Oral Primo-Colonizing Bacteria Modulate Inflammation and Gene Expression in Bronchial Epithelial Cells

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Media and Growth Conditions
2.2. Bacterial Strain Identification
2.3. Bacterial Strains Preparation for Challenge Assay
2.4. BEAS-2B Human Bronchial Epithelial Cell Challenge
2.5. Human Cytokine and Chemokine Profiling
2.6. RNA Extraction and RNA Cleanup
2.7. RNA Reverse Transcription and Fluorescent Labeling
2.8. Microarrays Analysis
2.9. LDH Cytotoxicity Assay
2.10. Data and Statistical Analysis
2.10.1. Human Cytokine and Chemokine Profiling
2.10.2. Microarray Data Processing
3. Results
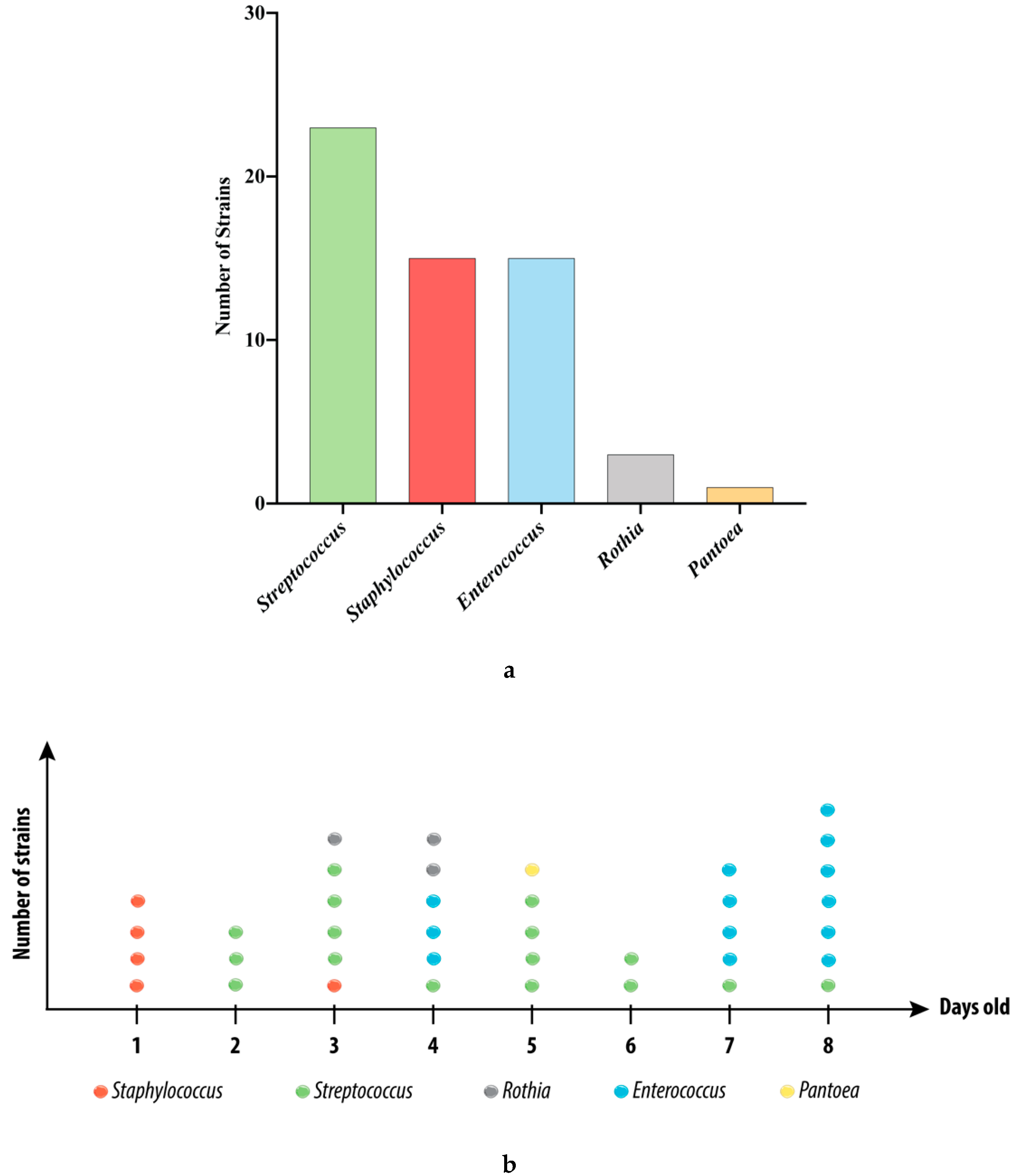
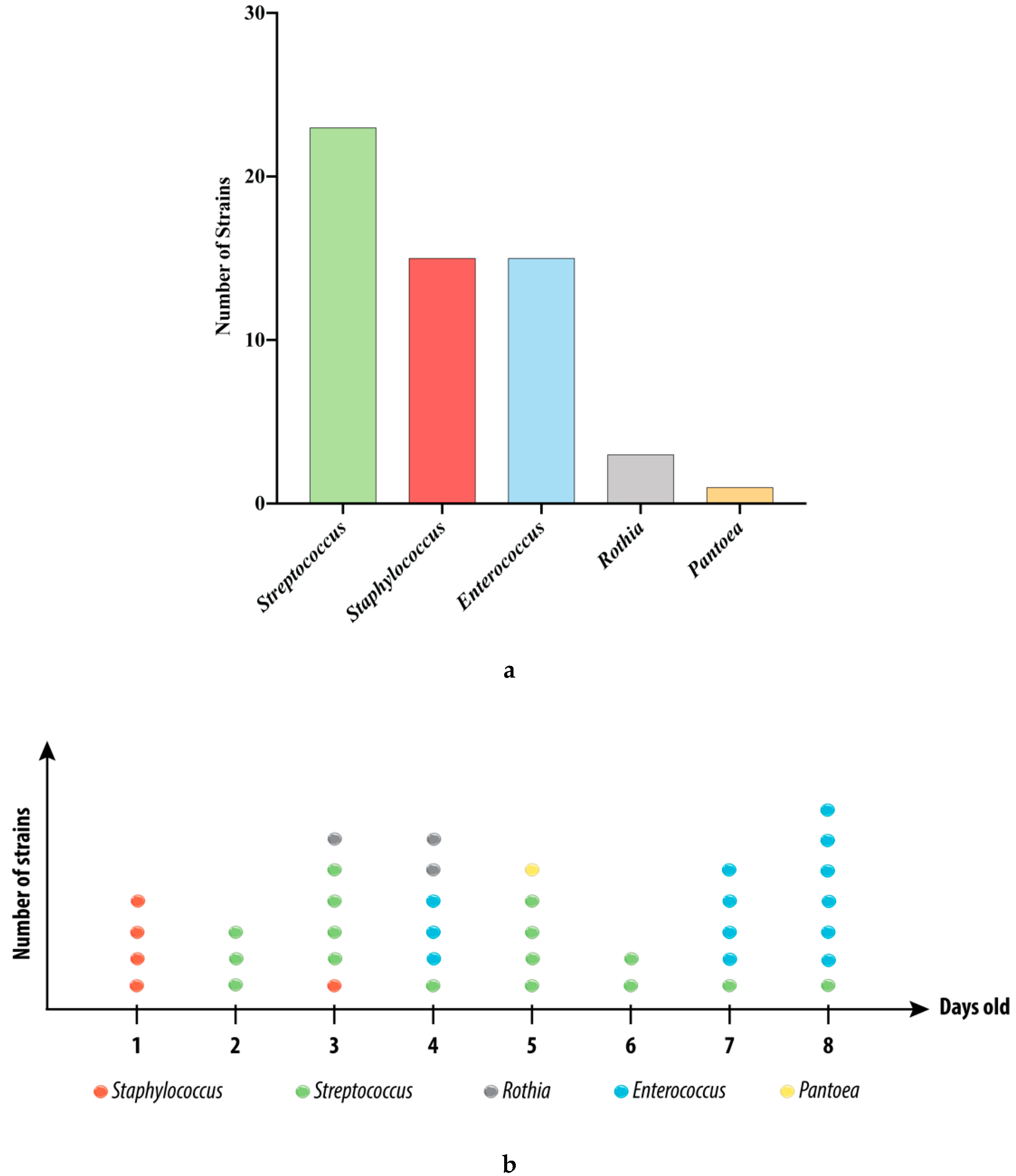
3.1. Strains Isolated from the Swabs
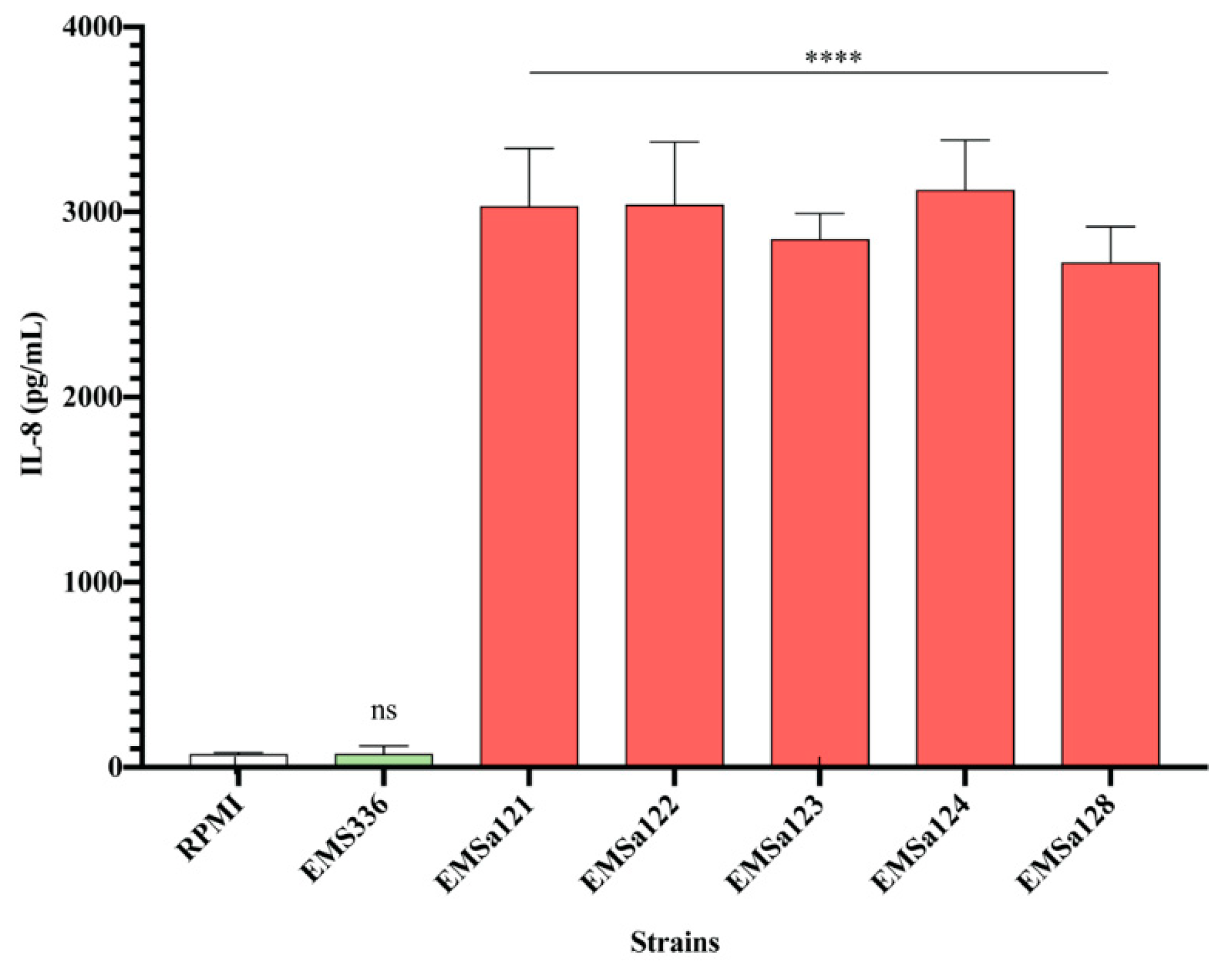
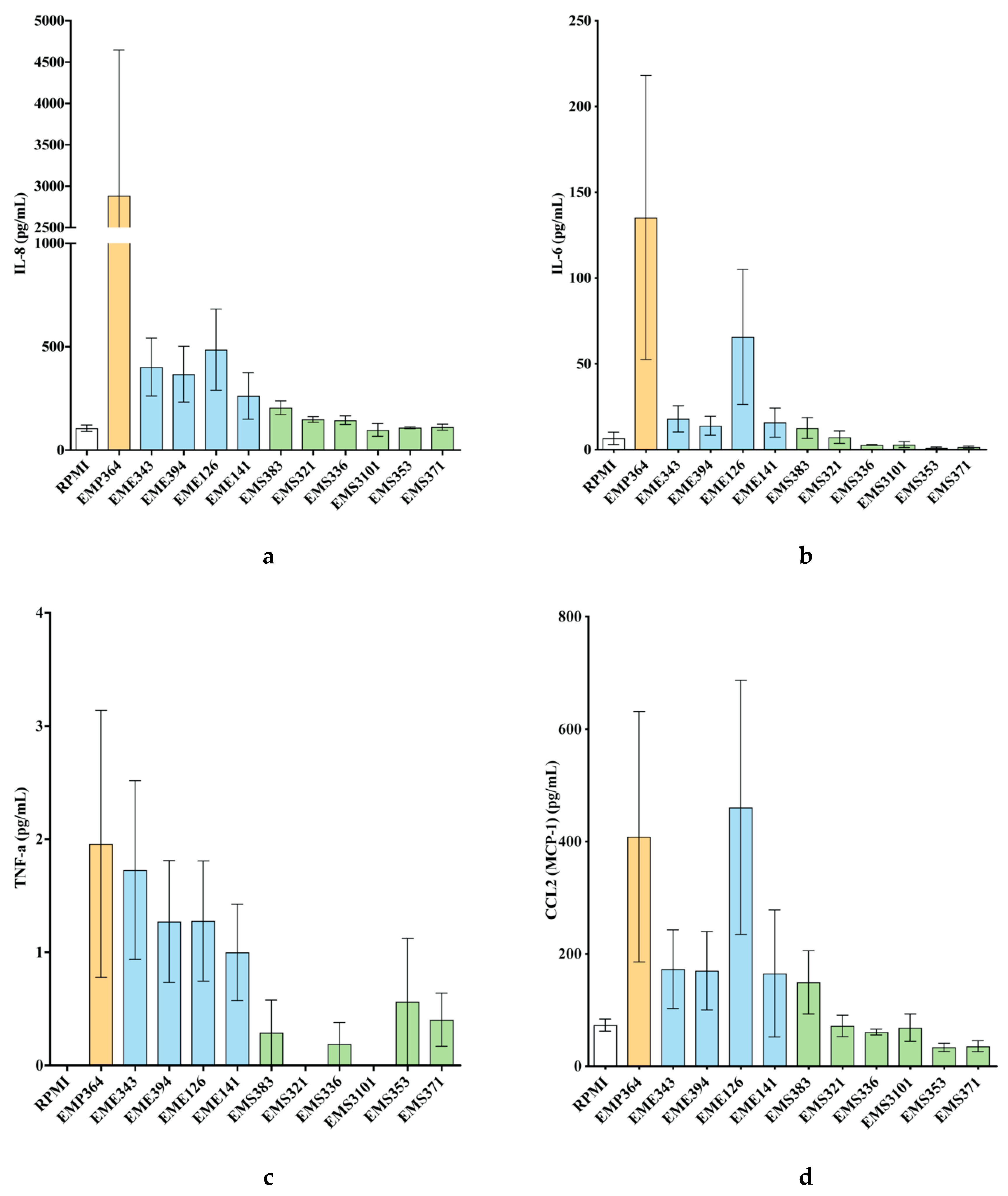
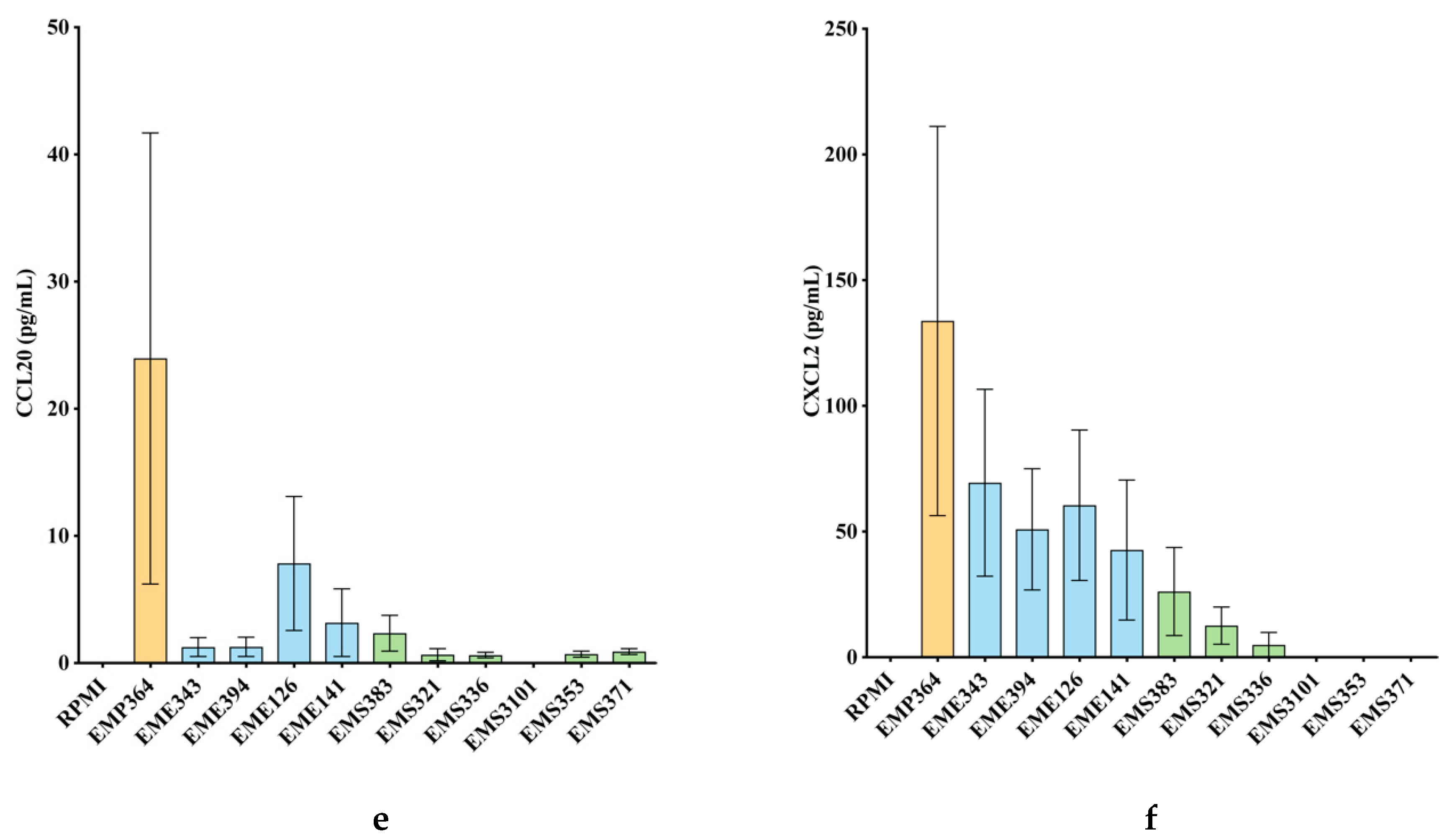
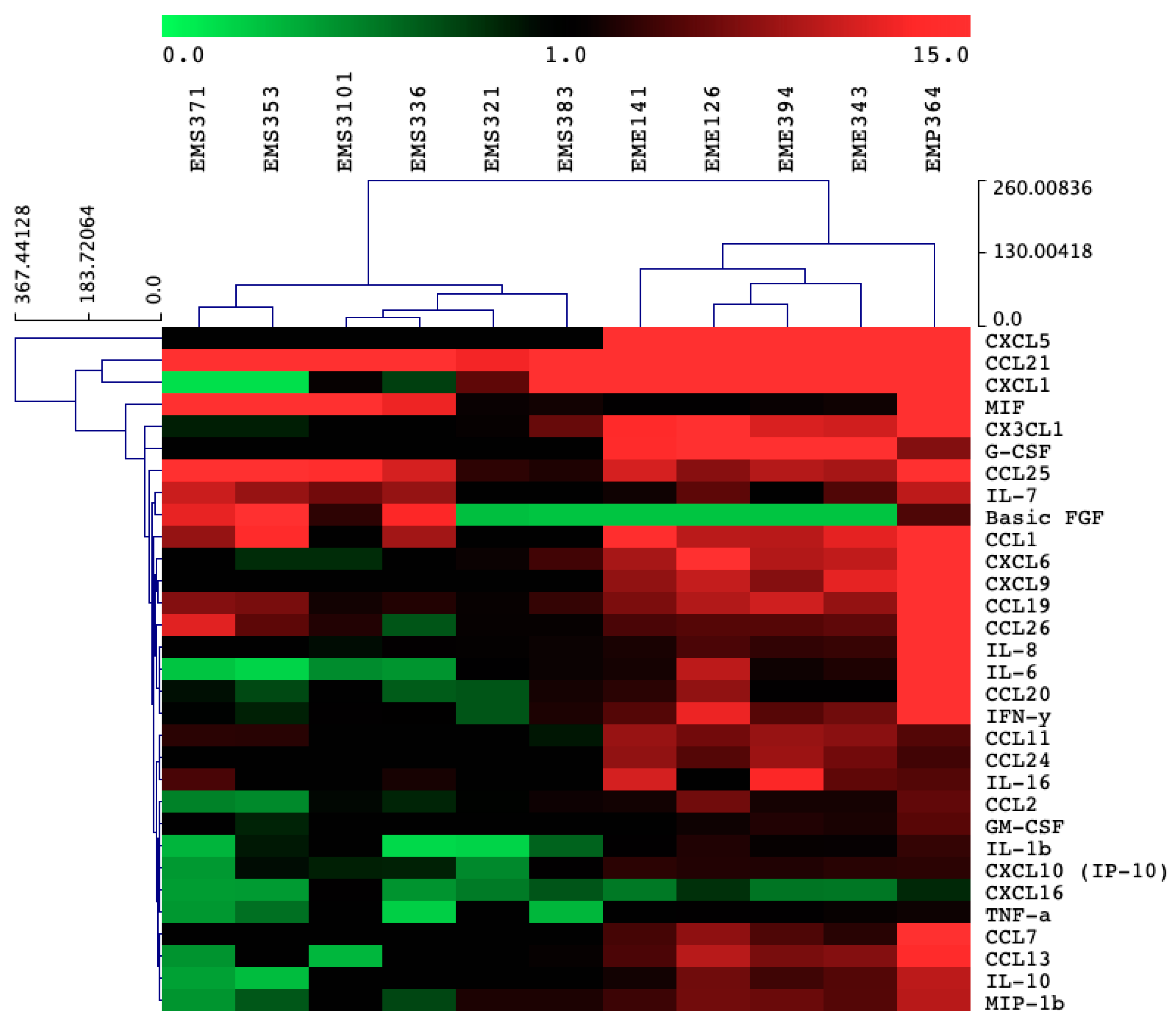
3.2. Cytokines and Chemokines Production Profiling in BEAS-2B Cells, Following Bacterial Treatments
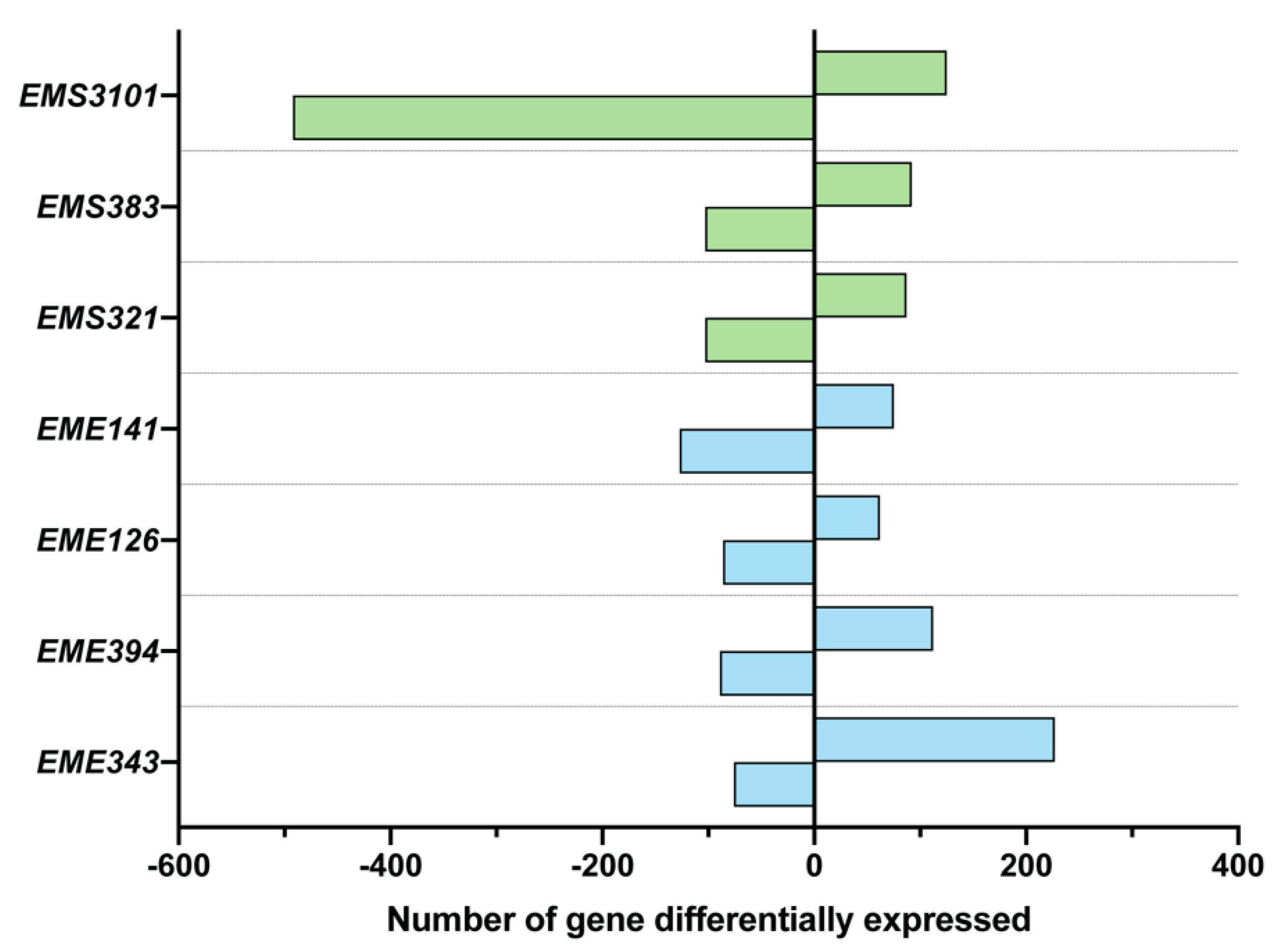
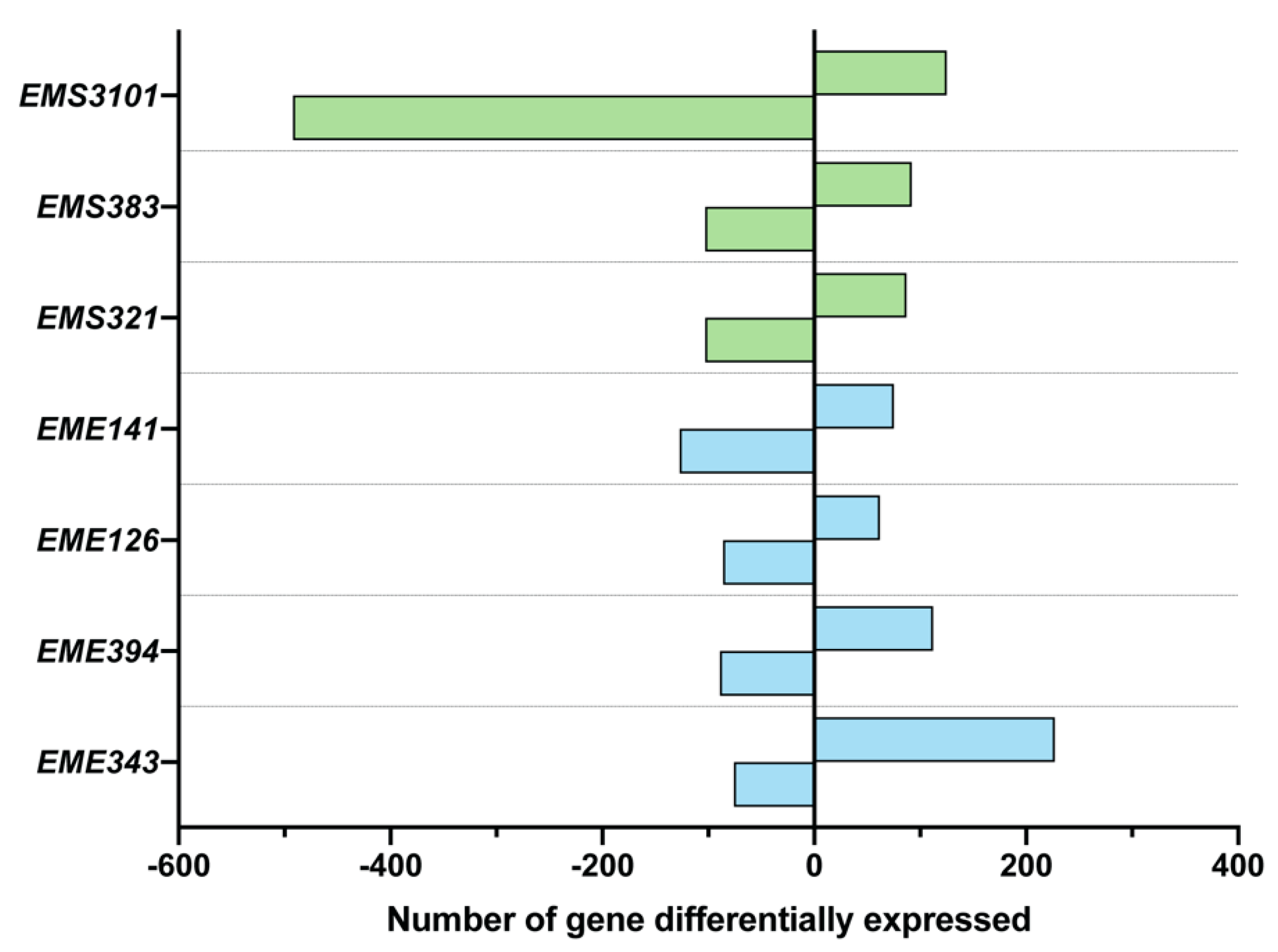
3.3. Analysis of Differential Gene Expression of BEAS-2B Cells, Using Microarray Analysis
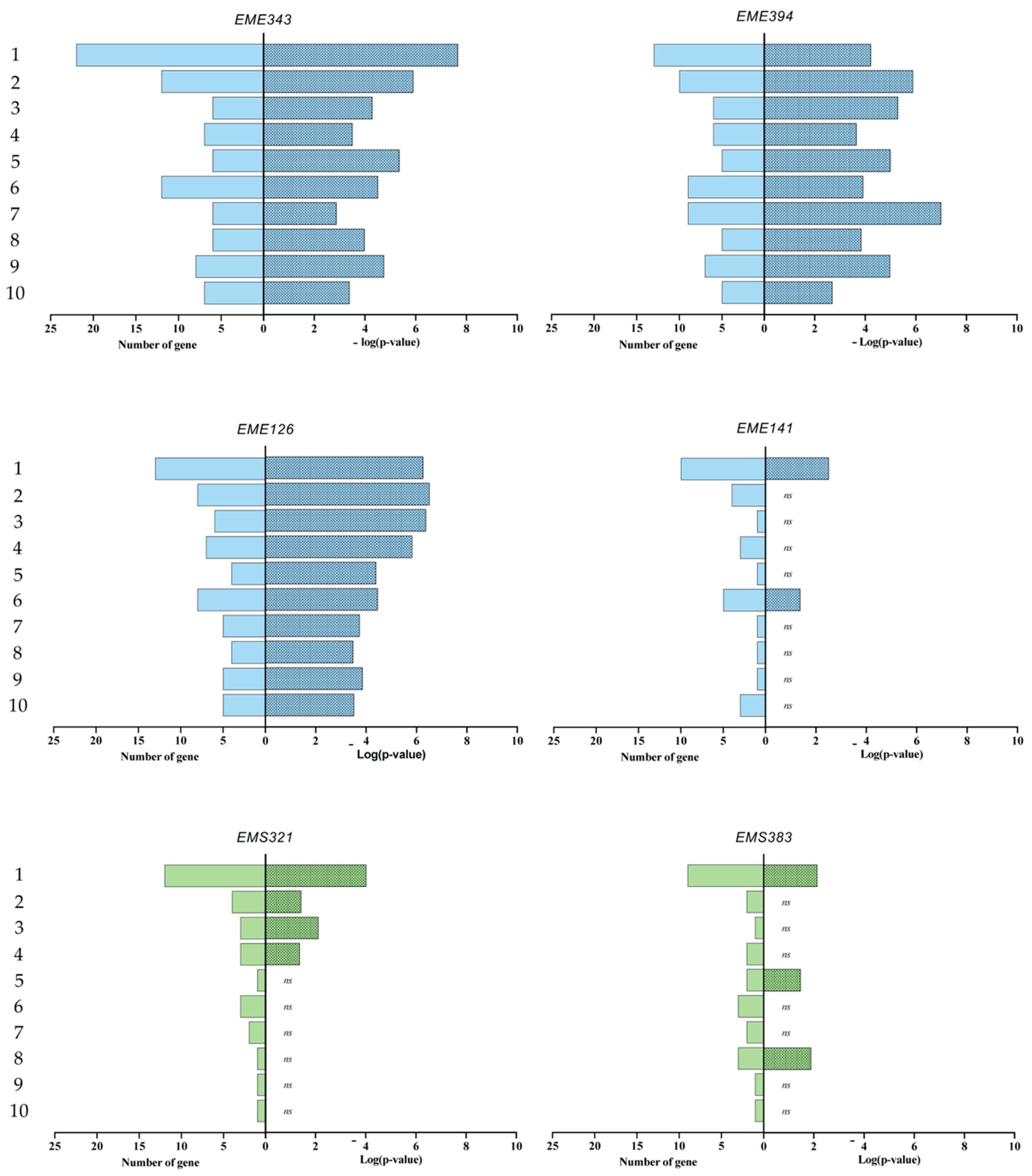
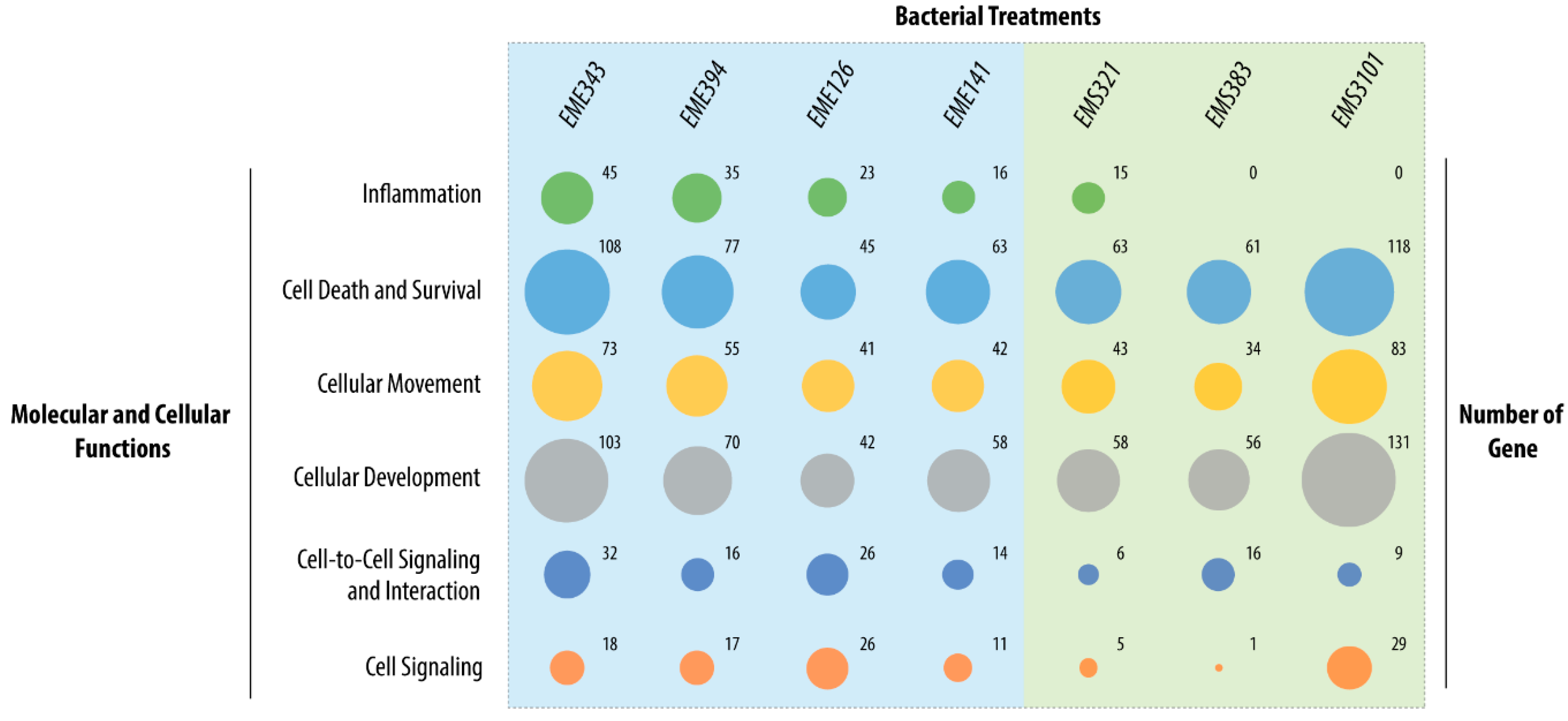
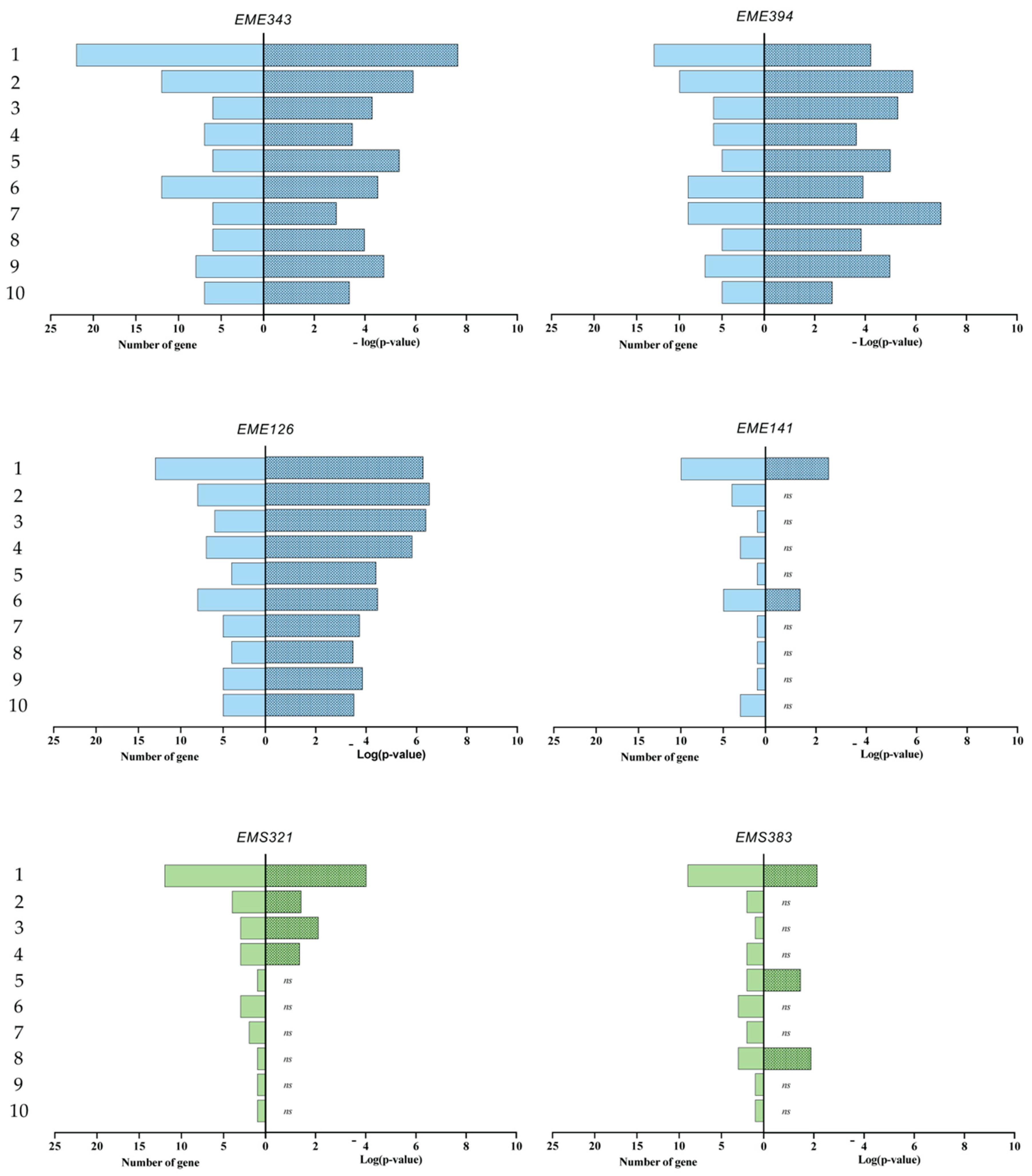
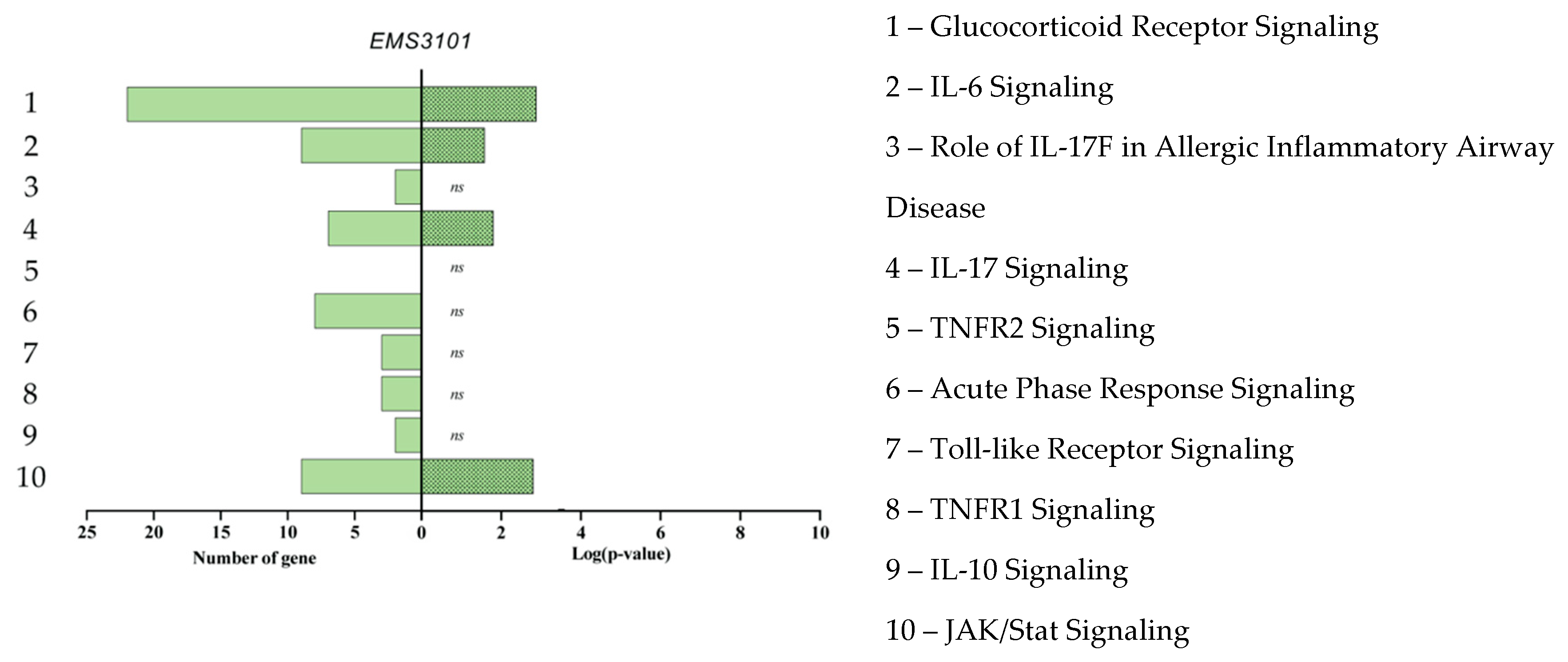
