Duodenal Metatranscriptomics to Define Human and Microbial Functional Alterations Associated with Severe Obesity: A Pilot Study


 ,
,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Controls
2.2. Sample Collection
2.3. RNA Isolation and Sequencing
2.4. Metatranscriptomics Analysis/Processing
2.4.1. Human Transcriptome Analysis
2.4.2. Bacterial Transcriptome Analysis
3. Results
3.1. Severely Obese Subjects Show Significant Alterations in Blood Parameters Compared with Lean Controls
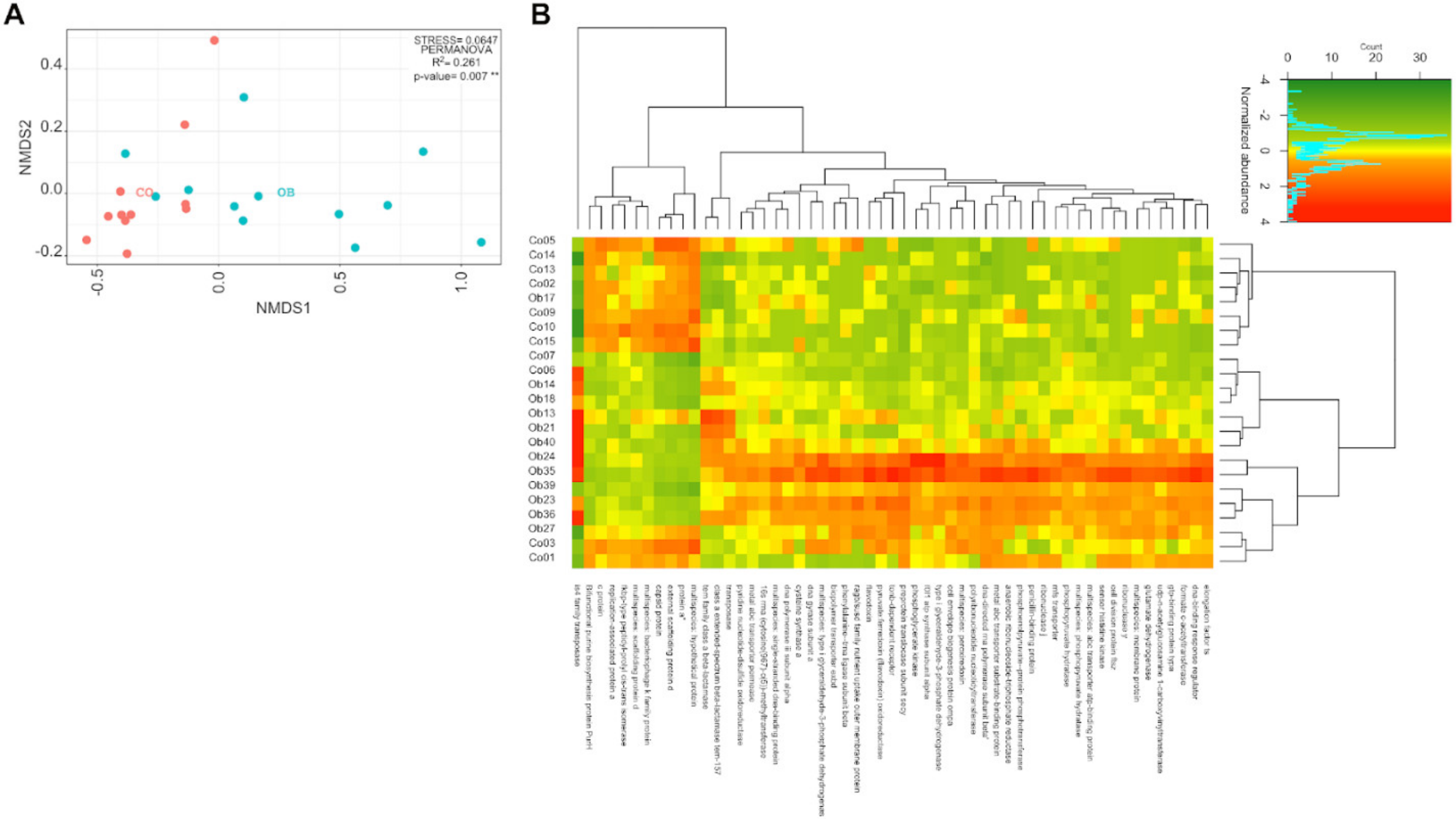
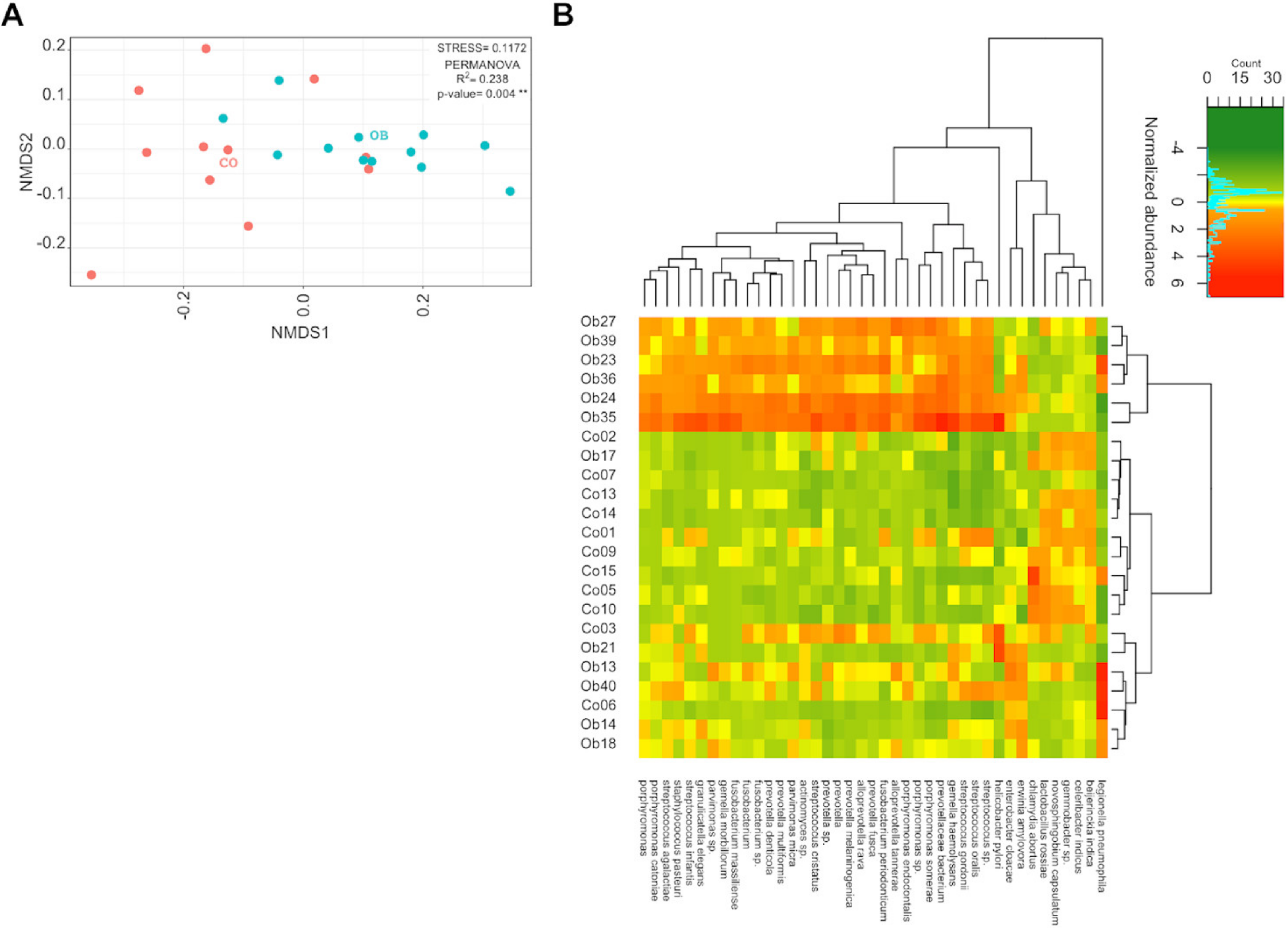
3.2. The Variability of the Bacterial Transcriptome Profile Is Higher in Obese than in Control Samples
3.3. Prevotella and Streptococcus Are Likely the Major Contributors to Some of the Altered Microbiota Metabolic Functions in Obese Subjects
3.4. Taxa Abundance May Not Correspond to Transcriptional Activity
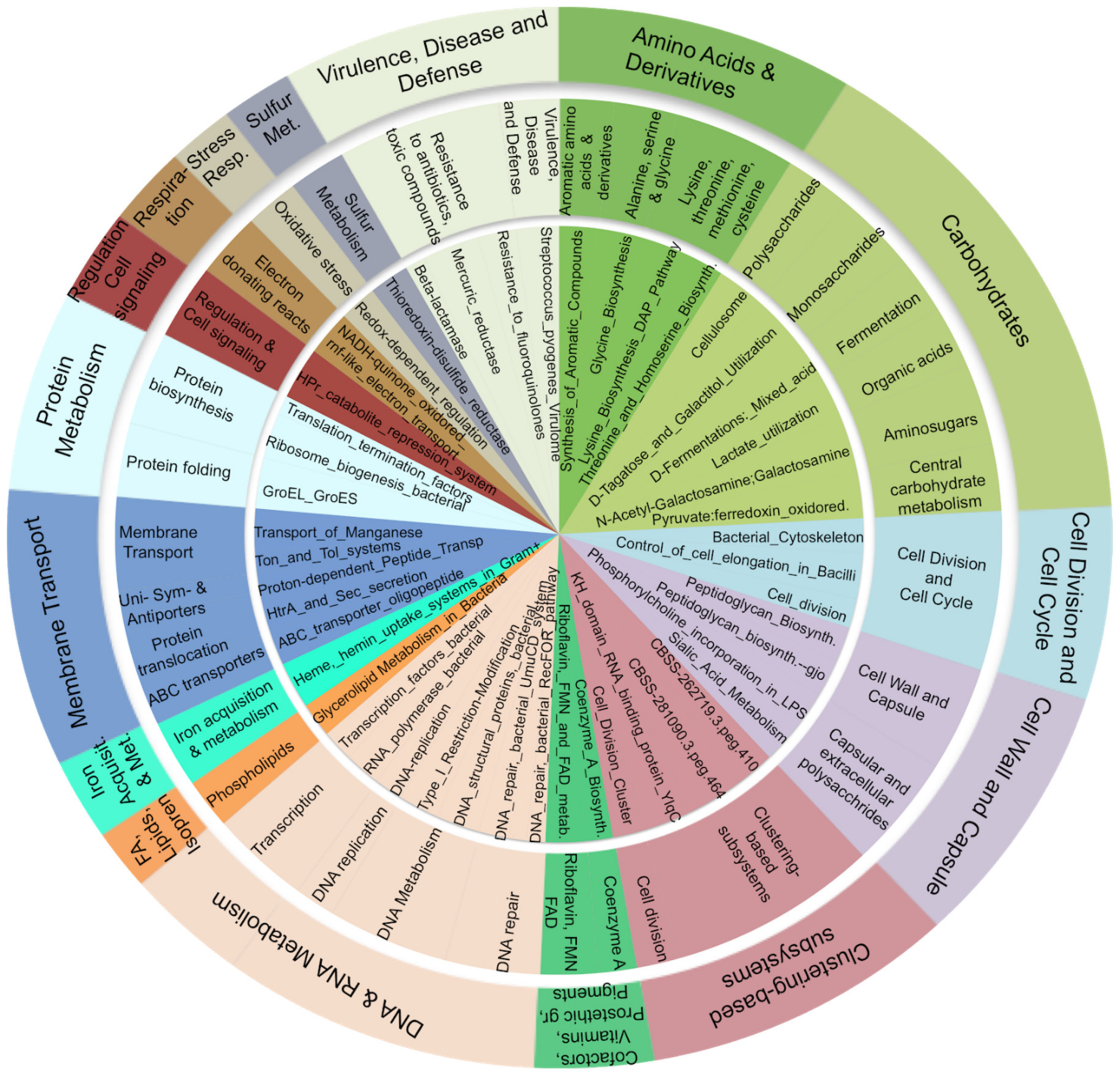
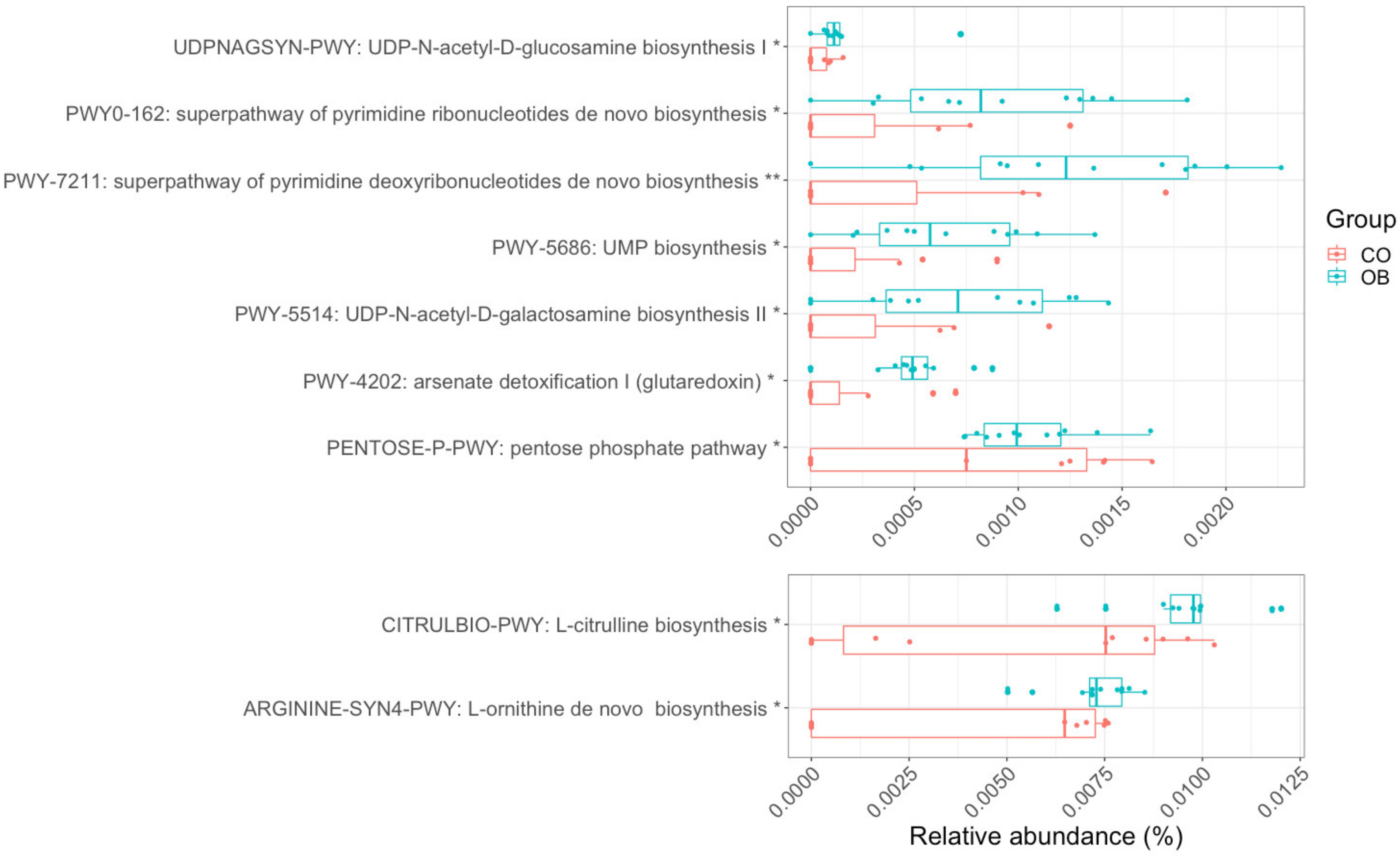
3.5. Carbohydrate, Amino-Acid, and Nucleotide Metabolism Are Impaired in Obese Microbiota
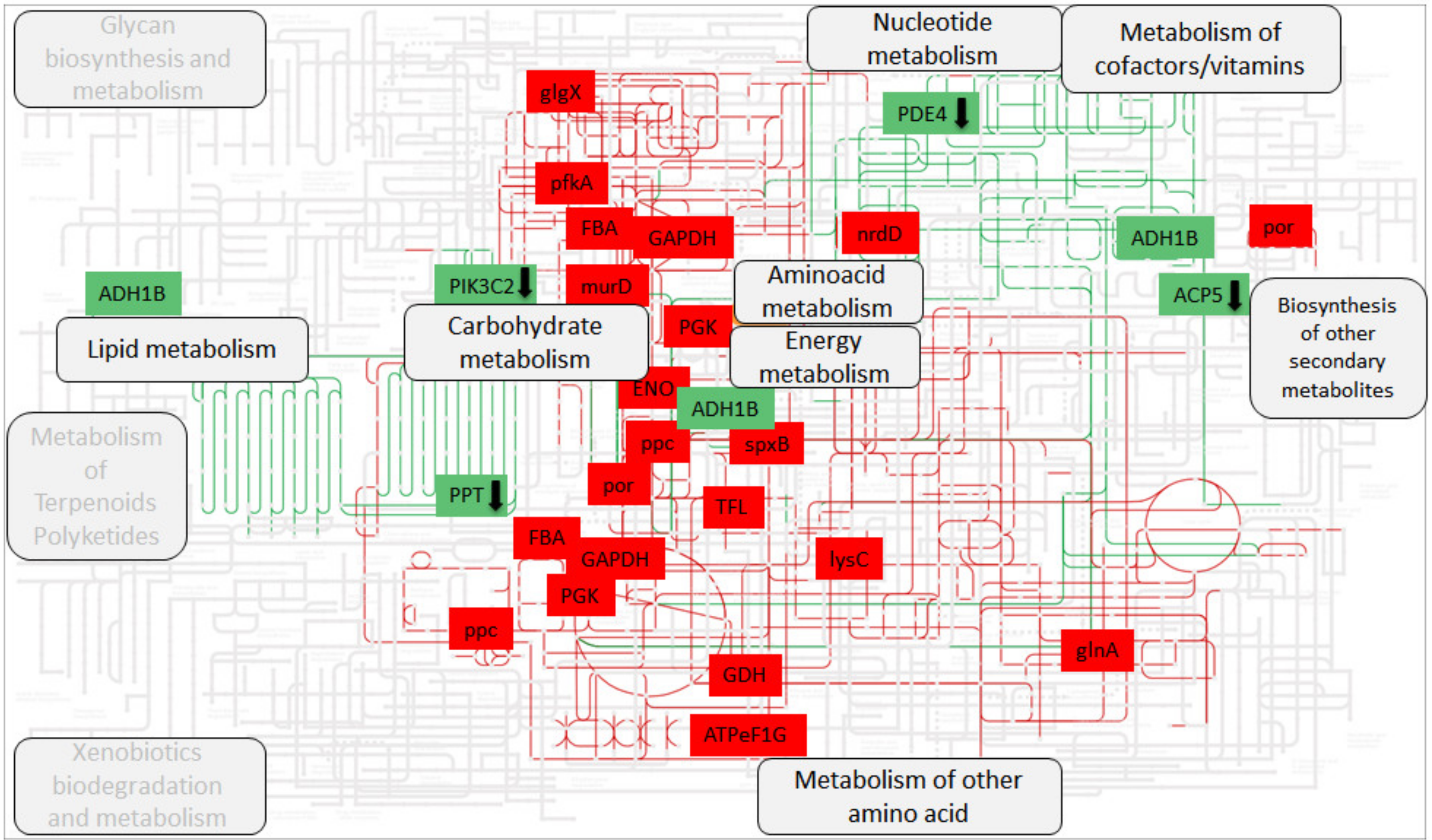
3.6. Differentially Expressed Host Genes Share the Same Metabolic Pathways as Microbial Genes
4. Discussion
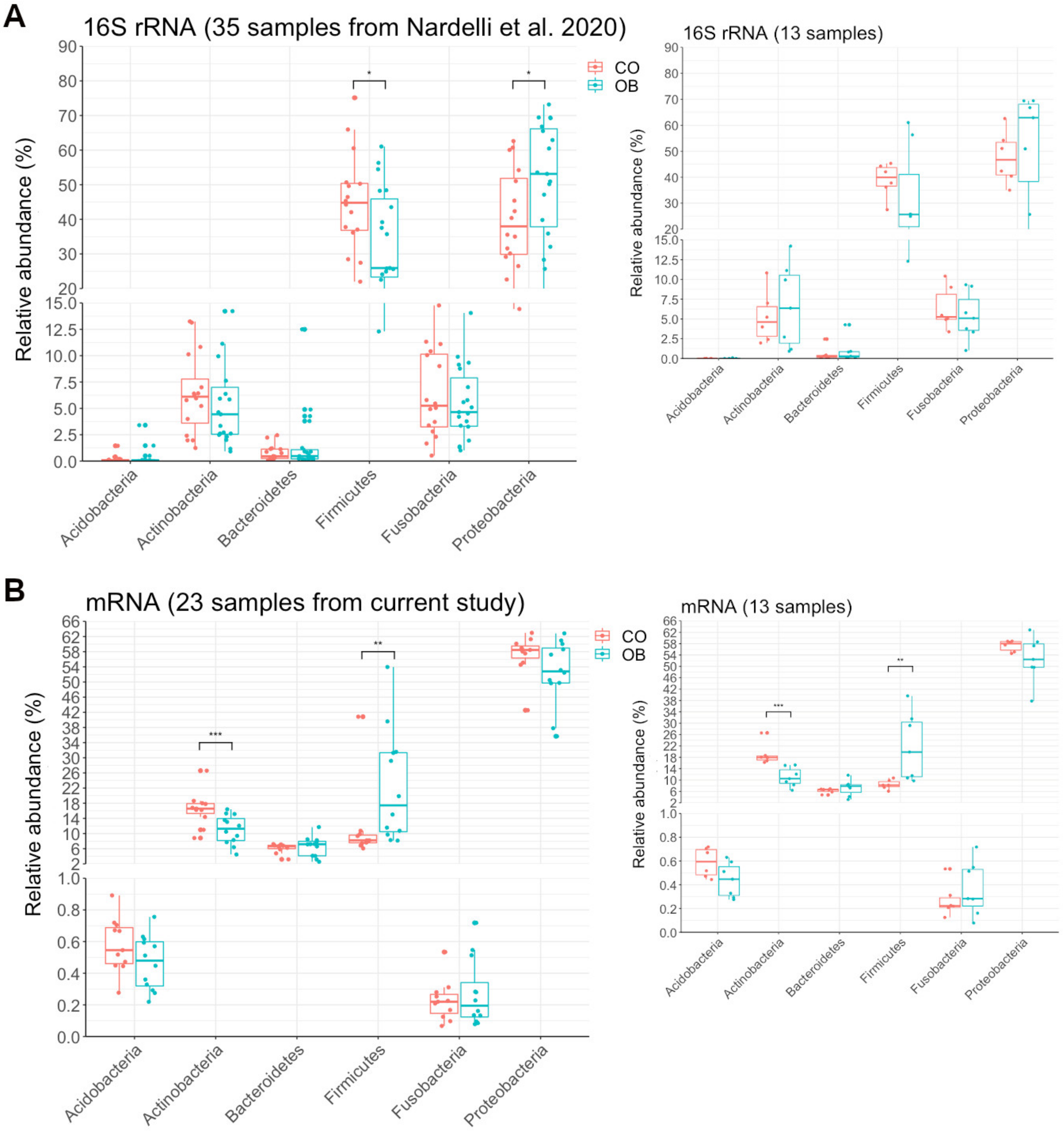
4.1. Microbiome Composition by 16S rRNA Compared to Microbiome Inferred by Transcriptome
4.2. An Increased Prevotella/Bacteroides Activity Ratio Is Associated with Obesity
4.3. The Bacterial Functional Profile in Duodenum Differs between Obese and Lean Control Subjects
4.3.1. Carbohydrate Metabolism
4.3.2. Cell Wall
4.3.3. Defense Mechanisms: The Arsenate Detoxification Pathway
4.3.4. Amino Acids and Derivatives
4.3.5. Nucleic Acids
4.4. The Human Tanscriptional Profile in the Duodenum Differs between Obese and Lean Subjects
4.5. Putative Convergence between Host and Microbial Transcriptomes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GAPDH | NAD-dependent glyceraldehyde-3-phosphate dehydrogenase (EC 1.2.1.12) |
| spxB | Pyruvate oxidase (EC 1.2.3.3) |
| GDH | NADP-specific glutamate dehydrogenase (EC 1.4.1.4) |
| PFL | Pyruvate formate-lyase (EC 2.3.1.54) |
| pfkA | 6-phosphofructokinase (EC 2.7.1.11) |
| PGK | Phosphoglycerate kinase (EC 2.7.2.3) |
| lysC | Aspartokinase (EC 2.7.2.4) |
| ppc | Phosphoenolpyruvate carboxylase (EC 4.1.1.31) |
| FBA | Fructose-bisphosphate aldolase class II (EC 4.1.2.13) |
| ENO | Enolase (EC 4.2.1.11) |
| glnA | Glutamine synthetase type I (EC 6.3.1.2) |
| murD | UDP-N-acetylmuramoylalanine--D-glutamate ligase (EC 6.3.2.9) |
| ATPeF1G | ATP synthase gamma chain (EC 3.6.3.14) |
| glgX | Glycogen debranching enzyme (EC 3.2.1.196) |
| por | Pyruvate-flavodoxin oxidoreductase (EC 1.2.7.1) |
| nrdD | Ribonucleotide reductase of class III (anaerobic), large subunit (EC 1.17.4.2) |
| PIK3C2B | phosphatidylinositol-4-phosphate 3-kinase catalytic subunit type 2 beta (EC 2.7.1.154) |
| PPT | palmitoyl-protein thioesterase 1 (EC 3.1.2.22) |
| PDE4 | phosphodiesterase 4 (EC 3.1.4.53) |
| ADH1B | alcohol dehydrogenase 1B (class I), beta polypeptide (ADH1B) (EC 1.1.1.1) |
