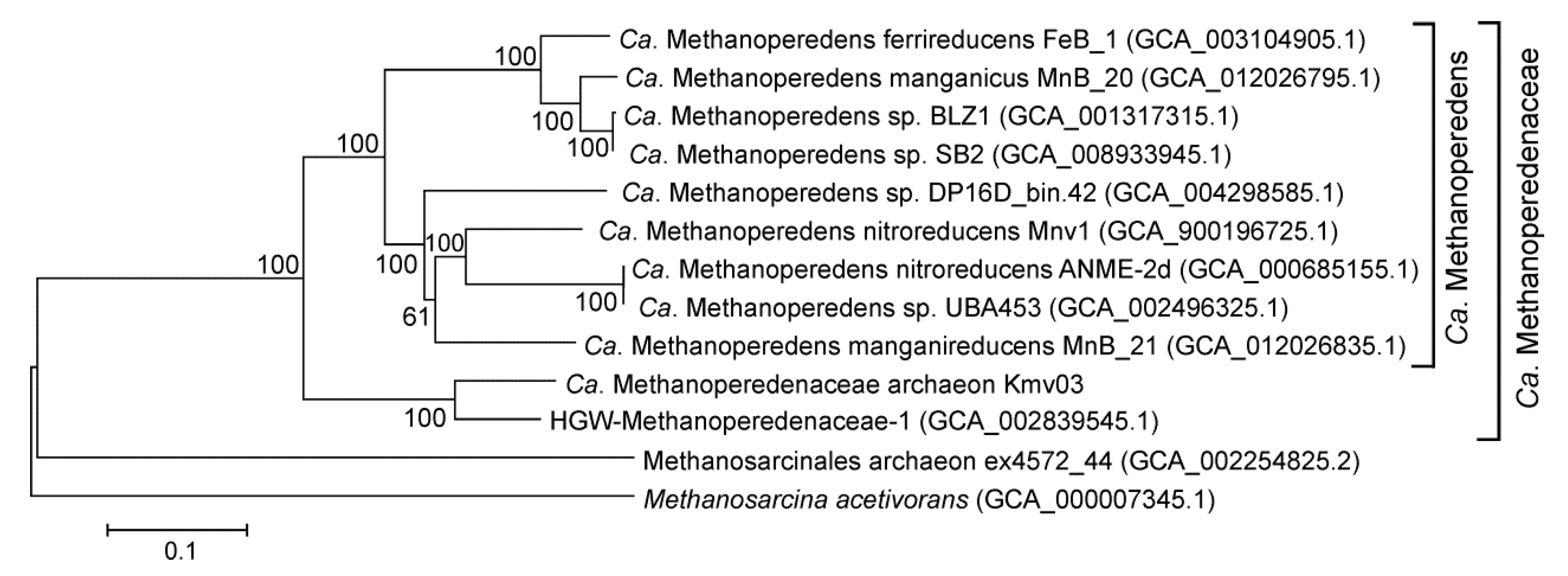
3.6. Bacteria and Archaea Involved in the Methane Cycle
3.7. Iron-Reducing Deltaproteobacteria
3.8. Bathyarchaeota
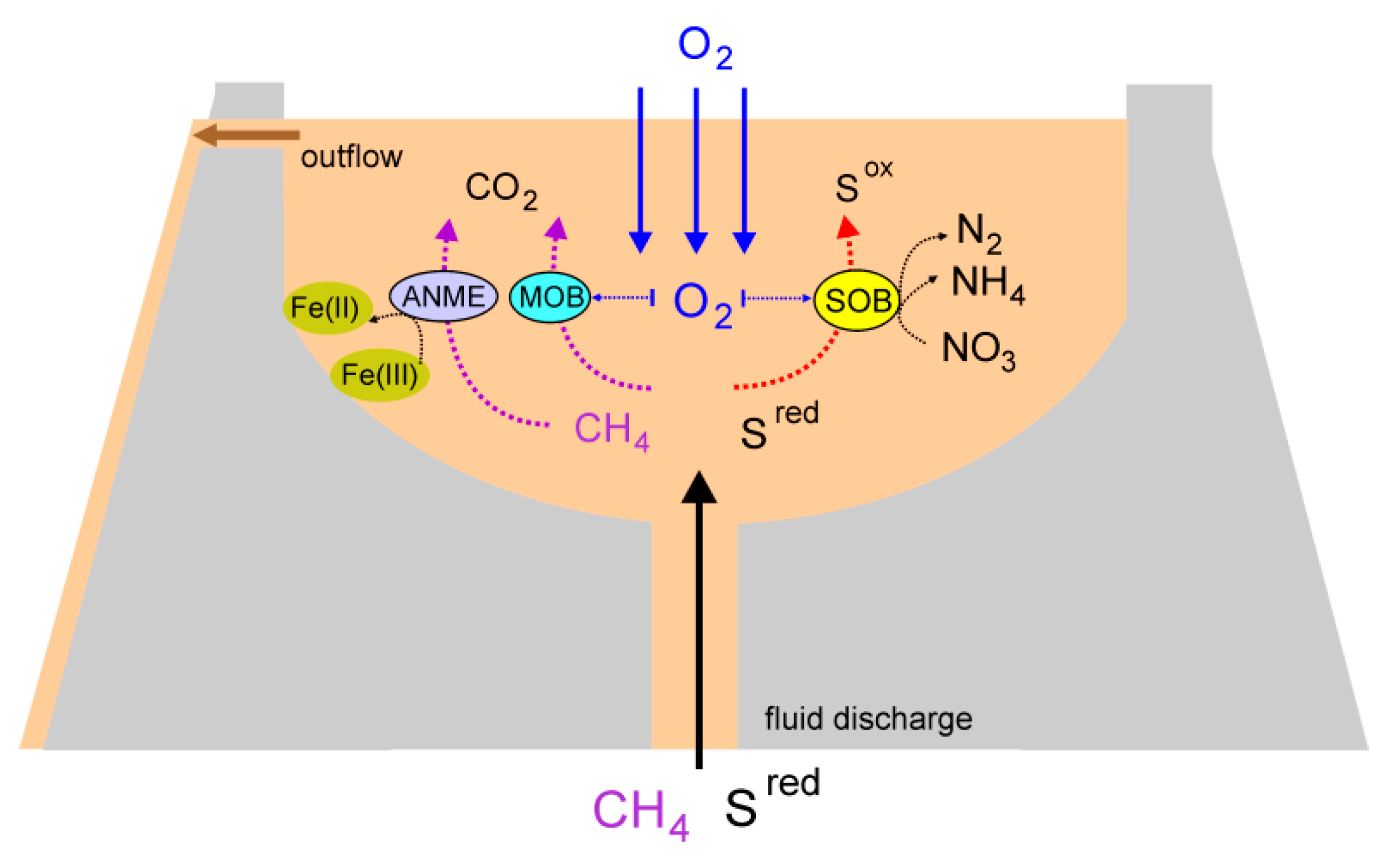
3.9. An overview of Microbial Processes in the Mud Volcano
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mazzini, A.; Etiope, G. Mud volcanism: An updated review. Earth Sci. Rev. 2017, 168, 81–112. [Google Scholar] [CrossRef]
- Dimitrov, L.I. Mud volcanoes—The most important pathway for degassing deeply buried sediments. Earth Sci. Rev. 2002, 59, 49–76. [Google Scholar] [CrossRef]
- Kopf, A.J. Significance of mud volcanism. Rev. Geophys. 2002, 40, 1–52. [Google Scholar] [CrossRef]
- Kioka, A.; Ashi, J. Episodic massive mud eruptions from submarine mud volcanoes examined through topographical signatures. Geophys. Res. Lett. 2015, 42, 8406–8414. [Google Scholar] [CrossRef]
- Mazzini, A.; Svensen, H.; Planke, S.; Guliyev, I.; Akhmanov, G.G.; Fallik, T.; Banks, D. When mud volcanoes sleep: Insight from seep geochemistry at the Dashgil mud volcano, Azerbaijan. Mar. Pet. Geol. 2009, 26, 1704–1715. [Google Scholar] [CrossRef]
- Etiope, G.; Lassey, K.R.; Klusman, R.W.; Boschi, E. Reappraisal of the fossil methane budget and related emission from geologic sources. Geophys. Res. Lett. 2008, 35, L09307. [Google Scholar] [CrossRef]
- Martinez, R.J.; Mills, H.J.; Story, S.; Sobecky, P.A. Prokaryotic diversity and metabolically active microbial populations in sediments from an active mud volcano in the Gulf of Mexico. Environ. Microbiol. 2006, 8, 1783–1796. [Google Scholar] [CrossRef]
- Niemann, H.; Lösekann, T.; de Beer, D.; Elvert, M.; Nadalig, T.; Knittel, K.; Amann, R.; Sauter, E.J.; Schlüter, M.; Klages, M.; et al. Novel microbial communities of the Haakon Mosby mud volcano and their role as methane sink. Nature 2006, 443, 854–858. [Google Scholar] [CrossRef]
- Lösekann, T.; Knittel, K.; Nadalig, T.; Fuchs, B.; Niemann, H.; Boetius, A.; Amann, R. Diversity and abundance of aerobic and anaerobic methane oxidizers at the Haakon Mosby mud volcano, Barents Sea. Appl. Environ. Microbiol. 2007, 73, 3348–3362. [Google Scholar] [CrossRef]
- Klasek, S.A.; Torres, M.E.; Loher, M.; Bohrmann, G.; Pape, T.; Colwell, F.S. Deep-Sourced Fluids from a Convergent Margin Host Distinct Subseafloor Microbial Communities That Change Upon Mud Flow Expulsion. Front. Microbiol. 2019, 10, 1436. [Google Scholar] [CrossRef]