3.4. Gene Enrichment Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, Y.; Wang, X.; Li, H.; Ni, C.; Du, Z.; Yan, F. Human oral microbiota and its modulation for oral health. Biomed. Pharmacother. 2018, 99, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Arweiler, N.B.; Netuschil, L. The Oral Microbiota. Adv. Exp. Med. Biol. 2016, 902, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Rosier, B.T.; Marsh, P.D.; Mira, A. Resilience of the Oral Microbiota in Health: Mechanisms That Prevent Dysbiosis. Crit. Rev. Oral Biol. Med. 2018, 97, 371–380. [Google Scholar] [CrossRef]
- Escapa, I.F.; Chen, T.; Huang, Y.; Gajare, P.; Dewhirst, F.E.; Lemon, K.P. New insights into human nostril microbiome from the expanded Human Oral Microbiome Database (eHOMD): A resource for species-level identification of microbiome data from the aerodigestive tract. mSystems 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Human Microbiome Project Consortium. Structure, Function and Diversity of the Healthy Human Microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siqueira, J.F.; Roôças, I.N. The Oral Microbiota in Health and Disease: An Overview of Molecular Findings. Methods Mol. Biol. 2017, 1537, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Suda, W.; Luo, C.; Kawaguchi, T.; Motoo, I.; Narushima, S.; Kiguchi, Y.; Yasuma, K.; Watanabe, E.; Tanoue, T.; et al. Ectopic colonization of oral bacteria in the intestine drives TH1 cell induction and inflammation. Science 2017, 358, 359–365. [Google Scholar] [CrossRef]