| ACP5 | acid phosphatase 5, tartrate resistant (EC 3.1.3.2) |
References
- Finkelstein, E.A.; Khavjou, O.A.; Thompson, H.; Trogdon, J.G.; Pan, L.; Sherry, B.; Dietz, W. Obesity and Severe Obesity Forecasts Through 2030. Am. J. Prev. Med. 2012, 42, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Meijnikman, A.S.; Gerdes, V.E.; Nieuwdorp, M.; Herrema, H. Evaluating Causality of Gut Microbiota in Obesity and Diabetes in Humans. Endocr. Rev. 2018, 39, 133–153. [Google Scholar] [CrossRef] [PubMed]
- Sacchetti, L.; Nardelli, C. Gut microbiome investigation in celiac disease: From methods to its pathogenetic role. Clin. Chem. Lab. Med. 2020, 58, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, B.D.; Goncalves, M.D.; Cantley, L.C. Obesity and Cancer Mechanisms: Cancer Metabolism. J. Clin. Oncol. 2016, 34, 4277–4283. [Google Scholar] [CrossRef] [PubMed]
- Biener, A.I.; Cawley, J.; Meyerhoefer, C. The Impact of Obesity on Medical Care Costs and Labor Market Outcomes in the US. Clin. Chem. 2018, 64, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, E.A.; Trogdon, J.G.; Brown, D.S.; Allaire, B.T.; Dellea, P.S.; Kamal-Bahl, S.J. The Lifetime Medical Cost Burden of Overweight and Obesity: Implications for Obesity Prevention. Obesity 2008, 16, 1843–1848. [Google Scholar] [CrossRef]
- Blakemore, A.I.; Froguel, P. Investigation of Mendelian forms of obesity holds out the prospect of personalized medicine. Ann. N. Y. Acad. Sci. 2010, 1214, 180–189. [Google Scholar] [CrossRef]
- Rohde, K.; Keller, M.; Poulsen, L.L.C.; Blüher, M.; Kovacs, P.; Böttcher, Y. Genetics and epigenetics in obesity. Metabolism 2019, 92, 37–50. [Google Scholar] [CrossRef]
- Mathes, W.F.; Kelly, S.A.; Pomp, D. Advances in comparative genetics: Influence of genetics on obesity. Br. J. Nutr. 2011, 106, S1–S10. [Google Scholar] [CrossRef]
- Capobianco, V.; Nardelli, C.; Ferrigno, M.; Iaffaldano, L.; Pilone, V.; Forestieri, P.; Sacchetti, L. MiRNA and protein expression profiles of visceral adipose tissue reveal miR-141/ywhag and miR-520e/rab11a as two potential miRNA/protein target pairs associated with severe obesity. J. Proteome Res. 2012, 11, 3358–3369. [Google Scholar] [CrossRef]
- Nardelli, C.; Granata, I.; Iaffaldano, L.; D’Argenio, V.; Del Monaco, V.; Maruotti, G.M.; Guarracino, M.R. MiR-138/miR-222 overexpression characterizes the miRNome of amniotic mesenchymal stem cells in obesity. Stem Cells Dev. 2017, 26, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nat. Cell Biol. 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- John, G.K.; Mullin, G.E. The Gut Microbiome and Obesity. Curr. Oncol. Rep. 2016, 18, 1–7. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell 2016, 164, 337–340. [Google Scholar] [CrossRef]
- Alonso, V.R.; Guarner, F. Linking the gut microbiota to human health. Br. J. Nutr. 2013, 109, S21–S26. [Google Scholar] [CrossRef]
- Kho, Z.Y.; Lal, S.K. The Human Gut Microbiome—A Potential Controller of Wellness and Disease. Front. Microbiol. 2018, 9, 1835. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Kuczynski, J.; Lauber, C.L.; Walters, W.A.; Parfrey, L.W.; Clemente, J.C.; Gevers, D.; Knight, R. Experimental and analytical tools for studying the human microbiome. Nat. Rev. Genet. 2012, 13, 47–58. [Google Scholar] [CrossRef]
