The Porcine Nasal Microbiota with Particular Attention to Livestock-Associated Methicillin-Resistant Staphylococcus aureus in Germany—A Culturomic Approach



Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Culturome Composition and Novel Taxa
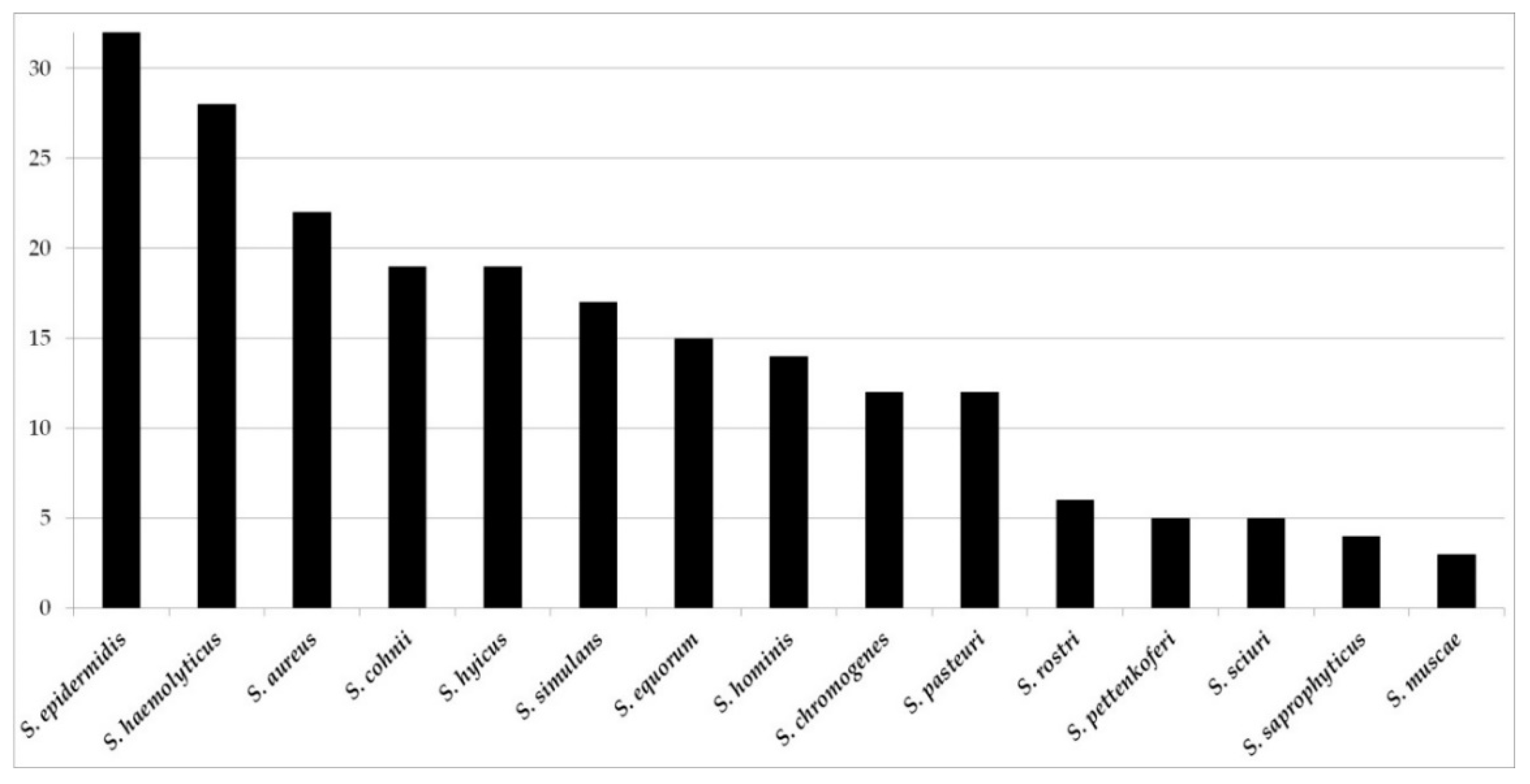
3.1.1. Firmicutes
3.1.2. Actinobacteria
3.1.3. Proteobacteria and Bacteroidetes
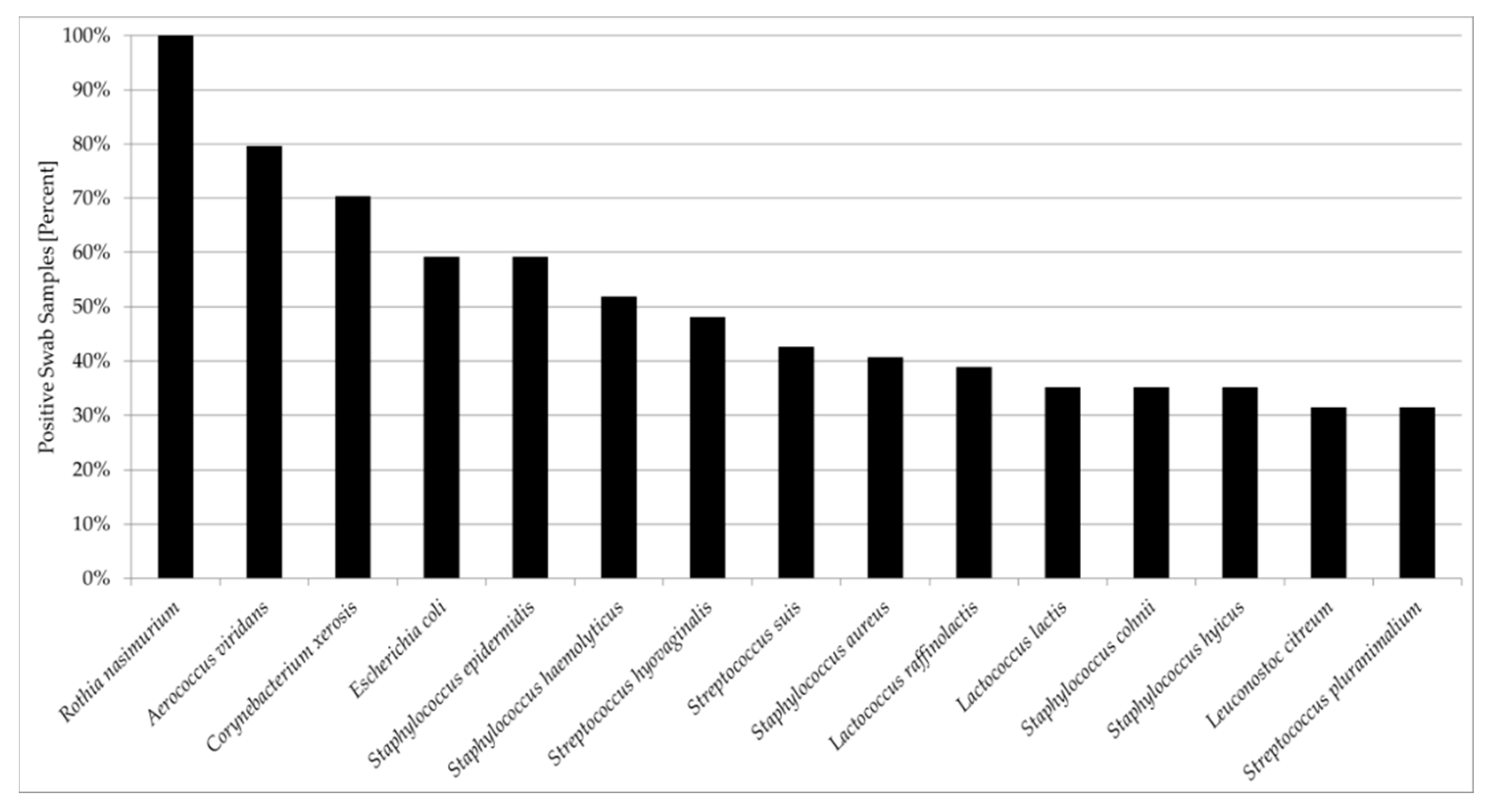
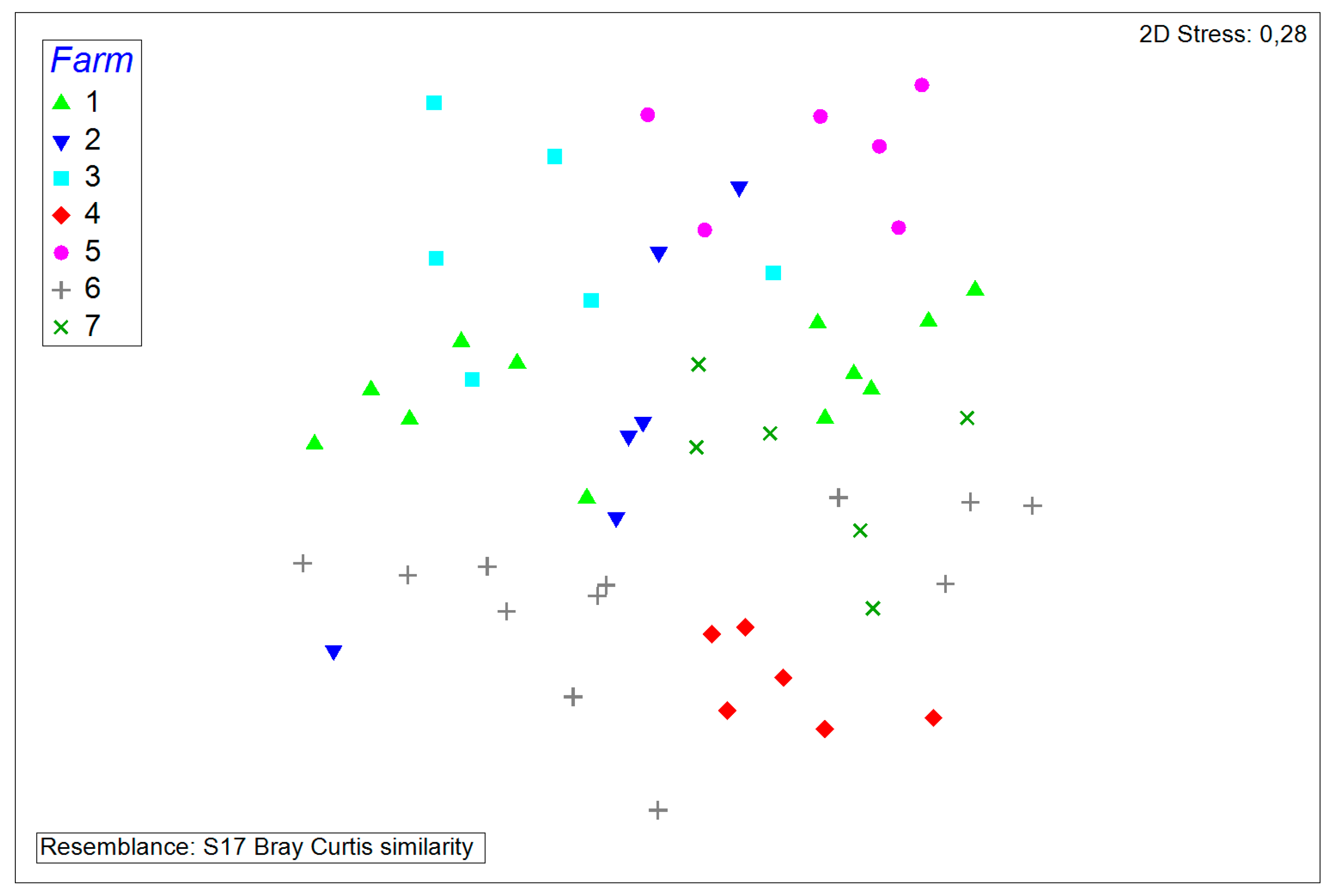
3.2. Comparisons and Correlations of the Porcine Nasal Microbiota
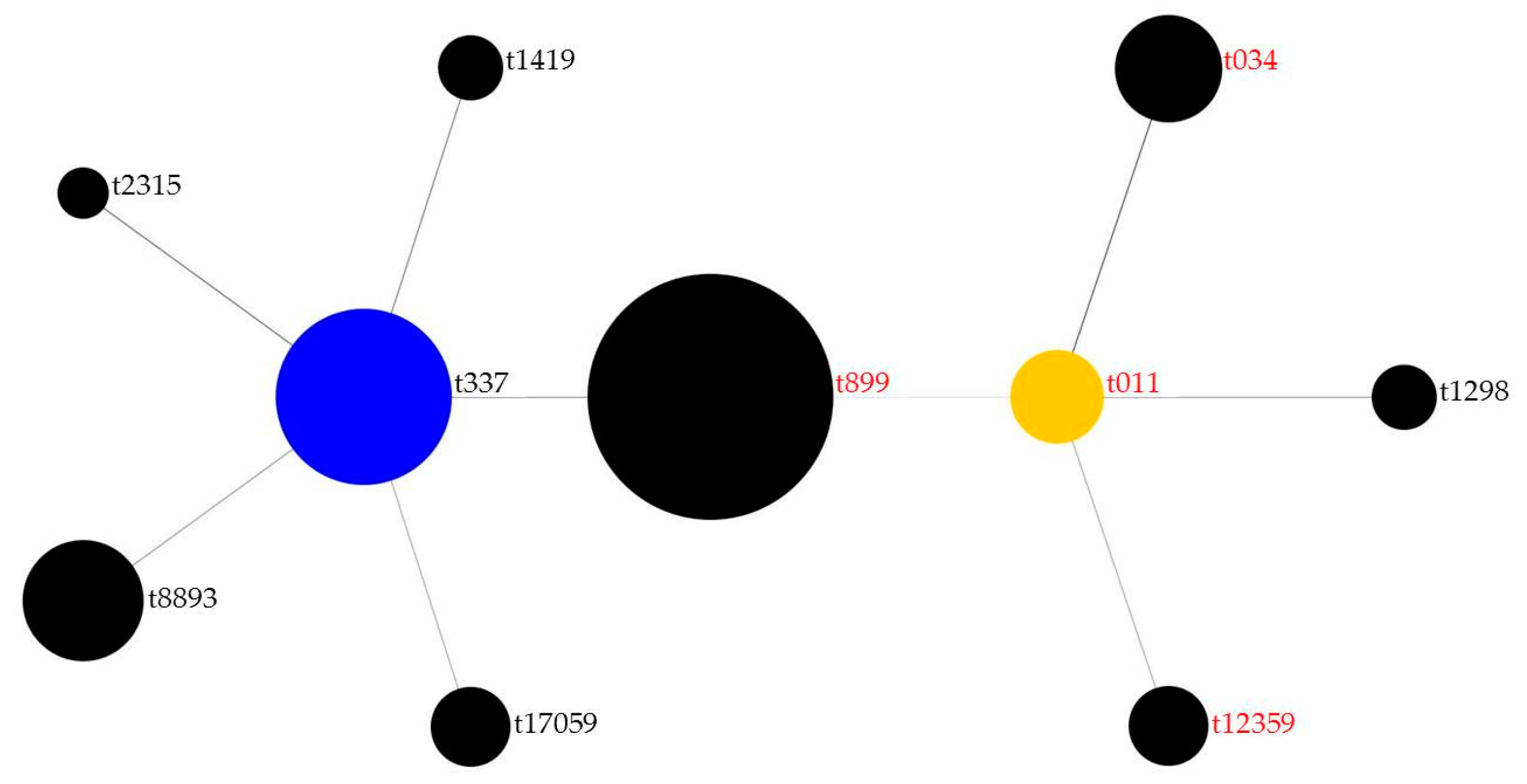
3.3. Analysis of Resistance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Interagency Coordination Group on Antimicrobial Resistance. No Time To Wait: Infections From Drug-Resistant Securing the Future; United Nations Foundation for the IACG: Geneva, Swizerland, 2019. [Google Scholar]
- Kourtis, A.P.; Hatfield, K.; Baggs, J.; Mu, Y.; See, I.; Epson, E.; Nadle, J.; Kainer, M.A.; Dumyati, G.; Petit, S.; et al. Vital Signs: Epidemiology and Recent Trends in Methicillin-Resistant and in Methicillin-Susceptible Staphylococcus aureus Bloodstream Infections — United States. MMWR. Morb. Mortal. Wkly. Rep. 2019, 68, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Robert Koch-Institut. Infektionsepidemiologisches Jahrbuch meldepflichtiger Krankheiten für 2017; Krankenhaus-Hygiene + Infekt: Berlin, Germany, 2018. [Google Scholar]