- Pu, C.Y.; Seshadri, M.; Manuballa, S.; Yendamuri, S. The Oral Microbiome and Lung Diseases. Current Oral Health Rep. 2020, 7, 79–86. [Google Scholar] [CrossRef]
- Dzidic, M.; Abrahamsson, T.; Artacho, A.; Collado, M.C.; Mira, A.; Jenmalm, M.C. Oral microbiota maturation during the first 7 years of life in relation to allergy development. Allergy 2018, 73, 2000–2011. [Google Scholar] [CrossRef] [Green Version]
- Heo, S.M.; Haase, E.M.; Lesse, A.J.; Gill, S.R.; Scannapieco, F.A. Genetic Relationships between Respiratory Pathogens Isolated from Dental Plaque and Bronchoalveolar Lavage Fluid from Patients in the Intensive Care Unit Undergoing Mechanical Ventilation. Clin. Infect. Dis. 2008, 47, 1562–1570. [Google Scholar] [CrossRef] [Green Version]
- Morris, A.; Beck, M.; Schloss, P.D.; Campbell, T.B.; Crothers, K.; Curtis, J.L. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am. J. Respir. Crit. Care Med. 2013, 187, 1067–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, R.P.; Huffnagle, G.B. The lung microbiome: New principles for respiratory bacteriology in health and disease. PLoS Pathog. 2015, 11, e1004923. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, A.; Bassis, C.M.; Beck, J.M.; Young, V.B.; Curtis, J.L.; Huffnagle, G.B. Application of a neutral community model to assess structuring of the human lung microbiome. mBio 2015, 