- Coelho, F.J.; Louvado, A.; Domingues, P.M.; Cleary, D.F.; Ferreira, M.; Almeida, A.; Cunha, M.R.; Cunha, Â.; Gomes, N.C. Integrated analysis of bacterial and microeukaryotic communities from differentially active mud volcanoes in the Gulf of Cadiz. Sci. Rep. 2016, 6, 35272. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, T.; Toki, T.; Ijiri, A.; Morono, Y.; Machiyama, H.; Ashi, J.; Okamura, K.; Inagaki, F. Atribacteria from the Subseafloor Sedimentary Biosphere Disperse to the Hydrosphere through Submarine Mud Volcanoes. Front. Microbiol. 2017, 8, 1135. [Google Scholar] [CrossRef] [PubMed]
- Alain, K.; Holler, T.; Musat, F.; Elvert, M.; Treude, T.; Krüger, M. Microbiological investigation of methane- and hydrocarbon-discharging mud volcanoes in the Carpathian Mountains, Romania. Environ. Microbiol. 2006, 8, 574–590. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.W.; Chang, Y.H.; Tang, S.L.; Tseng, C.H.; Chiang, P.W.; Chang, K.T.; Sun, C.H.; Chen, Y.G.; Kuo, H.C.; Wang, C.H.; et al. Metabolic stratification driven by surface and subsurface interactions in a terrestrial mud volcano. ISME J. 2012, 6, 2280–2290. [Google Scholar] [CrossRef]
- Wrede, C.; Brady, S.; Rockstroh, S.; Dreier, A.; Kokoschka, S.; Heinzelmann, S.M.; Heller, C.; Reitner, J.; Taviani, M.; Daniel, R.; et al. Aerobic and anaerobic methane oxidation in terrestrial mud volcanoes in the Northern Apennines. Sediment. Geol. 2012, 263–264, 210–219. [Google Scholar] [CrossRef]
- Lin, Y.T.; Tu, T.H.; Wei, C.L.; Rumble, D.; Lin, L.H.; Wang, P.L. Steep redox gradient and biogeochemical cycling driven by deeply sourced fluids and gases in a terrestrial mud volcano. FEMS Microbiol. Ecol. 2018, 94. [Google Scholar] [CrossRef]
- Ren, G.; Ma, A.; Zhang, Y.; Deng, Y.; Zheng, G.; Zhuang, X.; Zhuang, G.; Fortin, D. Electron acceptors for anaerobic oxidation of methane drive microbial community structure and diversity in mud volcanoes. Environ. Microbiol. 2018, 20, 2370–2385. [Google Scholar] [CrossRef]
- Tu, T.H.; Wu, L.W.; Lin, Y.S.; Imachi, H.; Lin, L.H.; Wang, P.L. Microbial Community Composition and Functional Capacity in a Terrestrial Ferruginous, Sulfate-Depleted Mud Volcano. Front. Microbiol. 2017, 8, 2137. [Google Scholar] [CrossRef]
- Wang, P.L.; Chiu, Y.P.; Cheng, T.W.; Chang, Y.H.; Tu, W.X.; Lin, L.H. Spatial variations of community structures and methane cycling across a transect of Lei-Gong-Hou mud volcanoes in eastern Taiwan. Front. Microbiol. 2014, 5, 121. [Google Scholar] [CrossRef]