- Wos-Oxley, M.L.; Plumeier, I.; Von Eiff, C.; Taudien, S.; Platzer, M.; Vilchez-Vargas, R.; Becker, K.; Pieper, D.H. A poke into the diversity and associations within human anterior nare microbial communities. ISME J. 2010, 4, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Camelo-Castillo, A.; Henares, D.; Brotons, P.; Galiana, A.; Rodríguez, J.C.; Mira, A.; Muñoz-Almagro, C. Nasopharyngeal Microbiota in Children With Invasive Pneumococcal Disease: Identification of Bacteria With Potential Disease-Promoting and Protective Effects. Front. Microbiol. 2019, 10, 11. [Google Scholar] [CrossRef] [PubMed]
- van Alen, S.; Ballhausen, B.; Peters, G.; Friedrich, A.W.; Mellmann, A.; Köck, R.; Becker, K. In the centre of an epidemic: Fifteen years of LA-MRSA CC398 at the University Hospital Münster. Vet. Microbiol. 2017, 200, 19–24. [Google Scholar] [CrossRef] [PubMed]
- van Cleef, B.A.; Verkade, E.J.M.; Wulf, M.W.; Buiting, A.G.; Voss, A.; Huijsdens, X.W.; van Pelt, W.; Mulders, M.N.; Kluytmans, J.A. Prevalence of Livestock-Associated MRSA in Communities with High Pig-Densities in The Netherlands. PLoS ONE 2010, 5, e9385. [Google Scholar] [CrossRef] [PubMed]