6, e02284-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathieu, E.; Escribano-Vazquez, U.; Descamps, D.; Cherbuy, C.; Langella, P.; Riffault, S.; Remot, A.; Thomas, M. Paradigms of Lung Microbiota Functions in Health and Disease, Particularly, in Asthma. Front. Physiol. 2018, 9, 1168. [Google Scholar] [CrossRef] [PubMed]
- Hilty, M.; Burke, C.; Pedro, H.; Cardenas, P.; Bush, A.; Bossley, C. Disordered microbial communities in asthmatic airways. PLoS ONE 2010, 5, e8578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segal, L.N.; Clemente, J.C.; Tsay, J.C.; Koralov, S.B.; Keller, B.C.; Wu., B.G. Enrichment of the lung microbiome with oral taxa is associated with lung inflammation of a Th17 phenotype. Nat. Microbiol. 2016, 1, 16031. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Gail, M.H.; Consonni, D.; Carugno, M.; Humphrys, M.; Pesatori, A.C. Characterizing human lung tissue microbiota and its relationship to epidemiological and clinical features. Genome Biol. 2016, 17, 163. [Google Scholar] [CrossRef] [Green Version]
- Pattaroni, C.; Watzenboeck, M.L.; Schneidegger, S.; Kieser, S.; Wong, N.C.; Bernasconi, E.; Pernot, J.; Mercier, L.; Knapp, S.; Nicod, L.P.; et al. Early-Life Formation of the Microbial and Immunological Environment of the Human Airways. Cell Host Microb. 2018, 24, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Vats, A.; Sharma, A.; Arora, A.; Kumar, A. The development of lower respiratory tract microbiome in mice. Microbiome 2017, 5, 61. [Google Scholar] [CrossRef] [Green Version]