- Schulze-Makuch, D.; Haque, S.; Beckles, D.; Schmitt-Kopplin, P.; Harir, M.; Schneider, B.; Stumpp, C.; Wagner, D.A. A chemical and microbial characterization of selected mud volcanoes in Trinidad reveals pathogens introduced by surface water and rain water. Sci. Total Environ. 2020, 707, 136087. [Google Scholar] [CrossRef]
- Boetius, A.; Ravenschlag, K.; Schubert, C.J.; Rickert, D.; Widdel, F.; Gieseke, A.; Amann, R.; Jørgensen, B.B.; Witte, U.; Pfannkuche, O. A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature 2000, 407, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Raghoebarsing, A.A.; Pol, A.; van de Pas-Schoonen, K.T.; Smolders, A.J.; Ettwig, K.F.; Rijpstra, W.I.; Schouten, S.; Damsté, J.S.; Op den Camp, H.J.; Jetten, M.S.; et al. A microbial consortium couples anaerobic methane oxidation to denitrification. Nature 2006, 440, 918–921. [Google Scholar] [CrossRef] [PubMed]
- Beal, E.J.; House, C.H.; Orphan, V.J. Manganese- and iron-dependent marine methane oxidation. Science 2009, 325, 184–187. [Google Scholar] [CrossRef]
- Ettwig, K.F.; Butler, M.K.; Le Paslier, D.; Pelletier, E.; Mangenot, S.; Kuypers, M.M.; Schreiber, F.; Dutilh, B.E.; Zedelius, J.; de Beer, D.; et al. Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 2010, 464, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Akhmetjanov, A.; Akhmanov, G.; Krylov, O.; Basov, E.; Kozlova, E.; Stadnitskaya, A. Mud volcanoes of the Kerch Peninsula: General review. MarinF (UNESCO) 1996, 100, 23–24. [Google Scholar]
- Frey, B.; Rime, T.; Phillips, M.; Stierli, B.; Hajdas, I.; Widmer, F.; Hartmann, M. Microbial diversity in European alpine permafrost and active layers. FEMS Microbiol. Ecol. 2016, 92, fiw018. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Sickle v.1.33. Available online: https://github.com/najoshi/sickle (accessed on 30 August 2020).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Alneberg, J.; Bjarnason, B.S.; de Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Lahti, L.; Loman, N.J.; Andersson, A.F.; Quince, C. Binning metagenomic contigs by coverage and composition. Nat. Methods 2014, 11, 1144–1146. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015, 3, e1165. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef]
