Composition and Metabolic Functions of the Microbiome in Fermented Grain during Light-Flavor Baijiu Fermentation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Microbiological and Physicochemical Analysis
2.3. Polar Non-Volatile Metabolite Analysis
2.4. Volatile Compound Analysis
2.5. DNA Extraction and Shotgun Metagenomic Sequencing
2.6. Sequence Assembly, Taxonomic and Functional Annotation
2.7. Statistical Analyses
3. Results
3.1. General Investigation of Microbiota and Physicochemical Properties
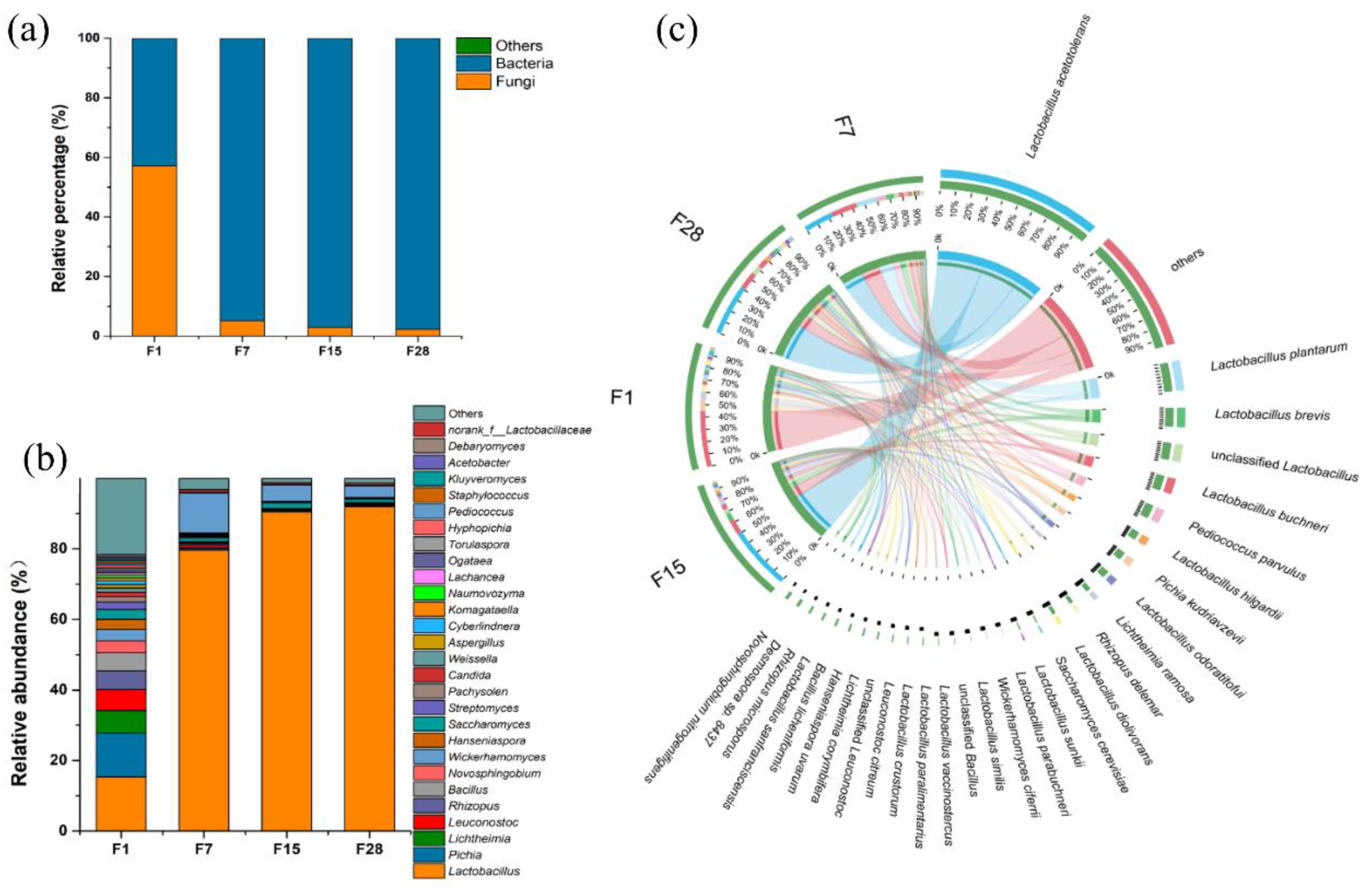
3.2. Microbial Composition and Dynamics Based on Shotgun Metagenomics
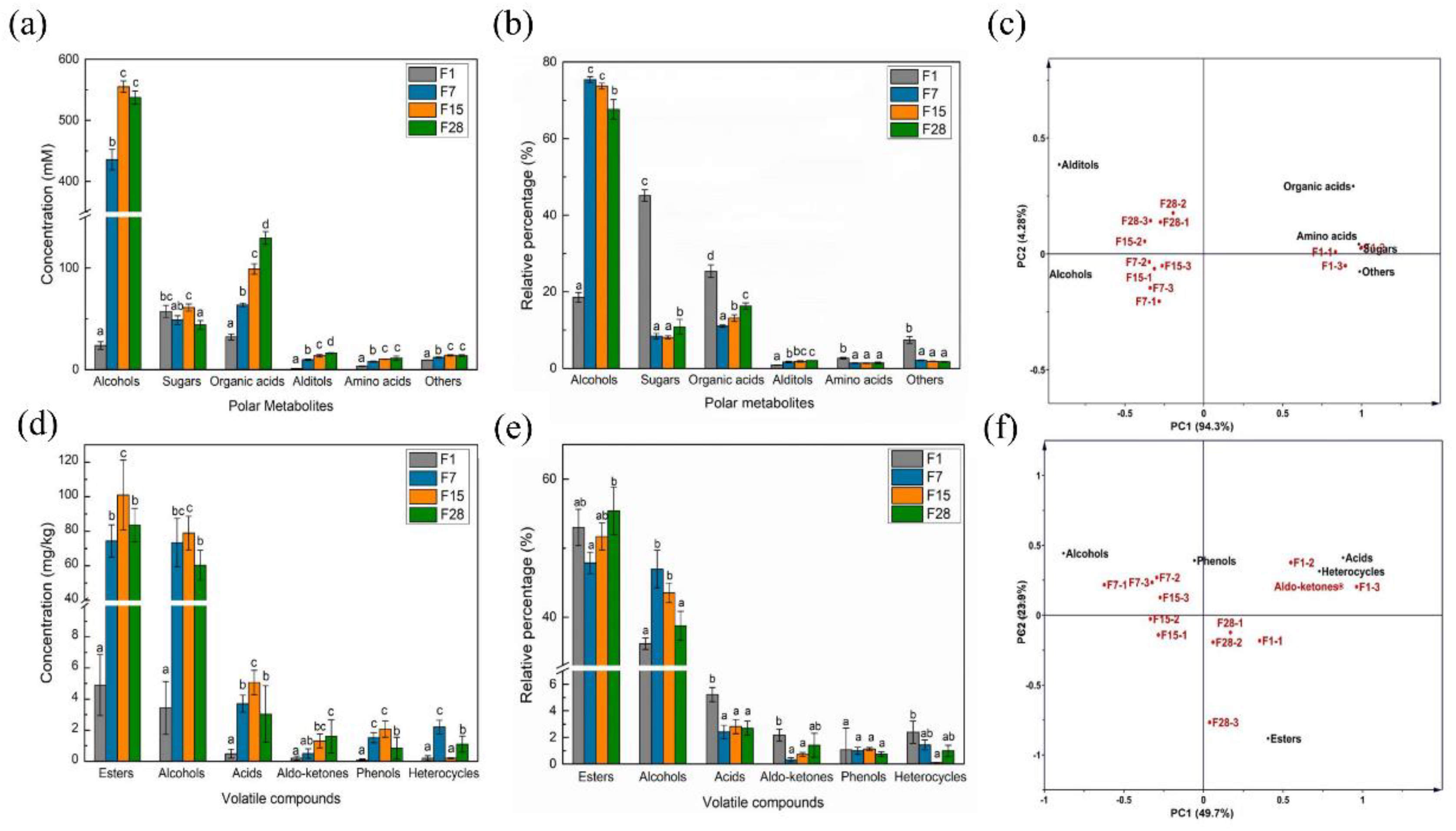
3.3. Metabolic Succession during Light-Flavor Baijiu Fermentation
3.4. Correlations of Microorganisms and Metabolites in Fermented Grain
3.5. Functional Gene Categories of Microbiota in Fermented Grain
3.6. Metabolic Potential of Microbiota in Fermented Grain
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jin, G.; Zhu, Y.; Xu, Y. Mystery behind Chinese liquor fermentation. Trends Food Sci. Technol. 2017, 63, 18–28. [Google Scholar] [CrossRef]
- Zhao, M.; Su, X.Q.; Nian, B.; Chen, L.J.; Zhang, D.L.; Duan, S.M.; Wang, L.Y.; Shi, X.Y.; Jiang, B.; Jiang, W.W. Integrated meta-omics approaches to understand the microbiome of spontaneous fermentation of traditional Chinese Pu-erh tea. MSystems 2019, 4, e00680-19. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, B.E.; Dutton, R.J. Fermented foods as experimentally tractable microbial ecosystems. Cell 2015, 161, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Blasche, S.; Kim, Y.; Mars, R.; Kafkia, E.; Maansson, M.; Machado, D.; Teusink, B.; Nielsen, J.; Benes, V.; Neves, R. Emergence of stable coexistence in a complex microbial community through metabolic cooperation and spatio-temporal niche partitioning. bioRxiv 2019, 541870. [Google Scholar] [CrossRef]
- Cosetta, C.M.; Wolfe, B.E. Deconstructing and reconstructing cheese rind microbiomes for experiments in microbial ecology and evolution. Curr. Protoc. Microbiol. 2020, 56, e95. [Google Scholar] [CrossRef]
- Pang, X.N.; Han, B.Z.; Huang, X.N.; Zhang, X.; Hou, L.F.; Cao, M.; Gao, L.J.; Hu, G.H.; Chen, J.Y. Effect of the environment microbiota on the flavour of light-flavour Baijiu during spontaneous fermentation. Sci. Rep. 2018, 8, 3396. [Google Scholar] [CrossRef]