- Remot, A.; Descamps, D.; Noordine, M.L.; Boukadiri, A.; Mathieu, E.; Robert, V.; Riffault, S.; Lambrecht, B.; Langella, P.; Hammad, H.; et al. Bacteria isolated from lung modulate asthma susceptibility in mice. ISME J. 2017, 11, 1061–1107. [Google Scholar] [CrossRef] [Green Version]
- Gollwitzer, E.S.; Saglani, S.; Trompette, A.; Yadava, K.; Sherburn, R.; McCoy, K.D. Lung microbiota promotes tolerance to allergens in neonates via PD-L1. Nat. Med. 2014, 20, 642–647. [Google Scholar] [CrossRef]
- Segal, L.N.; Rom, W.N.; Weiden, M.D. Lung microbiome for clinicians. New discoveries about bugs in healthy and diseased lungs. Ann. Am. Thorac. Soc. 2014, 11, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Ege, M.J.; Mayer, M.; Normand, A.-C.; Genuneit, J.; Cookson, W.O.C.M.; Phil, D.; Braun-Fahrländer, C.; Heederik, R.; Piarroux, R.; Von Mutius, E.; et al. Exposure to Environmental Microorganisms and Childhood Asthma. N. Engl. J. Med. 2011, 364, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Marsland, B.J. Influences of the microbiome on the early origins of allergic asthma. Ann. Am. Thorac. Soc. 2013, 10, S165–S169. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.V.; Wood, R.A.; Boushey, H.; Bacharier, L.B.; Bloomberg, G.R.; Kattan, M.; O’Connor, G.T.; Sandel, M.T.; Calatroni, A.; Matsui, E.; et al. Effects of Early Life Exposure to Allergens and Bacteria on Recurrent Wheeze and Atopy in Urban Children. J. Allergy Clin. Immunol. 