- Rodriguez-R, L.M.; Konstantinidis, K.T. The enveomics collection: A toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Prepr. 2016, 4, e1900v1. [Google Scholar]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Inagaki, F.; Takai, K.; Kobayashi, H.; Nealson, K.H.; Horikoshi, K. Sulfurimonas autotrophica gen. nov., sp. nov., a novel sulfur-oxidizing epsilon-proteobacterium isolated from hydrothermal sediments in the Mid-Okinawa Trough. Int. J. Syst. Evol. Microbiol. 2003, 53 Pt 6, 1801–1805. [Google Scholar] [CrossRef]
- Han, Y.; Perner, M. The globally widespread genus Sulfurimonas: Versatile energy metabolisms and adaptations to redox clines. Front. Microbiol. 2015, 6, 989. [Google Scholar] [CrossRef] [PubMed]
- van der Stel, A.X.; Wösten, M.M.S.M. Regulation of Respiratory Pathways in Campylobacterota: A Review. Front. Microbiol. 2019, 10, 1719. [Google Scholar] [CrossRef] [PubMed]
- Brinkhoff, T.; Muyzer, G. Increased species diversity and extended habitat range of sulfur-oxidizing Thiomicrospira spp. Appl. Environ. Microbiol. 1997, 63, 3789–3796. [Google Scholar] [CrossRef]
- Sorokin, D.Y.; Tourova, T.P.; Kolganova, T.V.; Sjollema, K.A.; Kuenen, J.G. Thioalkalispira microaerophila gen. nov., sp. nov., a novel lithoautotrophic, sulfur-oxidizing bacterium from a soda lake. Int. J. Syst. Evol. Microbiol 2002, 52 Pt 6, 2175–2182. [Google Scholar]
- Tavormina, P.L.; Hatzenpichler, R.; McGlynn, S.; Chadwick, G.; Dawson, K.S.; Connon, S.A.; Orphan, V.J. Methyloprofundus sedimenti gen. nov., sp. nov., an obligate methanotroph from ocean sediment belonging to the ‘deep sea-1’ clade of marine methanotrophs. Int. J. Syst. Evol. Microbiol. 2015, 65 Pt 1, 251–259. [Google Scholar] [CrossRef]
- Zhang, W.; Xue, Y.; Ma, Y.; Grant, W.D.; Ventosa, A.; Zhou, P. Marinospirillum alkaliphilum sp. nov., a new alkaliphilic helical bacterium from Haoji soda lake in Inner Mongolia Autonomous Region of China. Extremophiles 2002, 6, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Meng, X.; Du, Z.Z.; Du, Z.J.; Mu, D.S. Marinospirillum perlucidum sp. nov., a novel helical bacterium isolated from a sea cucumber culture pond. Int. J. Syst. Evol. Microbiol. 2020, 70, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhong, X.C.; Xu, W.; Lu, D.C.; Zhao, J.X.; Du, Z.J. Marinobacter vulgaris sp. nov., a moderately halophilic bacterium isolated from a marine solar saltern. Int. J. Syst. Evol. Microbiol. 2020, 70, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Lee, J.S.; Stevens, D.A. Microbiology and epidemiology of Halomonas species. Future Microbiol. 2013, 8, 1559–1573. [Google Scholar] [CrossRef]