- Zheng, X.W.; Tabrizi, M.R.; Nout, M.R.; Han, B.Z. Daqu-a traditional Chinese liquor fermentation starter. J. Inst. Brew. 2011, 117, 82–90. [Google Scholar] [CrossRef]
- Li, X.R.; Ma, E.B.; Yan, L.Z.; Meng, H.; Du, X.W.; Zhang, S.W.; Quan, Z.X. Bacterial and fungal diversity in the traditional Chinese liquor fermentation process. Int. J. Food Microbiol. 2011, 146, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Du, H.; Zhang, Y.; Xu, Y. Environmental microbiota drives microbial succession and metabolic profiles during Chinese liquor fermentation. Appl. Environ. Microbiol. 2018, 84, e02369-17. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xu, Y. Microbial succession and metabolite changes during the fermentation of Chinese light aroma-style liquor. J. Inst. Brew. 2019, 125, 162–170. [Google Scholar]
- Zheng, X.W.; Yan, Z.; Han, B.Z.; Zwietering, M.H.; Samson, R.A.; Boekhout, T.; Robert Nout, M.J. Complex microbiota of a Chinese “Fen” liquor fermentation starter (Fen-Daqu), revealed by culture-dependent and culture-independent methods. Food Microbiol. 2012, 31, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.W.; Yan, Z.; Nout, M.R.; Smid, E.J.; Zwietering, M.H.; Boekhout, T.; Han, J.S.; Han, B.Z. Microbiota dynamics related to environmental conditions during the fermentative production of Fen-Daqu, a Chinese industrial fermentation starter. Int. J. Food Microbiol. 2014, 182, 57–62. [Google Scholar] [CrossRef] [PubMed]
- De Roy, K.; Marzorati, M.; Van Den Abbeele, P.; Van De Wiele, T.; Boon, N. Synthetic microbial ecosystems: An exciting tool to understand and apply microbial communities. Environ. Microbiol. 2014, 16, 1472–1481. [Google Scholar] [CrossRef]
- Tan, J.; Zuniga, C.; Zengler, K. Unraveling interactions in microbial communities-from co-cultures to microbiomes. J. Microbiol. 2015, 53, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Sun, B.; Fu, Z.; Xia, Y.; Huang, M.; Xu, C.; Li, X. Analysis of physicochemical indices, volatile flavor components, and microbial community of a light-flavor Daqu. J. Am. Soc. Brew. Chem. 2018, 76, 209–218. [Google Scholar] [CrossRef]
- Ellegaard, K.M.; Engel, P. Genomic diversity landscape of the honey bee gut microbiota. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Tedersoo, L.; Anslan, S.; Bahram, M.; Põlme, S.; Riit, T.; Liiv, I.; Kõljalg, U.; Kisand, V.; Nilsson, R.H.; Hildebrand, F. Shotgun metagenomes and multiple primer pair-barcode combinations of amplicons reveal biases in metabarcoding analyses of fungi. MycoKeys 2015, 10, 1–43. [Google Scholar] [CrossRef]