2014, 134, 593–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilpeläinen, M.; Terho, E.O.; Helenius, H.; Koskenvuo, M. Farm environment in childhood prevents the development of allergies. Clin. Exp. Allergy 2000, 30, 201–208. [Google Scholar] [CrossRef]
- Riedler, J.; Eder, W.; Oberfeld, G.; Schreuer, M. Austrian children living on a farm have less hay fever, asthma and allergic sensitization. Clin. Exp. Allergy 2000, 30, 194–200. [Google Scholar] [CrossRef]
- Douwes, J.; Travier, N.; Huang, K.; Cheng, S.; McKenzie, J.; Le Gros, G.; von Mutius, E.; Pearce, N. Lifelong farm exposure may strongly reduce the risk of asthma in adults. Allergy 2007, 62, 1158–1165. [Google Scholar] [CrossRef]
- Zou, J.; Zhou, L.; Hu, C.; Jing, P.; Guo, X.; Liu, S.; Lei, Y.; Yang, S.; Deng, J.; Zhang, H. IL-8 and IP-10 expression from human bronchial epithelial cells BEAS-2B are promoted by Streptococcus pneumoniae endopeptidase O (PepO). BMC Microbiol. 2017, 17, 187. [Google Scholar] [CrossRef]
- Wang, Y.; Bai, C.; Li, K.; Adler, K.B.; Wang, X. Role of airway epithelial cells in development of asthma and allergic rhinitis. Respir. Med. 2008, 102, 949–955. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, R.; Gappa-Fahlenkamp, H. Cells and Culture Systems Used to Model the Small Airway Epithelium. Lung 2016, 194, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Hammad, H.; Chieppa, M.; Perros, F.; Willart, M.A.; Germain, R.N.; Lambrecht, B.N. House dust mite allergen induces asthma via Toll- like receptor 4 triggering of airway structural cells. Nat. Med. 2009, 15, 410–416. [Google Scholar] [CrossRef] [Green Version]
- Di Stefano, A.; Ricciardolo, F.L.; Caramori, G.; Adcock, I.M.; Chung, K.F.; Barnes, P.J. Bronchial inflammation and bacterial load in stable COPD is associated with TLR4 overexpression. Eur. Respir. J. 2017, 49, 1602006. [Google Scholar] [CrossRef] [Green Version]
- Carlini, F.; Picard, C.; Garulli, C.; Piquemal, D.; Roubertoux, P.; Chiaroni, J.; Chanez, P.; Gras, D.; Di Cristofaro, J. Bronchial Epithelial Cells from Asthmatic Patients Display Less Functional HLA-G Isoform Expression. Front. Immunol. 2017, 8, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, J.M.; Niccum, D.; Dunitz, J.M.; Hunter, R.C. Evidence and role for bacterial mucin degradation in cystic fibrosis airway disease. PLoS Pathog. 2016, 12, e1005846. [Google Scholar] [CrossRef] [Green Version]
- Ni, K.; Wang, Y.; Li, D.; Cai, Y.; Pang, H. Characterization, Identification and Application of Lactic Acid Bacteria Isolated from Forage Paddy Rice Silage. PLoS ONE 2015, 10, e0121967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medicine, U.S. National Library of. n.d. National Center for Biotechnology Information. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 17 July 2020).