- Sievert, S.M.; Scott, K.M.; Klotz, M.G.; Chain, P.S.; Hauser, L.J.; Hemp, J.; Hügler, M.; Land, M.; Lapidus, A.; Larimer, F.W.; et al. Genome of the epsilonproteobacterial chemolithoautotroph Sulfurimonas denitrificans. Appl. Environ. Microbiol. 2008, 74, 1145–1156. [Google Scholar] [CrossRef]
- Lahme, S.; Callbeck, C.M.; Eland, L.E.; Wipat, A.; Enning, D.; Head, I.M.; Hubert, C.R.J. Comparison of sulfide-oxidizing Sulfurimonas strains reveals a new mode of thiosulfate formation in subsurface environments. Environ. Microbiol. 2020, 22, 1784–1800. [Google Scholar] [CrossRef] [PubMed]
- Preisig, O.; Zufferey, R.; Thöny-Meyer, L.; Appleby, C.A.; Hennecke, H. A high-affinity cbb3-type cytochrome oxidase terminates the symbiosis-specific respiratory chain of Bradyrhizobium japonicum. J. Bacteriol. 1996, 178, 1532–1538. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.E.; Marshall, C.W.; May, H.D.; Norman, R.S. Comparative Genomic Analysis of Sulfurospirillum cavolei MES Reconstructed from the Metagenome of an Electrosynthetic Microbiome. PLoS ONE 2016, 11, e0151214. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Squier, T.C.; Zachara, J.M.; Fredrickson, J.K. Respiration of metal (hydr)oxides by Shewanella and Geobacter: A key role for multihaem c-type cytochromes. Mol. Microbiol. 2007, 65, 12–20. [Google Scholar] [CrossRef]
- Goris, T.; Schubert, T.; Gadkari, J.; Wubet, T.; Tarkka, M.; Buscot, F.; Adrian, L.; Diekert, G. Insights into organohalide respiration and the versatile catabolism of Sulfurospirillum multivorans gained from comparative genomics and physiological studies. Environ. Microbiol. 2014, 16, 3562–3580. [Google Scholar] [CrossRef]
- Scott, K.M.; Sievert, S.M.; Abril, F.N.; Ball, L.A.; Barrett, C.J.; Blake, R.A.; Boller, A.J.; Chain, P.S.; Clark, J.A.; Davis, C.R.; et al. The genome of deep-sea vent chemolithoautotroph Thiomicrospira crunogena XCL-2. PLoS Biol. 2006, 4, e383. [Google Scholar] [CrossRef]
- Watsuji, T.O.; Hada, E.; Miyazaki, M.; Ichimura, M.; Takai, K. Thiomicrospira hydrogeniphila sp. nov., an aerobic, hydrogen- and sulfur-oxidizing chemolithoautotroph isolated from a seawater tank containing a block of beef tallow. Int. J. Syst. Evol. Microbiol. 2016, 66, 3688–3693. [Google Scholar] [CrossRef]