- Verce, M.; De Vuyst, L.; Weckx, S. Shotgun metagenomics of a water kefir fermentation ecosystem reveals a novel Oenococcus species. Front. Microbiol. 2019, 10, 479. [Google Scholar] [CrossRef]
- De Filippo, C.; Ramazzotti, M.; Fontana, P.; Cavalieri, D. Bioinformatic approaches for functional annotation and pathway inference in metagenomics data. Brief. Bioinform. 2012, 13, 696–710. [Google Scholar] [CrossRef]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef]
- Ferrocino, I.; Bellio, A.; Giordano, M.; Macori, G.; Romano, A.; Rantsiou, K.; Decastelli, L.; Cocolin, L. Shotgun metagenomics and volatilome profile of the microbiota of fermented sausages. Appl. Environ. Microbiol. 2018, 84, e02120-17. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Feng, C.; Luo, X.; Yao, H.; Zhang, D.; Zhang, T. Revealing the influence of microbiota on the quality of Pu-erh tea during fermentation process by shotgun metagenomic and metabolomic analysis. Food Microbiol. 2018, 76, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, B.; Button, J.; Santarelli, M.; Dutton, R. Cheese rind communities provide tractable systems for in situ and in vitro studies of microbial diversity. Cell 2014, 158, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Zepeda, A.; Sanchez-Flores, A.; Baruch, M.Q. Metagenomic analysis of a Mexican ripened cheese reveals a unique complex microbiota. Food Microbiol. 2016, 57, 116–127. [Google Scholar] [CrossRef]
- Zheng, X.W.; Han, B.Z. Baijiu, Chinese liquor: History, classification and manufacture. J. Ethn. Foods 2016, 3, 19–25. [Google Scholar] [CrossRef]
- Le, V.D.; Zheng, X.W.; Chen, J.Y.; Han, B.Z. Characterization of volatile compounds in Fen-Daqu–a traditional Chinese liquor fermentation starter. J. Inst. Brew. 2012, 18, 107–113. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Li, R.; Li, Y.; Kristiansen, K.; Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar] [CrossRef]