- Lewis, H.C.; Mølbak, K.; Reese, C.; Aarestrup, F.M.; Selchau, M.; Sørum, M.; Skov, R.L. Pigs as Source of Methicillin-Resistant Staphylococcus aureus CC398 Infections in Humans, Denmark. Emerg. Infect. Dis. 2008, 14, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, U.; von Lützau, A.; Schlattmann, A.; Rösler, U.; Köck, R.; Becker, K. Zoonotic multidrug-resistant microorganisms among small companion animals in Germany. PLoS ONE 2018, 13, e0208364. [Google Scholar] [CrossRef]
- Kaspar, U.; von Lützau, K.; Schlattmann, A.; Rösler, U.; Köck, R.; Becker, K. Zoonotic multidrug-resistant microorganisms among non-hospitalized horses from Germany. One Health 2019, 7, 100091. [Google Scholar] [CrossRef]
- Cuny, C.; Friedrich, A.; Kozytska, S.; Layer, F.; Nübel, U.; Ohlsen, K.; Strommenger, B.; Walther, B.; Wieler, L.; Witte, W. Emergence of methicillin-resistant Staphylococcus aureus (MRSA) in different animal species. Int. J. Med. Microbiol. 2010, 300, 109–117. [Google Scholar] [CrossRef]
- Becker, K.; Ballhausen, B.; Kahl, B.C.; Köck, R. The clinical impact of livestock-associated methicillin-resistant Staphylococcus aureus of the clonal complex 398 for humans. Vet. Microbiol. 2017, 200, 33–38. [Google Scholar] [CrossRef]