- MacPherson, C.W.; Shastri, P.; Mathieu, O.; Tompkins, T.A.; Burguieère, P. Genome-Wide Immune Modulation of TLR3- Mediated Inflammation in Intestinal Epithelial Cells Differs between Single and Multi-Strain Probiotic Combination. PLoS ONE 2017, 12, e0169847. [Google Scholar] [CrossRef] [PubMed]
- Dashper, S.G.; Mitchell, H.L.; Leêcao, K.A.; Carpenter, L.; Gussy, M.G.; Calache, H.; Gladman, S.L.; Bulach, D.M.; Hoffmann, B.; Catmull, D.V.; et al. Temporal development of the oral microbiome and prediction of early childhood caries. Sci. Rep. 2019, 9, 19732. [Google Scholar] [CrossRef]
- Komiyama, E.Y.; Lepesqueu, L.S.S.; Yassuda, C.G.; Samaranayake, L.P.; Parahitiyawa, N.B.; Balducci, I.; Koga-Ito, C.Y. Enterococcus Species in the Oral Cavity: Prevalence, Virulence Factors and Antimicrobial Susceptibility. PLoS ONE 2016, 11, e0163001. [Google Scholar] [CrossRef] [Green Version]
- Abranches, J.; Zeng, L.; Kajfasz, J.K.; Palmer, S.R.; Chakraborty, B.; Wen, Z.T.; Richards, V.P.; Brady, L.J.; Lemos, J.A. Biology of Oral Streptococci. Microbiol. Spectr. 2018, 6, GPP3-0042-201. [Google Scholar] [CrossRef]
- Kennedy, B.; Peura, S.; Hammar, U.; Vicenzi, S.; Hedman, A.; Almqvist, C.; Andolf, E.; Pershagen, G.; Dicksved, J.; Bertilsonn, S.; et al. Oral Microbiota Development in early childhood. Sci. Rep. 2019, 9, 19025. [Google Scholar] [CrossRef]
- Dzidic, M.; Collado, M.C.; Abrahamsson, T.; Artacho, A.; Stensson, M.; Jenmalm, M.C.; Mira, A. Oral microbiome development during childhood: An ecological succession influenced by postnatal factors and associated with tooth decay. ISME J. 2018, 12, 2292–2306. [Google Scholar] [CrossRef]
- Kononen, E. Development of oral bacterial flora in young children. Ann. Med. 2000, 32, 107–112. [Google Scholar] [CrossRef]
- Arrieta, M.C.; Stiemsma, L.T.; Dimitriu, P.A.; Thorson, L.; Russell, S.; Yurist-Doutsch, S.; Kuzeljevic, B. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci. Transl. Med. 2015, 7, 307ra152. [Google Scholar] [CrossRef]
- Yamaguchi, E.; Nishihira, J.; Shimizu, T.; Takahashi, T.; Kitashiro, N.; Hizawa, N.; Kamishima, K.; Kawakami, Y. Macrophage migration inhibitory factor (MIF) in bronchial asthma. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2000, 30, 1244–1249. [Google Scholar] [CrossRef]
- Lan, H.; Luo, L.; Chen, Y.; Wang, M.; Yu, Z.; Gong, Y. MIF signaling blocking alleviates airway inflammation and airway epithelial barrier disruption in a HDM-induced asthma model. Cell. Immunol. 2020, 347. [Google Scholar] [CrossRef]
- Jeffrey, M.P.; MacPherson, C.W.; Mathieu, O.; Tompkins, T.A.; Green-Johnson, J.M. Secretome-Mediated Interactions with Intestinal Epithelial Cells: A Role for Secretome Components from Lactobacillus rhamnosus R0011 in the Attenuation of Salmonella enterica Serovar Typhimurium Secretome and TNF- α−Induced Proinflammatory Responses. J. Immunol. Publ. Online 2020, 204, 2523–2534. [Google Scholar] [CrossRef]
- Rossetti, V.; Ammann, T.W.; Thurnheer, T.; Bagheri, H.C.; Belibasakis, G.N. Phenotypic Diversity of Multicellular Filamentation in Oral Streptococci. PLoS ONE 2013, 8, e76221. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, M.; Kokubu, F.; Fujita, J.; Huang, S.K.; Hizawa, N. Role of interleukin-17F in asthma. Inflamm. Allergy Drug Targets 2009, 8, 383–389. [Google Scholar] [CrossRef]
- Dempsey, P.W.; Doyle, S.E.; He, J.Q.; Cheng, G. The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev. 2003, 14, 193–209. [Google Scholar] [CrossRef]
- Warburton, D.; Bellusci, S.; Del Moral, P.-M.; Kaartinen, V.; Lee, M.; Tefft, D.; Shi, W. Growth factor signaling in lung morphogenetic centers: Automaticity, stereotypy and symmetry. Respir. Res. 2003, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Been, J.V.; Debeer, A.; Van Iwaarden, J.F.; Kloosterboer, N.; Passos, V.L.; Naulaers, G.; Zimmermann, L.J. Early Alterations of Growth Factor Patterns in Bronchoalveolar Lavage Fluid From Preterm Infants Developing Bronchopulmonary Dysplasia. Pediatr. Res. 2010, 67, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Bird, A.D.; McDougall, A.R.A.; Seow, B.; Hooper, S.B.; Cole, T.J. Glucocorticoid regulation of lung development: Lessons learned from conditional GR knockout mice. Mol. Endocrinol. 2014, 29, 158–171. [Google Scholar] [CrossRef] [Green Version]
- Bolt, R.J.; van Weissenbruch, M.M.; Lafeber, H.N.; Delemarre-van de Waal., H.A. Glucocorticoids and Lung Development in the Fetus and Preterm Infant. Pediatr. Pulmonol. 2001, 32, 76–91. [Google Scholar] [CrossRef]
- Scott, S.M.; Rose, S.R. Use of glucocorticoid for treatment of fetus and preterm infant. Clin. Perinatol. 2017, 45, 93–102. [Google Scholar] [CrossRef]
- Bry, K.; Whitsett, J.A.; Lappalainen, U. IL-1b Disrupts Postnatal Lung Morphogenesis in the Mouse. Am. J. Respir. Cell Mol. Biol. 2007, 36, 32–42. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EME343 | EME394 | EME126 | EME141 | EMS321 | EMS383 | EMS3101 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Fold Change | Gene | Fold Change | Gene | Fold Change | Gene | Fold Change | Gene | Fold Change | Gene | Fold Change | Gene | Fold Change |
| IL8 | 14.502 | IL8 | 12.219 | IL8 | 8.043 | CCL2 | 4.718 | ATF3 | 5.989 | CCL2 | 6.741 | SNORD3B-1 | 4.091 |
| CCL2 | 9.667 | CCL20 | 11.190 | CCL2 | 6.391 | GADD45A | 3.706 | IL8 | 5.036 | C1QTNF9 | 6.711 | HIST2H2AA4 | 3.981 |
| ATF3 | 7.888 | IL1A | 9.573 | TNFAIP3 | 4.777 | PTX3 | 3.620 | CCL2 | 3.968 | SULT1B1 | 5.156 | CCL2 | 3.108 |
| TNFAIP3 | 6.536 | CXCL2 | 7.994 | CLDN1 | 3.609 | DDIT3 | 3.497 | EGR1 | 3.734 | CLDN1 | 4.469 | SLC5A7 | 3.086 |
| PTX3 | 6.242 | CCL2 | 7.351 | IER3 | 3.527 | CLDN1 | 3.257 | STC2 | 3.732 | CHAC1 | 4.310 | HIST1H2BI | 3.074 |
| CDKN1A | 6.132 | PTX3 | 5.996 | NDRG1 | 3.296 | STC2 | 3.160 | PTX3 | 3.420 | ATF3 | 4.124 | HIST1H2AC | 2.910 |
| IL1A | 6.088 | EGR1 | 4.945 | CXCL1 | 3.220 | CXCL2 | 3.052 | DDIT3 | 3.307 | ASNS | 4.020 | HIST1H2BC | 2.848 |
| SLC7A11 | 5.094 | STC2 | 4.444 | PTX3 | 2.892 | MAFF | 2.901 | CHAC1 | 3.166 | TRIB3 | 3.930 | HIST2H4B | 2.759 |
| EGR1 | 4.936 | ATF3 | 4.391 | CSN1S1 | 2.471 | PPP1R15A | 2.601 | CLDN1 | 3.116 | STC2 | 3.792 | HIST1H4L | 2.676 |
| CXCL2 | 4.823 | CHAC1 | 4.344 | CEBPB | 2.432 | EGR1 | 2.573 | VEGFA | 2.963 | SLC7A11 | 3.502 | HIST4H4 | 2.593 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathieu, E.; MacPherson, C.W.; Belvis, J.; Mathieu, O.; Robert, V.; Saint-Criq, V.; Langella, P.; Tompkins, T.A.; Thomas, M. Oral Primo-Colonizing Bacteria Modulate Inflammation and Gene Expression in Bronchial Epithelial Cells. Microorganisms 2020, 8, 1094. https://doi.org/10.3390/microorganisms8081094
Mathieu E, MacPherson CW, Belvis J, Mathieu O, Robert V, Saint-Criq V, Langella P, Tompkins TA, Thomas M. Oral Primo-Colonizing Bacteria Modulate Inflammation and Gene Expression in Bronchial Epithelial Cells. Microorganisms. 2020; 8(8):1094. https://doi.org/10.3390/microorganisms8081094
Chicago/Turabian StyleMathieu, Elliot, Chad W. MacPherson, Jocelyn Belvis, Olivier Mathieu, Véronique Robert, Vinciane Saint-Criq, Philippe Langella, Thomas A. Tompkins, and Muriel Thomas. 2020. "Oral Primo-Colonizing Bacteria Modulate Inflammation and Gene Expression in Bronchial Epithelial Cells" Microorganisms 8, no. 8: 1094. https://doi.org/10.3390/microorganisms8081094
APA StyleMathieu, E., MacPherson, C. W., Belvis, J., Mathieu, O., Robert, V., Saint-Criq, V., Langella, P., Tompkins, T. A., & Thomas, M. (2020). Oral Primo-Colonizing Bacteria Modulate Inflammation and Gene Expression in Bronchial Epithelial Cells. Microorganisms, 8(8), 1094. https://doi.org/10.3390/microorganisms8081094