- Lawson, C.E.; Wu, S.; Bhattacharjee, A.S.; Hamilton, J.J.; McMahon, K.D.; Goel, R.; Daniel, R. Metabolic network analysis reveals microbial community interactions in anammox granules. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.J.; Julien, P.; Kuhn, M.; von Mering, C.; Muller, J.; Doerks, T.; Bork, P. EggNOG: Automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 2007, 36 (Suppl. 1), D250–D254. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Rui, J.; Li, X.; Li, J.; Dong, L.; Huang, Q.; Huang, C.; Wang, Z.; Li, L.; Xuan, P.; et al. Bacterial community succession and metabolite changes during doubanjiang-meju fermentation, a Chinese traditional fermented broad bean (Vicia faba L.) paste. Food Chem. 2017, 218, 534–542. [Google Scholar] [CrossRef]
- Quan, J.; Cai, G.; Yang, M.; Zeng, Z.H.; Ding, R.R.; Wang, X.W.; Zhuang, Z.; Zhou, S.; Li, S.; Yang, H.; et al. Exploring the fecal microbial composition and metagenomic functional capacities associated with feed efficiency in commercial DLY pigs. Front. Microbiol. 2019, 10, 52. [Google Scholar] [CrossRef]
- Fan, G.; Du, Y.; Fu, Z.; Chen, M.; Wang, Z.; Liu, P.; Li, X. Characterisation of physicochemical properties, flavour components and microbial community in Chinese Guojing roasted sesame-like flavor Daqu. J. Inst. Brew. 2020, 126, 105–115. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Garrido-Oter, R.; Münch, P.C.; Weiman, A.; Dröge, J.; Pan, Y.; Mchardy, A.C.; Schulze-Lefert, P. Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe 2015, 17, 392–403. [Google Scholar] [CrossRef]
- Gan, S.-H.; Yang, F.; Sahu, S.K.; Luo, R.Y.; Liao, S.L.; Wang, H.Y.; Jin, T.; Wang, L.; Zhang, P.F.; Liu, X. Deciphering the composition and function profile of the microbial communities in Chinese Moutai liquor starters. Front. Microbiol. 2019, 10, 1540. [Google Scholar] [CrossRef]
- Wang, S.; Wu, Q.; Nie, Y.; Wu, J.; Xu, Y. Construction of synthetic microbiota for reproducible flavor compound metabolism in Chinese light-aroma-type liquor produced by solid-state fermentation. Appl. Environ. Microbiol. 2019, 85, e03090-18. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Zhang, X.J.; Zhao, L.P.; Xu, Y. Analysis and comparison of the bacterial community in fermented grains during the fermentation for two different styles of Chinese liquor. J. Ind. Microbiol. Biot. 2008, 35, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Du, H.; Zhang, Y.; Xu, Y. Unraveling core functional microbiota in traditional solid-state fermentation by high-throughput amplicons and meta-transcriptomics sequencing. Front. Microbiol. 2017, 8, 1294–1308. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wu, Q.; Xu, Y. Filamentous fungal diversity and community structure associated with the solid-state fermentation of Chinese Maotai-flavor liquor. Int. J. Food Microbiol. 2014, 179, 80–84. [Google Scholar] [CrossRef]