- Shakya, M.; Lo, C.-C.; Chain, P.S. Advances and Challenges in Metatranscriptomic Analysis. Front. Genet. 2019, 10, 904. [Google Scholar] [CrossRef]
- Kootte, R.S.; Vrieze, A.; Holleman, F.; Dallinga-Thie, G.M.; Zoetendal, E.G.; De Vos, W.M.; Groen, A.K.; Hoekstra, J.B.L.; Stroes, E.S.; Nieuwdorp, M. The therapeutic potential of manipulating gut microbiota in obesity and type 2 diabetes mellitus. Diabetes Obes. Metab. 2011, 14, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Moran-Ramos, S.; López-Contreras, B.E.; Canizales-Quinteros, S. Gut Microbiota in Obesity and Metabolic Abnormalities: A Matter of Composition or Functionality? Arch. Med Res. 2017, 48, 735–753. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Ma, L.; Ma, Y.; Zhang, F.; Zhao, C.; Nie, Y. Insights into the role of gut microbiota in obesity: Pathogenesis, mechanisms, and therapeutic perspectives. Protein Cell 2018, 9, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Nardelli, C.; Granata, I.; D’Argenio, V.; Tramontano, S.; Compare, D.; Guarracino, M.R.; Nardone, G.; Pilone, V.; Sacchetti, L. Characterization of the Duodenal Mucosal Microbiome in Obese Adult Subjects by 16S rRNA Sequencing. Microorganisms 2020, 8, 485. [Google Scholar] [CrossRef]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Shilo, S. Environment dominates over host genetics in shaping human gut microbiota. Nature 2018, 555, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, M.; Franzosa, E.A.; Lloyd-Price, J.; McIver, L.J.; Schwager, R.; Poon, T.W.; Ananthakrishnan, A.N.; Andrews, E.; Barron, G.; Lake, K.; et al. Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat. Microbiol. 2018, 3, 337–346. [Google Scholar] [CrossRef]
- Abu-Ali, G.S.; Mehta, R.S.; Lloyd-Price, J.; Mallick, H.; Branck, T.; Ivey, K.L.; Drew, D.A.; Dulong, C.; Rimm, E.; Izard, J.; et al. Metatranscriptome of human faecal microbial communities in a cohort of adult men. Nat. Microbiol. 2018, 3, 356–366. [Google Scholar] [CrossRef]
- Wang, M.; Ahrné, S.; Jeppsson, B.; Molin, G. Comparison of bacterial diversity along the human intestinal tract by direct cloning and sequencing of 16S rRNA genes. FEMS Microbiol. Ecol. 2005, 54, 219–231. [Google Scholar] [CrossRef]
- Vasapolli, R.; Schütte, K.; Schulz, C.; Vital, M.; Schomburg, D.; Pieper, D.H.; Vilchez-Vargas, R.; Malfertheiner, P. Analysis of Transcriptionally Active Bacteria Throughout the Gastrointestinal Tract of Healthy Individuals. Gastroenterology 2019, 157, 1081–1092. [Google Scholar] [CrossRef]
- Zoetendal, E.G.; Raes, J.; Bogert, B.V.D.; Arumugam, M.; Booijink, C.C.G.M.; Troost, F.J.; Bork, P.; Wels, M.; De Vos, W.M.; Kleerebezem, M. The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. ISME J. 2012, 6, 1415–1426. [Google Scholar] [CrossRef]