- Köck, R.; Ballhausen, B.; Bischoff, M.; Cuny, C.; Eckmanns, T.; Fetsch, A.; Harmsen, D.; Goerge, T.; Oberheitmann, B.; Schwarz, S.; et al. The impact of zoonotic MRSA colonization and infection in Germany. Berl. Munch. Tierarztl. Wochenschr. 2014, 127, 384–398. [Google Scholar] [PubMed]
- Köck, R.; Schaumburg, F.; Mellmann, A.; Köksal, M.; Jurke, A.; Becker, K.; Friedrich, A.W. Livestock-Associated Methicillin-Resistant Staphylococcus aureus (MRSA) as Causes of Human Infection and Colonization in Germany. PLoS ONE 2013, 8, e55040. [Google Scholar] [CrossRef] [PubMed]
- Schaumburg, F.; Köck, R.; Mellmann, A.; Richter, L.; Hasenberg, F.; Kriegeskorte, A.; Friedrich, A.W.; Gatermann, S.; Peters, G.; Von Eiff, C.; et al. Population dynamics among methicillin-resistant Staphylococcus aureus isolates in Germany during a 6-year period. J. Clin. Microbiol. 2012, 50, 3186–3192. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kaiser, A.M.; Haenen, A.J.P.; de Neeling, A.J.; Vandenbroucke-Grauls, C.M.J.E. Prevalence of Methicillin-Resistant Staphylococcus aureus and Risk Factors for Carriage in Dutch Hospitals. Infect. Control Hosp. Epidemiol. 