- Tomita, S.; Nakamura, T.; Okada, S. NMR-and GC/MS-based metabolomic characterization of sunki, an unsalted fermented pickle of turnip leaves. Food Chem. 2018, 258, 25–34. [Google Scholar] [CrossRef]
- Zang, X.; Liu, M.; Fan, Y.; Xu, J.; Xu, X.; Li, H. The structural and functional contributions of β-glucosidase-producing microbial communities to cellulose degradation in composting. Biotechnol. Biofuel. 2018, 11, 51. [Google Scholar] [CrossRef]
- Mateo, J.; Maicas, S. Application of non-Saccharomyces yeasts to wine-making process. Fermentation 2016, 2, 14. [Google Scholar] [CrossRef]
- Sanni, A.; Morlon-Guyot, J.; Guyot, J. New efficient amylase-producing strains of Lactobacillus plantarum and L. fermentum isolated from different Nigerian traditional fermented foods. Int. J. Food Microbiol. 2002, 72, 53–62. [Google Scholar] [CrossRef]
- Liu, S.Q. Practical implications of lactate and pyruvate metabolism by lactic acid bacteria in food and beverage fermentations. Int. J. Food Microbiol. 2003, 83, 115–131. [Google Scholar] [CrossRef]
- Liu, S.; Skinner-Nemec, K.A.; Leathers, T.D. Lactobacillus buchneri strain NRRL B-30929 converts a concentrated mixture of xylose and glucose into ethanol and other products. J. Ind. Microbiol. Biot. 2008, 35, 75–81. [Google Scholar] [CrossRef]
- Yang, X.; Teng, K.; Zhang, J.; Wang, F.; Zhang, T.; Ai, G.; Han, P.; Bai, F.; Zhong, J. Transcriptome responses of Lactobacillus acetotolerans F28 to a short and long term ethanol stress. Sci. Rep. 2017, 7, 2650. [Google Scholar] [CrossRef]
- Sood, S.; Sharma, A.; Sharma, N.; Kanwar, S. Carboxylesterases: Sources, characterization and broader applications. Insights Enzym. Res. 2016, 1, 1–11. [Google Scholar] [CrossRef]
- Liu, H.; Sun, B. Effect of fermentation processing on the flavor of Baijiu. J. Agric. Food Chem. 2018, 66, 5425–5432. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, M.K.; Wong, J.W.; Kumar, S.; Awasthi, S.K.; Wang, Q.; Wang, M.; Ren, X.; Zhao, J.; Chen, H.; Zhang, Z. Biodegradation of food waste using microbial cultures producing thermostable α-amylase and cellulase under different pH and temperature. Bioresour. Technol. 