- Krajmalnik-Brown, R.; Ilhan, Z.-E.; Kang, D.-W.; DiBaise, J.K. Effects of Gut Microbes on Nutrient Absorption and Energy Regulation. Nutr. Clin. Pract. 2012, 27, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Tomé, D. Digestibility Issues of Vegetable versus Animal Proteins: Protein and Amino Acid Requirements—Functional Aspects. Food Nutr. Bull. 2013, 34, 272–274. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FASTQC. A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.scienceopen.com/document?vid=de674375-ab83-4595-afa9-4c8aa9e4e736 (accessed on 16 November 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Love, M.H.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Westreich, S.T.; Treiber, M.L.; Mills, D.A.; Korf, I.; Lemay, D.G. SAMSA2: A standalone metatranscriptome analysis pipeline. BMC Bioinform. 2018, 19, 175. [Google Scholar] [CrossRef]
- Franzosa, E.A.; McIver, L.J.; Rahnavard, G.; Thompson, L.R.; Schirmer, M.; Weingart, G.; Lipson, K.S.; Knight, R.; Caporaso, J.G.; Segata, N.; et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 2018, 15, 962–968. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; E Midford, P.; Ong, W.K.; Paley, S.; Subhraveti, P.; Karp, P.D. The MetaCyc database of metabolic pathways and enzymes—A 2019 update. Nucleic Acids Res. 2020, 48, D445–D453. [Google Scholar] [CrossRef] [PubMed]
- Mallick, H.; Rahnavard, A.; McIver, L. Maaslin2: Maaslin2. R Package Version 1.2.0. Bioconductor. 2019. Available online: http://huttenhower.sph.harvard.edu/maaslin2 (accessed on 10 November 2020). [CrossRef]
- Sanchez, A.; Golding, I. Genetic Determinants and Cellular Constraints in Noisy Gene Expression. Science 2013, 342, 1188–1193. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, G.; Kost, C. Experimental Evolution of Metabolic Dependency in Bacteria. PLoS Genet. 2016, 12, e1006364. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Human gut microbes associated with obesity. Nat. Cell Biol. 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Ley, R.E. Prevotella in the gut: Choose carefully. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 69–70. [Google Scholar] [CrossRef]
- Gorvitovskaia, A.; Holmes, S.P.; Huse, S.M. Interpreting Prevotella and Bacteroides as biomarkers of diet and lifestyle. Microbiome 2016, 4, 15. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- De Filippis, F.; Pellegrini, N.; Vannini, L.; Jeffery, I.B.; La Storia, A.; Laghi, L.; I Serrazanetti, D.; Di Cagno, R.; Ferrocino, I.; Lazzi, C.; et al. High-level adherence to a Mediterranean diet beneficially impacts the gut microbiota and associated metabolome. Gut 2015, 65, 1812–1821. [Google Scholar] [CrossRef]
- Hjorth, M.F.; Christensen, L.; Kjølbæk, L.; Larsen, L.H.; Roager, H.M.; Kiilerich, P.; Kristiansen, K.; Astrup, A. Pretreatment Prevotella-to-Bacteroides ratio and markers of glucose metabolism as prognostic markers for dietary weight loss maintenance. Eur. J. Clin. Nutr. 2019, 74, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Franke, T.; Deppenmeier, U. Physiology and central carbon metabolism of the gut bacterium Prevotella copri. Mol. Microbiol. 2018, 109, 528–540. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, M.; Ferrocino, I.; Calabrese, F.M.; De Filippis, F.; Cavallo, N.; Siragusa, S.; Rampelli, S.; Di Cagno, R.; Rantsiou, K.; Vannini, L.; et al. Diet influences the functions of the human intestinal microbiome. Sci. Rep. 2020, 10, 4247. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, F.; Pellegrini, N.; Laghi, L.; Gobbetti, M.; Ercolini, D. Unusual sub-genus associations of faecal Prevotella and Bacteroides with specific dietary patterns. Microbiome 2016, 4, 57. [Google Scholar] [CrossRef] [PubMed]
- Dadashipour, M.; Iwamoto, M.; Hossain, M.M.; Akutsu, J.-I.; Zhang, Z.; Kawarabayasi, Y.; Hossain, M. Identification of a Direct Biosynthetic Pathway for UDP–N-Acetylgalactosamine from Glucosamine-6-Phosphate in Thermophilic Crenarchaeon Sulfolobus tokodaii. J. Bacteriol. 2018, 200, e00048-18. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, R.P.; Lowary, T.L. New insights into lipopolysaccharide assembly and export. Curr. Opin. Chem. Biol. 2019, 53, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Possemiers, S.; Van De Wiele, T.; Guiot, Y.; Everard, A.; Rottier, O.; Geurts, L.; Naslain, D.; Neyrinck, A.; Lambert, D.M.; et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 2009, 58, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.E.; Snow, J.; Li, J.; Zola, T.A.; Weiser, J.N. Phosphorylcholine Allows for Evasion of Bactericidal Antibody by Haemophilus influenzae. PLoS Pathog. 2012, 8, e1002521. [Google Scholar] [CrossRef] [PubMed]
- Muscogiuri, G.; Cantone, E.; Cassarano, S.; Tuccinardi, D.; Barrea, L.; Savastano, S.; Colao, A. Gut microbiota: A new path to treat obesity. Int. J. Obes. Suppl. 2019, 9, 10–19. [Google Scholar] [CrossRef]
- Zhao, F.-J. A novel pathway of arsenate detoxification. Mol. Microbiol. 2016, 100, 928–930. [Google Scholar] [CrossRef]
- Chen, J.; Yoshinaga, M.; Garbinski, L.D.; Rosen, B.P. Synergistic interaction of glyceraldehydes-3-phosphate dehydrogenase and ArsJ, a novel organoarsenical efflux permease, confers arsenate resistance. Mol. Microbiol. 2016, 100, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Bekebrede, A.F.; Keijer, J.; Gerrits, W.J.J.; De Boer, V.C.J. The Molecular and Physiological Effects of Protein-Derived Polyamines in the Intestine. Nutrients 2020, 12, 197. [Google Scholar] [CrossRef] [PubMed]
- Lipowska, J.; Miks, C.D.; Kwon, K.; Shuvalova, L.; Zheng, H.; Lewiński, K.; Cooper, D.R.; Shabalin, I.G.; Minor, W. Pyrimidine biosynthesis in pathogens—Structures and analysis of dihydroorotases from Yersinia pestis and Vibrio cholerae. Int. J. Biol. Macromol. 2019, 136, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Almohazey, D.; Lo, Y.-H.; Vossler, C.V.; Simmons, A.J.; Hsieh, J.J.; Bucar, E.B.; Schumacher, M.A.; E Hamilton, K.; Lau, K.S.; Shroyer, N.F.; et al. The ErbB3 receptor tyrosine kinase negatively regulates Paneth cells by PI3K-dependent suppression of Atoh1. Cell Death Differ. 2017, 24, 855–865. [Google Scholar] [CrossRef]