2010, 31, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Sieber, R.N.; Skov, R.L.; Nielsen, J.; Schulz, J.; Price, L.B.; Aarestrup, F.M.; Larsen, A.R.; Stegger, M.; Larsen, J. Drivers and dynamics of methicillin-resistant livestock-associated Staphylococcus aureus CC398 in pigs and humans in Denmark. MBio 2018, 9, e02142-18. [Google Scholar] [CrossRef]
- Schlattmann, A.; von Lützau, K.; Kaspar, U.; Becker, K. ‘Rothia nasisuis’ sp. nov., ‘Dermabacter porcinasus’ sp. nov., ‘Propionibacterium westphaliense’ sp. nov. and ‘Tessaracoccus nasisuum’ sp. nov., isolated from porcine nasal swabs in the Münster region, Germany. New Microbes New Infect. 2018, 26, 114–117. [Google Scholar] [CrossRef]
- Idelevich, E.A.; Schüle, I.; Grünastel, B.; Wüllenweber, J.; Peters, G.; Becker, K. Rapid identification of microorganisms from positive blood cultures by MALDI-TOF mass spectrometry subsequent to very short-term incubation on solid medium. Clin. Microbiol. Infect. 2014, 20, 1001–1006. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Ebers, J. Taxonomic Parameters Revisited: Tarnished Gold Standards. Microbiol. Today 2006, 33, 152–155. [Google Scholar]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef]
- European Committee on Antimicrobial Susceptibility Testing. Breakpoint Tables for Interpretation of MICs and Zone Diameters. Version 8.0. 2018. Available online: http//www.eucast.org (accessed on 1 January 2018).