2018, 248, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Chi, Z.; Chi, Z.; Liu, G.; Wang, F.; Ju, L.; Zhang, T. Saccharomycopsis fibuligera and its applications in biotechnology. Biotechnol. Adv. 2009, 27, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Isono, N.; Hayakawa, H.; Usami, A.; Mishima, T.; Hisamatsu, M. A comparative study of ethanol production by Issatchenkia orientalis strains under stress conditions. J. Biosci. Bioeng. 2012, 113, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wu, Q.; Jiang, X.; Wang, Z.; Tang, J.; Xu, Y. Bacillus licheniformis affects the microbial community and metabolic profile in the spontaneous fermentation of Daqu starter for Chinese liquor making. Int. J. Food Microbiol. 2017, 250, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Carroll, E.; Trinh, T.N.; Son, H.; Lee, Y.W.; Seo, J.A. Comprehensive analysis of fungal diversity and enzyme activity in nuruk, a Korean fermenting starter, for acquiring useful fungi. J. Microbiol. 2017, 55, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Garcia, N.F.L.; Da Silva Santos, F.R.; Bocchini, D.A.; Da Paz, M.F.; Fonseca, G.G.; Leite, R.S.R. Catalytic properties of cellulases and hemicellulases produced by Lichtheimia ramosa: Potential for sugarcane bagasse saccharification. Ind. Crop. Prod. 2018, 122, 49–56. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, X.; Fan, Y.; Lu, T.; Kang, J.; Pang, X.; Han, B.; Chen, J. Composition and Metabolic Functions of the Microbiome in Fermented Grain during Light-Flavor Baijiu Fermentation. Microorganisms 2020, 8, 1281. https://doi.org/10.3390/microorganisms8091281
Huang X, Fan Y, Lu T, Kang J, Pang X, Han B, Chen J. Composition and Metabolic Functions of the Microbiome in Fermented Grain during Light-Flavor Baijiu Fermentation. Microorganisms. 2020; 8(9):1281. https://doi.org/10.3390/microorganisms8091281
Chicago/Turabian StyleHuang, Xiaoning, Yi Fan, Ting Lu, Jiamu Kang, Xiaona Pang, Beizhong Han, and Jingyu Chen. 2020. "Composition and Metabolic Functions of the Microbiome in Fermented Grain during Light-Flavor Baijiu Fermentation" Microorganisms 8, no. 9: 1281. https://doi.org/10.3390/microorganisms8091281
APA StyleHuang, X., Fan, Y., Lu, T., Kang, J., Pang, X., Han, B., & Chen, J. (2020). Composition and Metabolic Functions of the Microbiome in Fermented Grain during Light-Flavor Baijiu Fermentation. Microorganisms, 8(9), 1281. https://doi.org/10.3390/microorganisms8091281