- Sehgal, A.; Donaldson, D.S.; Pridans, C.; Sauter, K.A.; Hume, D.A.; Mabbott, N.A. The role of CSF1R-dependent macrophages in control of the intestinal stem-cell niche. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Sato, T.; Van Es, J.H.; Snippert, H.J.; Stange, D.E.; Vries, R.G.; Born, M.V.D.; Barker, N.; Shroyer, N.F.; Van De Wetering, M.; Clevers, H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nat. Cell Biol. 2011, 469, 415–418. [Google Scholar] [CrossRef]
- Grewal, T.; Enrich, C.; Rentero, C.; Buechler, C. Annexins in Adipose Tissue: Novel Players in Obesity. Int. J. Mol. Sci. 2019, 20, 3449. [Google Scholar] [CrossRef]
- Akasheh, R.T.; Pini, M.; Pang, J.; Fantuzzi, G. Increased Adiposity in Annexin A1-Deficient Mice. PLoS ONE 2013, 8, e82608. [Google Scholar] [CrossRef]
- Kosicka, A.; Cunliffe, A.D.; MacKenzie, R.; Zariwala, M.G.; Perretti, M.; Flower, R.J.; Renshaw, D. Attenuation of plasma annexin A1 in human obesity. FASEB J. 2012, 27, 368–378. [Google Scholar] [CrossRef]
- Pietrani, N.T.; Ferreira, C.N.; Rodrigues, K.F.; Perucci, L.O.; Carneiro, F.S.; Bosco, A.A.; Oliveira, M.C.; Pereira, S.S.; Teixeira, A.L.; Alvarez-Leite, J.I.; et al. Proresolving protein Annexin A1: The role in type 2 diabetes mellitus and obesity. Biomed. Pharmacother. 2018, 103, 482–489. [Google Scholar] [CrossRef]
- Purvis, G.S.D.; Collino, M.; Loiola, R.A.; Baragetti, A.; Chiazza, F.; Brovelli, M.; Sheikh, M.H.; Collotta, D.; Cento, A.; Mastrocola, R.; et al. Identification of AnnexinA1 as an Endogenous Regulator of RhoA, and Its Role in the Pathophysiology and Experimental Therapy of Type-2 Diabetes. Front. Immunol. 2019, 10, 571. [Google Scholar] [CrossRef] [PubMed]
- Nelson, W.J. Epithelial cell polarity from the outside looking in. Physiology 2003, 18, 143–146. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tanos, B.E.; Yeaman, C.; Rodriguez-Boulan, E. An emerging role for IQGAP1 in tight junction control. Small GTPases 2018, 9, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Kim, S.H.; Higgins, J.M.G.; Brenner, M.B.; Sacks, D.B. IQGAP1 and Calmodulin Modulate E-cadherin Function. J. Biol. Chem. 1999, 274, 37885–37892. [Google Scholar] [CrossRef] [PubMed]
- Lechuga, S.; Baranwal, S.; Ivanov, A.I. Actin-interacting protein 1 controls assembly and permeability of intestinal epithelial apical junctions. Am. J. Physiol. Liver Physiol. 2015, 308, G745–G756. [Google Scholar] [CrossRef] [PubMed]
- Moir, R.; Spann, T. The structure and function of nuclear lamins: Implications for disease. Cell. Mol. Life Sci. 2001, 58, 1748–1757. [Google Scholar] [CrossRef]
- Wong, C.-M.; Xu, L.; Yau, M.Y.-C. Alternative mRNA Splicing in the Pathogenesis of Obesity. Int. J. Mol. Sci. 2018, 19, 632. [Google Scholar] [CrossRef]
- Winnier, D.A.; Fourcaudot, M.; Norton, L.; Abdul-Ghani, M.A.; Hu, S.L.; Farook, V.S.; Coletta, D.K.; Kumar, S.; Puppala, S.; Chittoor, G.; et al. Transcriptomic Identification of ADH1B as a Novel Candidate Gene for Obesity and Insulin Resistance in Human Adipose Tissue in Mexican Americans from the Veterans Administration Genetic Epidemiology Study (VAGES). PLoS ONE 2015, 10, e0119941. [Google Scholar] [CrossRef]