- Kriegeskorte, A.; Ballhausen, B.; Idelevich, E.A.; Köck, R.; Friedrich, A.W.; Karch, H.; Peters, G.; Becker, K. Human MRSA isolates with novel genetic homolog, Germany. Emerg. Infect. Dis. 2012, 18, 1016–1018. [Google Scholar] [CrossRef]
- Becker, K.; van Alen, S.; Idelevich, E.A.; Schleimer, N.; Seggewiß, J.; Mellmann, A.; Kaspar, U.; Peters, G. Plasmid-Encoded Transferable mecB-Mediated Methicillin Resistance in Staphylococcus aureus. Emerg. Infect. Dis. 2018, 24, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Schwendener, S.; Cotting, K.; Perreten, V. Novel methicillin resistance gene mecD in clinical Macrococcus caseolyticus strains from bovine and canine sources. Sci. Rep. 2017, 7, 43797. [Google Scholar] [CrossRef] [PubMed]
- Mellmann, A.; Weniger, T.; Berssenbrügge, C.; Keckevoet, U.; Friedrich, A.W.; Harmsen, D.; Grundmann, H. Characterization of Clonal Relatedness among the Natural Population of Staphylococcus aureus Strains by Using spa Sequence Typing and the BURP (Based upon Repeat Patterns) Algorithm. J. Clin. Microbiol. 2008, 46, 2805–2808. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Clarke, K.R. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Clarke, K.R.; Gorley, R.N. PRIMER v6: User Manual/Tutorial; PRIMER-E: Plymouth, UK, 2006; p. 190. [Google Scholar]
- Lowe, B.A.; Marsh, T.L.; Isaacs-Cosgrove, N.; Kirkwood, R.N.; Kiupel, M.; Mulks, M.H. Defining the “core microbiome” of the microbial communities in the tonsils of healthy pigs. BMC Microbiol. 2012, 12, 20. [Google Scholar] [CrossRef]
- Weese, J.S.; Slifierz, M.; Jalali, M.; Friendship, R. Evaluation of the nasal microbiota in slaughter-age pigs and the impact on nasal methicillin-resistant Staphylococcus aureus (MRSA) carriage. BMC Vet. Res. 2014, 10, 69. [Google Scholar] [CrossRef]
- Strube, M.L.; Hansen, J.E.; Rasmussen, S.; Pedersen, K. A detailed investigation of the porcine skin and nose microbiome using universal and Staphylococcus specific primers. Sci. Rep. 2018, 8, 12751. [Google Scholar] [CrossRef]
- Bhargava, K.; Zhang, Y. Characterization of methicillin-resistant coagulase-negative staphylococci (MRCoNS) in retail meat. Food Microbiol. 2014, 42, 56–60. [Google Scholar] [CrossRef]
- Zong, Z.; Peng, C.; Lü, X. Diversity of SCCmec elements in methicillin-resistant coagulase-negative staphylococci clinical isolates. PLoS ONE 2011, 6, e20191. [Google Scholar] [CrossRef]
- Nemeghaire, S.; Vanderhaeghen, W.; Argudin, M.A.; Haesebrouck, F.; Butaye, P. Characterization of methicillin-resistant Staphylococcus sciuri isolates from industrially raised pigs, cattle and broiler chickens. J. Antimicrob. Chemother. 2014, 69, 2928–2934. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, U.; Kriegeskorte, A.; Schubert, T.; Peters, G.; Rudack, C.; Pieper, D.H.; Wos-Oxley, M.; Becker, K. The culturome of the human nose habitats reveals individual bacterial fingerprint patterns. Environ. Microbiol. 2016, 18, 2130–2142. [Google Scholar] [CrossRef] [PubMed]
- Sahibzada, S.; Hernández-Jover, M.; Jordan, D.; Thomson, P.C.; Heller, J. Emergence of highly prevalent CA-MRSA ST93 as an occupational risk in people working on a pig farm in Australia. PLoS ONE 2018, 13, e0195510. [Google Scholar] [CrossRef] [PubMed]
- Parada, J.L.; Caron, C.R.; Medeiros, A.B.P.; Soccol, C.R. Bacteriocins from lactic acid bacteria: Purification, properties and use as biopreservatives. Braz. Arch. Biol. Technol. 2007, 50, 521–542. [Google Scholar] [CrossRef]
- Zipperer, A.; Konnerth, M.C.; Laux, C.; Berscheid, A.; Janek, D.; Weidenmaier, C.; Burian, M.; Schilling, N.A.; Slavetinsky, C.; Marschal, M.; et al. Human commensals producing a novel antibiotic impair pathogen colonization. Nature 2016, 535, 511–516. [Google Scholar] [CrossRef]
- Schmithausen, R.M.; Schulze-Geisthoevel, S.V.; Stemmer, F.; El-Jade, M.; Reif, M.; Hack, S.; Meilaender, A.; Montabauer, G.; Fimmers, R.; Parcina, M.; et al. Analysis of Transmission of MRSA and ESBL-E among Pigs and Farm Personnel. PLoS ONE 2015, 10, e0138173. [Google Scholar] [CrossRef]
- Fetsch, A.; Roesler, U.; Kraushaar, B.; Friese, A. Co-colonization and clonal diversity of methicillin-sensitive and methicillin-resistant Staphylococcus aureus in sows. Vet. Microbiol. 2016, 185, 7–14. [Google Scholar] [CrossRef]
- Schoenfelder, S.M.K.; Dong, Y.; Feßler, A.T.; Schwarz, S.; Schoen, C.; Köck, R.; Ziebuhr, W. Antibiotic resistance profiles of coagulase-negative staphylococci in livestock environments. Vet. Microbiol. 2017, 200, 79–87. [Google Scholar] [CrossRef]
- Dahms, C.; Hübner, N.-O.; Kossow, A.; Mellmann, A.; Dittmann, K.; Kramer, A. Occurrence of ESBL-Producing Escherichia coli in Livestock and Farm Workers in Mecklenburg-Western Pomerania, Germany. PLoS ONE 2015, 10, e0143326. [Google Scholar] [CrossRef]
- Hooper, D.C.; Jacoby, G.A. Mechanisms of drug resistance: Quinolone resistance. Ann. N. Y. Acad. Sci. 2015, 1354, 12–31. [Google Scholar] [CrossRef]
- Federal Office of Consumer Protection and Food Safety (BVL). Erneut weniger Antibiotika in der Tiermedizin abgegeben. Available online: https://www.bvl.bund.de/SharedDocs/Pressemitteilungen/05_tierarzneimittel/2019/2019_07_25_PI_Antibiotikaabgabe.html (accessed on 10 December 2019).