- Henry, E.A.; Devereux, R.; Maki, J.S.; Gilmour, C.C.; Woese, C.R.; Mandelco, L.; Schauder, R.; Remsen, C.C.; Mitchell, R. Characterization of a new thermophilic sulfate-reducing bacterium Thermodesulfovibrio yellowstonii, gen. nov. and sp. nov.: Its phylogenetic relationship to Thermodesulfobacterium commune and their origins deep within the bacterial domain. Arch. Microbiol. 1994, 161, 62–69. [Google Scholar] [CrossRef]
- Chivian, D.; Brodie, E.L.; Alm, E.J.; Culley, D.E.; Dehal, P.S.; DeSantis, T.Z.; Gihring, T.M.; Lapidus, A.; Lin, L.H.; Lowry, S.R.; et al. Environmental genomics reveals a single-species ecosystem deep within Earth. Science 2008, 322, 275–278. [Google Scholar] [CrossRef]
- Karnachuk, O.V.; Frank, Y.A.; Lukina, A.P.; Kadnikov, V.V.; Beletsky, A.V.; Mardanov, A.V.; Ravin, N.V. Domestication of previously uncultivated Candidatus Desulforudis audaxviator from a deep aquifer in Siberia sheds light on its physiology and evolution. ISME J. 2019, 13, 1947–1959. [Google Scholar] [CrossRef]
- Jungbluth, S.P.; Glavina Del Rio, T.; Tringe, S.G.; Stepanauskas, R.; Rappé, M.S. Genomic comparisons of a bacterial lineage that inhabits both marine and terrestrial deep subsurface systems. PeerJ 2017, 5, e3134. [Google Scholar] [CrossRef]
- Haroon, M.F.; Hu, S.; Shi, Y.; Imelfort, M.; Keller, J.; Hugenholtz, P.; Yuan, Z.; Tyson, G.W. Anaerobic oxidation of methane coupled to nitrate reduction in a novel archaeal lineage. Nature 2013, 500, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Ettwig, K.F.; Zhu, B.; Speth, D.; Keltjens, J.T.; Jetten, M.S.M.; Kartal, B. Archaea catalyze iron-dependent anaerobic oxidation of methane. Proc. Natl. Acad. Sci. USA 2016, 113, 12792–12796. [Google Scholar] [CrossRef]
- Leu, A.O.; Cai, C.; McIlroy, S.J.; Southam, G.; Orphan, V.J.; Yuan, Z.; Hu, S.; Tyson, G.W. Anaerobic methane oxidation coupled to manganese reduction by members of the Methanoperedenaceae. ISME J. 2020, 14, 1030–1041. [Google Scholar] [CrossRef]
- Hernsdorf, A.W.; Amano, Y.; Miyakawa, K.; Ise, K.; Suzuki, Y.; Anantharaman, K.; Probst, A.; Burstein, D.; Thomas, B.C.; Banfield, J.F. Potential for microbial H2 and metal transformations associated with novel bacteria and archaea in deep terrestrial subsurface sediments. ISME J. 2017, 11, 1915–1929. [Google Scholar] [CrossRef] [PubMed]
- Arshad, A.; Speth, D.R.; de Graaf, R.M.; Op den Camp, H.J.; Jetten, M.S.; Welte, C.U. A Metagenomics-Based Metabolic Model of Nitrate-Dependent Anaerobic Oxidation of Methane by Methanoperedens-Like Archaea. Front. Microbiol. 2015, 6, 1423. [Google Scholar] [CrossRef]