- Ahtiainen, L.; Kolikova, J.; Mutka, A.-L.; Luiro, K.; Gentile, M.; Ikonen, E.; Khiroug, L.; Jalanko, A.; Kopra, O. Palmitoyl protein thioesterase 1 (Ppt1)-deficient mouse neurons show alterations in cholesterol metabolism and calcium homeostasis prior to synaptic dysfunction. Neurobiol. Dis. 2007, 28, 52–64. [Google Scholar] [CrossRef]
- Zhao, W.; Su, J.; Wang, Y.; Qian, T.; Liu, Y. Functional importance of palmitoyl protein thioesterase 1 (PPT1) expression by Sertoli cells in mediating cholesterol metabolism and maintenance of sperm quality. Mol. Reprod. Dev. 2019, 86, 984–998. [Google Scholar] [CrossRef]
- Margaria, J.P.; Ratto, E.; Gozzelino, L.; Li, H.; Hirsch, E. Class II PI3Ks at the Intersection between Signal Transduction and Membrane Trafficking. Biomolecules 2019, 9, 104. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcripts | Localization | Log2FC | Adjusted p-Value | Putative Role * | |
|---|---|---|---|---|---|
| BCL2 | Apoptosis regulator | Mitochondrial membrane | −1.82 | 0.004 | Immunity/Inflammation/ Apoptosis |
| TNFRSF13B | TNF receptor superfamily member 13B | Plasma membrane | −1.44 | 0.031 | |
| CSF1R | Colony stimulating factor 1 receptor | Membrane | −1.62 | 0.037 | |
| ACP5 | Acid phosphatase 5, tartrate resistant | Lysosome | −1.06 | 0.027 | |
| IGLL5 | Immunoglobulin lambda like polypeptide 5 | Extracellular localization | −1.75 | 0.027 | |
| ANXA1 | Annexin 1 | Different cellular localization | −2.12 | 0.039 | |
| ERBB3 | Erb-b2 receptor tyrosine kinase 3 | Plasma membrane | −24.56 | <0.0001 | |
| NGEF | Neuronal guanine nucleotide exchange factor | Membrane | 3.66 | 0.025 | |
| P4HB | Prolyl 4-hydroxylase subunit beta | Cytoplasm | 21.74 | <0.0001 | |
| PRMT5 | Protein arginine methyltransferase 5 | Nucleus and cytoplasm | −7.43 | 0.0001 | Nucleic acids structure and functions |
| TSEN34 | tRNA splicing endonuclease subunit 34 | Nucleus | −5.8 | 0.031 | |
| EIF3CL | Eukaryotic translation initiation factor 3 subunit C-like | Cytoplasm | 21.82 | <0.0001 | |
| ELF2 | E74 like ETS transcription factor 2 | Nucleus and cytoplasm | 6.94 | 0.008 | |
| WBP2 | WW domain binding protein 2 | Nucleus and cytoplasm | 2.88 | 0.0022 | |
| ZNF195 | Zinc finger protein 195 | Nucleus | 3.21 | 0.033 | |
| SRRT | Serrate, RNA effector molecule | Nucleus | 6.18 | <0.0001 | |
| PDE4A | Phosphodiesterase 4 | Cytoplasm | −1.22 | 0.015 | |
| ANXA2 | Annexin 2 | Different cellular localization | 1.69 | 0.012 | Cellular components/functions: membrane structure, cytoskeleton |
| IQGAP1 | IQ motif containing GTPase activating protein 1 | Plasma membrane | −1.73 | 0.008 | |
| WDR1 | WD repeat domain 1 | Different cellular localization | −24.6 | <0.0001 | |
| ANXA1 | Annexin 1 | Nucleus | −2.12 | 0.039 | |
| BANF1 | Barrier to autointegration factor 1 | Different cellular localization | −1.15 | 0.041 | |
| CDR1 | Cerebellar degeneration related protein 1 | Nucleus | −2.53 | 0.012 | |
| FAM192A | Family with sequence similarity 192 member A | Plasma membrane | −5.59 | 0.014 | |