- Lloyd, D.H.; Page, S.W. Antimicrobial Stewardship in Veterinary Medicine. Microbiol. Spectr. 2018, 6. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Farm | Total Number of Pigs | Additional Animals on Farm | Feed Consistency | Individual | Age in Months | Antibiotics |
|---|---|---|---|---|---|---|
| #1 | 1400 | Cattle | Solid | #1 | 0.5 | None |
| #2 | 0.5 | None | ||||
| #3 | 0.5 | None | ||||
| #4 | 65 | None | ||||
| #5 | 39 | None | ||||
| #6 | 52 | None | ||||
| #2 | 2000 | Dogs, cats | Liquid and pellets | #1 | 26 | None |
| #2 | 19.5 | None | ||||
| #3 | 26 | None | ||||
| #3 | 3800 | Dogs | Liquid | #1 | 52 | None |
| #2 | 39 | Enrofloxacin | ||||
| #3 | 51.5 | Enrofloxacin | ||||
| #4 | 1000 | Dogs, cats, horses | Liquid | #1 | 3 | None |
| #2 | 3 | None | ||||
| #3 | 3 | None | ||||
| #5 | 2200 | Dogs, horses | Liquid and solid | #1 | 2 | Amoxicillin |
| #2 | 2 | Amoxicillin | ||||
| #3 | 2 | Amoxicillin | ||||
| #6 | 1600 | Dogs | Liquid | #1 | 4 | None |
| #2 | 4 | None | ||||
| #3 | 4 | None | ||||
| #4 | 35.5 | None | ||||
| #5 | 9.5 | None | ||||
| #6 | 29.5 | None | ||||
| #7 | 1600 | Dogs, sheep | Liquid | #1 | 6 | None |
| #2 | 6 | None | ||||
| #3 | 6 | None |
| Farm | Individual | Sampling Site | spa Type | Resistance Gene mecA | Phenotypic Antimicrobial Susceptibility Test Profile a | # of Isolates | |
|---|---|---|---|---|---|---|---|
| CXI Screening | Other Resistances | ||||||
| #1 | #1 | Nasal cavity | t011 | + | + | OXA, BEN, TET | 2 |
| Snout surface | t011 | + | + | OXA, BEN, TET | 1 | ||
| #2 | Nasal cavity | t1298 | n/a | − | BEN, TET | 2 | |
| t337 | n/a | − | BEN, TET | 1 | |||
| Snout surface | t011 | + | + | OXA, BEN, TET | 1 | ||
| t337 | n/a | − | BEN, TET | 2 | |||
| #3 | Nasal cavity | t337 | n/a | − | BEN, TET | 1 | |
| Snout surface | t8893 | n/a | − | BEN, TET | 1 | ||
| t337 | n/a | − | BEN, TET | 4 | |||
| t1419 | n/a | − | BEN, TET, TRS | 2 | |||
| #2 | #2 | Nasal cavity | t337 | n/a | − | BEN | 2 |
| t8893 | n/a | − | BEN | 2 | |||
| Snout surface | t8893 | n/a | − | BEN | 2 | ||
| #3 | #1 | Snout surface | t17059 | n/a | − | LEV, BEN | 1 |
| #2 | Snout surface | t17059 | n/a | − | LEV, BEN | 2 | |
| #4 | #1 | Snout surface | t8893 | n/a | − | BEN | 1 |
| #2 | Snout surface | t034 | + | + | CLI, ERY, BEN, TRS, TET | 1 | |
| #5 | #1 | Nasal cavity | t034 | + | + | CLI, ERY, OXA, BEN, TRS, TET | 2 |
| t034 | + | + | CLI, ERY, OXA, BEN, TET | 1 | |||
| Snout surface | t034 | + | + | CLI, ERY, OXA, BEN, TRS, TET | 1 | ||
| #6 | #2 | Nasal cavity | t899 | + | + | CLI, LEV, OXA, BEN | 1 |
| #3 | Nasal cavity | t899 | n/a | − | LEV, BEN | 1 | |
| t899 | + | + | LEV, OXA, BEN | 3 | |||
| t899 | + | + | CLI, LEV, OXA, BEN | 1 | |||
| Snout surface | t899 | + | + | LEV, OXA, BEN | 9 | ||
| #4 | Snout surface | t2315 | n/a | − | BEN | 1 | |
| #7 | #1 | Nasal cavity | t12359 | + | + | CLI, ERY, OXA, BEN, TRS, TET | 1 |
| Snout surface | t12359 | + | + | CLI, ERY, OXA, BEN, TET | 2 | ||