- Cai, C.; Leu, A.O.; Xie, G.J.; Guo, J.; Feng, Y.; Zhao, J.X.; Tyson, G.W.; Yuan, Z.; Hu, S. A methanotrophic archaeon couples anaerobic oxidation of methane to Fe(III) reduction. ISME J. 2018, 12, 1929–1939. [Google Scholar] [CrossRef] [PubMed]
- Krukenberg, V.; Riedel, D.; Gruber-Vodicka, H.R.; Buttigieg, P.L.; Tegetmeyer, H.E.; Boetius, A.; Wegener, G. Gene expression and ultrastructure of meso- and thermophilic methanotrophic consortia. Environ. Microbiol. 2018, 20, 1651–1666. [Google Scholar] [CrossRef]
- Michaelis, W.; Seifert, R.; Nauhaus, K.; Treude, T.; Thiel, V.; Blumenberg, M.; Knittel, K.; Gieseke, A.; Peterknecht, K.; Pape, T.; et al. Microbial reefs in the Black Sea fueled by anaerobic oxidation of methane. Science 2002, 297, 1013–1015. [Google Scholar] [CrossRef]
- Pereira, I.A.C.; Haveman, S.A.; Voordouw, G. Biochemical, genetic and genomic characterization of anaerobic electron transport pathways in sulphate-reducing delta-proteobacteria. In Sulphate–Reducing Bacteria: Environmental and Engineered Systems; Barton, L.L., Hamilton, W.A.A., Eds.; Cambridge University Press: Cambridge, UK, 2007; pp. 215–240. [Google Scholar]
- Lazar, C.S.; Baker, B.J.; Seitz, K.; Hyde, A.S.; Dick, G.J.; Hinrichs, K.U.; Teske, A.P. Genomic evidence for distinct carbon substrate preferences and ecological niches of Bathyarchaeota in estuarine sediments. Environ. Microbiol. 2016, 18, 1200–1211. [Google Scholar] [CrossRef]
- He, Y.; Li, M.; Perumal, V.; Feng, X.; Fang, J.; Xie, J.; Sievert, S.M.; Wang, F. Genomic and enzymatic evidence for acetogenesis among multiple lineages of the archaeal phylum Bathyarchaeota widespread in marine sediments. Nat. Microbiol. 2016, 1, 16035. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.N.; Parks, D.H.; Chadwick, G.L.; Robbins, S.J.; Orphan, V.J.; Golding, S.D.; Tyson, G.W. Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genomecentric metagenomics. Science 2015, 350, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Borrel, G.; Adam, P.S.; McKay, L.J.; Chen, L.X.; Sierra-Garcıa, I.N.; Sieber, C.M.K.; Letourneur, Q.; Ghozlane, A.; Andersen, G.L.; Li, W.J.; et al. Wide diversity of methane and short-chain alkane metabolisms in uncultured archaea. Nat. Microbiol. 2019, 4, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Buckel, W.; Thauer, R.K. Energy conservation via electron bifurcating ferredoxin reduction and proton/Na(+) translocating ferredoxin oxidation. Biochim. Biophys. Acta 2013, 1827, 94–113. [Google Scholar] [CrossRef]