| PARD3 | Par-3 family cell polarity regulator | Cytoplasm and membrane | −8.32 | 0.023 | |
| PLEKHB1 | Pleckstrin homology domain containing B1 | Plasma membrane | −1.23 | 0.012 | |
| RGMA | Repulsive guidance molecule family member a | Cytoplasm | −4.68 | 0.037 | |
| CLASP1 | Cytoplasmic linker associated protein 1 | Plasma membrane | 3.80 | 0.014 | |
| CD82 | CD82 molecule | Membrane | 5.78 | 0.041 | |
| DAG1 | Dystroglycan 1 | Membrane | 4.47 | 0.009 | |
| ILDR1 | Immunoglobulin like domain containing receptor 1 | Membrane | 6.24 | 0.027 | |
| LMNA | Lamin A/C | Nucleus | −9.63 | 0.028 | |
| CASC4 | Cancer susceptibility candidate 4 | Membrane | 7.62 | <0.0001 | |
| MME | membrane metalloendopeptidase | Membrane | 20.61 (P) | <0.0001 | Metabolism: lipid (L), carbohydrates (C), protein (P) |
| PPT1 | palmitoyl-protein thioesterase 1 | Lysosome | −1.37 (L) | 0.014 | |
| MGEA5 | Meningioma expressed antigen 5 (hyaluronidase) | Nucleus | −24.08 (C) | <0.0001 | |
| RAP1GDS1 | Rap1 GTPase-GDP dissociation stimulator 1 | Cytoplasm | −4.02 (L) | 0.045 | |
| LMNA | Lamin A/C | Nucleus | −9.63 (L+C) | 0.028 | |
| PGA3 | Pepsinogen 3, group I | Different cellular localization | −5.92 (P) | 0.031 | |
| CALM2 | Calmodulin 2 | Different cellular localization | 10.91 (L+C) | <0.0001 | |
| PIK3C2B | Phosphatidylinositol-4-phosphate 3-kinase catalytic subunit type 2 beta | Cytosol | −7.39 (L) | 0.027 | |
| ADH1B | Alcohol dehydrogenase 1B | Different cellular localization | 3.41 (L+C) | 0.005 | |
| ANXA2 | Annexin 2 | Different cellular localization | 1.69 (L+C) | 0.012 | |
| BCL2 | Apoptosis regulator | Mitochondrial membrane | −1.82 (L) | 0.004 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Granata, I.; Nardelli, C.; D’Argenio, V.; Tramontano, S.; Compare, D.; Guarracino, M.R.; Nardone, G.; Pilone, V.; Sacchetti, L. Duodenal Metatranscriptomics to Define Human and Microbial Functional Alterations Associated with Severe Obesity: A Pilot Study. Microorganisms 2020, 8, 1811. https://doi.org/10.3390/microorganisms8111811
Granata I, Nardelli C, D’Argenio V, Tramontano S, Compare D, Guarracino MR, Nardone G, Pilone V, Sacchetti L. Duodenal Metatranscriptomics to Define Human and Microbial Functional Alterations Associated with Severe Obesity: A Pilot Study. Microorganisms. 2020; 8(11):1811. https://doi.org/10.3390/microorganisms8111811
Chicago/Turabian StyleGranata, Ilaria, Carmela Nardelli, Valeria D’Argenio, Salvatore Tramontano, Debora Compare, Mario Rosario Guarracino, Gerardo Nardone, Vincenzo Pilone, and Lucia Sacchetti. 2020. "Duodenal Metatranscriptomics to Define Human and Microbial Functional Alterations Associated with Severe Obesity: A Pilot Study" Microorganisms 8, no. 11: 1811. https://doi.org/10.3390/microorganisms8111811
APA StyleGranata, I., Nardelli, C., D’Argenio, V., Tramontano, S., Compare, D., Guarracino, M. R., Nardone, G., Pilone, V., & Sacchetti, L. (2020). Duodenal Metatranscriptomics to Define Human and Microbial Functional Alterations Associated with Severe Obesity: A Pilot Study. Microorganisms, 8(11), 1811. https://doi.org/10.3390/microorganisms8111811