| Farm | Individual | Sampling Site | Species | Resistance Gene | Resistances a |
|---|---|---|---|---|---|
| #4 | #1 | Nasal cavity | S. pasteuri | mecA | FOS, OXA, BEN, TET |
| S. haemolyticus | mecA | CLI, FOS, GEN, CXI, BEN, TET | |||
| S. pasteuri | mecA | FOS, OXA, BEN, TET | |||
| Snout surface | S. hominis | mecA | FOS, OXA, BEN, TET | ||
| S. pasteuri | mecA | CLI, FOS, OXA, CXI, BEN | |||
| S. pasteuri | mecA | CLI, FOS, OXA, CXI, BEN | |||
| #2 | Nasal cavity | S. saprophyticus | mecA | FOS, OXA, CXI, BEN, TET | |
| S. pasteuri | mecA | FOS, OXA, BEN, TET | |||
| S. epidermidis | mecA | CLI, OXA, CXI, TET | |||
| Snout surface | S. haemolyticus | mecA | CLI, FOS, GEN, CXI, BEN, TET | ||
| S. cohnii | mecA | CLI, ERY, FUS, OXA, CXI, BEN, TRS, TET | |||
| #3 | Nasal cavity | S. pasteuri | mecA | FOS, OXA, CXI, BEN, TET | |
| #5 | #1 | Nasal cavity | S. sciuri | mecA | CLI, DAP, FUS, OXA, CXI |
| S. sciuri | mecA | CLI, DAP, FUS, OXA, BEN, TET | |||
| M. goetzii | mecB | CLI, ERY, FOS, OXA, TET | |||
| Snout surface | M. goetzii | mecB | CLI, ERY, FOS, OXA, CXI, TET | ||
| S. epidermidis | mecA | CLI, ERY, FOS, OXA, CXI, BEN, TET | |||
| S. equorum | mecA | CLI, ERY, FOS, OXA, CXI, BEN, TET | |||
| #2 | Snout surface | S. epidermidis | mecA | CLI, ERY, FOS, OXA, CXI, BEN, TET | |
| S. sciuri | mecA | CLI, DAP, ERY, FUS, OXA, CXI, BEN, TET | |||
| S. sciuri | mecA | CLI, DAP, ERY, FUS, OXA, CXI, BEN, TET | |||
| S. sciuri | mecA | CLI, DAP, ERY, FUS, OXA, CXI, BEN, TET | |||
| S. haemolyticus | mecA | CLI, ERY, FOS, GEN, OXA, CXI, BEN, TET | |||
| #7 | #2 | Snout surface | S. pasteuri | mecA | CLI, ERY, FOS, OXA, CXI, BEN, TET |
| #3 | Snout surface | S. pasteuri | mecA | CLI, ERY, FOS, OXA, BEN, TET |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schlattmann, A.; von Lützau, K.; Kaspar, U.; Becker, K. The Porcine Nasal Microbiota with Particular Attention to Livestock-Associated Methicillin-Resistant Staphylococcus aureus in Germany—A Culturomic Approach. Microorganisms 2020, 8, 514. https://doi.org/10.3390/microorganisms8040514
Schlattmann A, von Lützau K, Kaspar U, Becker K. The Porcine Nasal Microbiota with Particular Attention to Livestock-Associated Methicillin-Resistant Staphylococcus aureus in Germany—A Culturomic Approach. Microorganisms. 2020; 8(4):514. https://doi.org/10.3390/microorganisms8040514
Chicago/Turabian StyleSchlattmann, Andreas, Knut von Lützau, Ursula Kaspar, and Karsten Becker. 2020. "The Porcine Nasal Microbiota with Particular Attention to Livestock-Associated Methicillin-Resistant Staphylococcus aureus in Germany—A Culturomic Approach" Microorganisms 8, no. 4: 514. https://doi.org/10.3390/microorganisms8040514
APA StyleSchlattmann, A., von Lützau, K., Kaspar, U., & Becker, K. (2020). The Porcine Nasal Microbiota with Particular Attention to Livestock-Associated Methicillin-Resistant Staphylococcus aureus in Germany—A Culturomic Approach. Microorganisms, 8(4), 514. https://doi.org/10.3390/microorganisms8040514