- Thomson, J.; Higgs, N.C.; Croudace, I.W.; Colley, S.; Hydes, D.J. Redox zonation of elements at an oxic/post-oxic boundary in deep-sea sediments. Geochim. Cosmochim. Acta 1993, 57, 579–595. [Google Scholar] [CrossRef]
- Frank, Y.A.; Kadnikov, V.V.; Lukina, A.P.; Banks, D.; Beletsky, A.V.; Mardanov, A.V.; Sen’kina, E.I.; Avakyan, M.R.; Karnachuk, O.V.; Ravin, N.V. Characterization and Genome Analysis of the First Facultatively Alkaliphilic Thermodesulfovibrio Isolated from the Deep Terrestrial Subsurface. Front. Microbiol. 2016, 7, 2000. [Google Scholar] [CrossRef]
- Moser, D.P.; Gihring, T.M.; Brockman, F.J.; Fredrickson, J.K.; Balkwill, D.L.; Dollhopf, M.E.; Lollar, B.S.; Pratt, L.M.; Boice, E.; Southam, G.; et al. Desulfotomaculum and Methanobacterium spp. dominate a 4- to 5-kilometer-deep fault. Appl. Environ. Microbiol. 2005, 71, 8773–8783. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter (Unit) | Value | Parameter (Unit) | Value |
|---|---|---|---|
| Temperature (°C) | 16 | Sr (mg L−1) | 2.4 |
| pH | 7.43 | Li (mg L−1) | 1.9 |
| Eh, mV | −123 | Ba (mg L−1) | 1.0 |
| Na (mg L−1) | 4165 | Fe (mg L−1) | 0.4 |
| B (mg L−1) | 337 | As (mg L−1) | 0.06 |
| K (mg L−1) | 26 | Cl− (mg L−1) | 1865 |
| Ca (mg L−1) | 58 | NO3− (mg L−1) | 12.0 |
| Mg (mg L−1) | 23 | PO43− (mg L−1) | 8.3 |
| Si (mg L−1) | 17 | SO42− (mg L−1) | 6.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mardanov, A.V.; Kadnikov, V.V.; Beletsky, A.V.; Ravin, N.V. Sulfur and Methane-Oxidizing Microbial Community in a Terrestrial Mud Volcano Revealed by Metagenomics. Microorganisms 2020, 8, 1333. https://doi.org/10.3390/microorganisms8091333
Mardanov AV, Kadnikov VV, Beletsky AV, Ravin NV. Sulfur and Methane-Oxidizing Microbial Community in a Terrestrial Mud Volcano Revealed by Metagenomics. Microorganisms. 2020; 8(9):1333. https://doi.org/10.3390/microorganisms8091333
Chicago/Turabian StyleMardanov, Andrey V., Vitaly V. Kadnikov, Alexey V. Beletsky, and Nikolai V. Ravin. 2020. "Sulfur and Methane-Oxidizing Microbial Community in a Terrestrial Mud Volcano Revealed by Metagenomics" Microorganisms 8, no. 9: 1333. https://doi.org/10.3390/microorganisms8091333
APA StyleMardanov, A. V., Kadnikov, V. V., Beletsky, A. V., & Ravin, N. V. (2020). Sulfur and Methane-Oxidizing Microbial Community in a Terrestrial Mud Volcano Revealed by Metagenomics. Microorganisms, 8(9), 1333. https://doi.org/10.3390/microorganisms8091333