Proteomics and Lipidomics Investigations to Decipher the Behavior of Willaertia magna C2c Maky According to Different Culture Modes

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Culture of Willaertia magna C2c Maky in Adhesion
2.2. Culture of Willaertia magna C2c Maky in Suspension
2.3. Protein Extraction
2.4. Label-Free Quantitative Nano-LC-MS/MS Proteomic Analysis
2.5. Analysis on Biological Function of Differentially Regulated Proteins
2.6. Quantitative Real-Time Reverse-Transcription-Polymerase Chain Reaction (qRT-PCR)
2.7. Analysis of Lipid Classes by Hydrophilic Interaction Liquid Chromatography-Mass Spectrometry (HILIC-MS)
2.8. Fatty Acid Methyl Ester (FAME) Analysis by Gas Chromatography/Mass Spectrometry
2.9. Electron Microscopy
3. Results
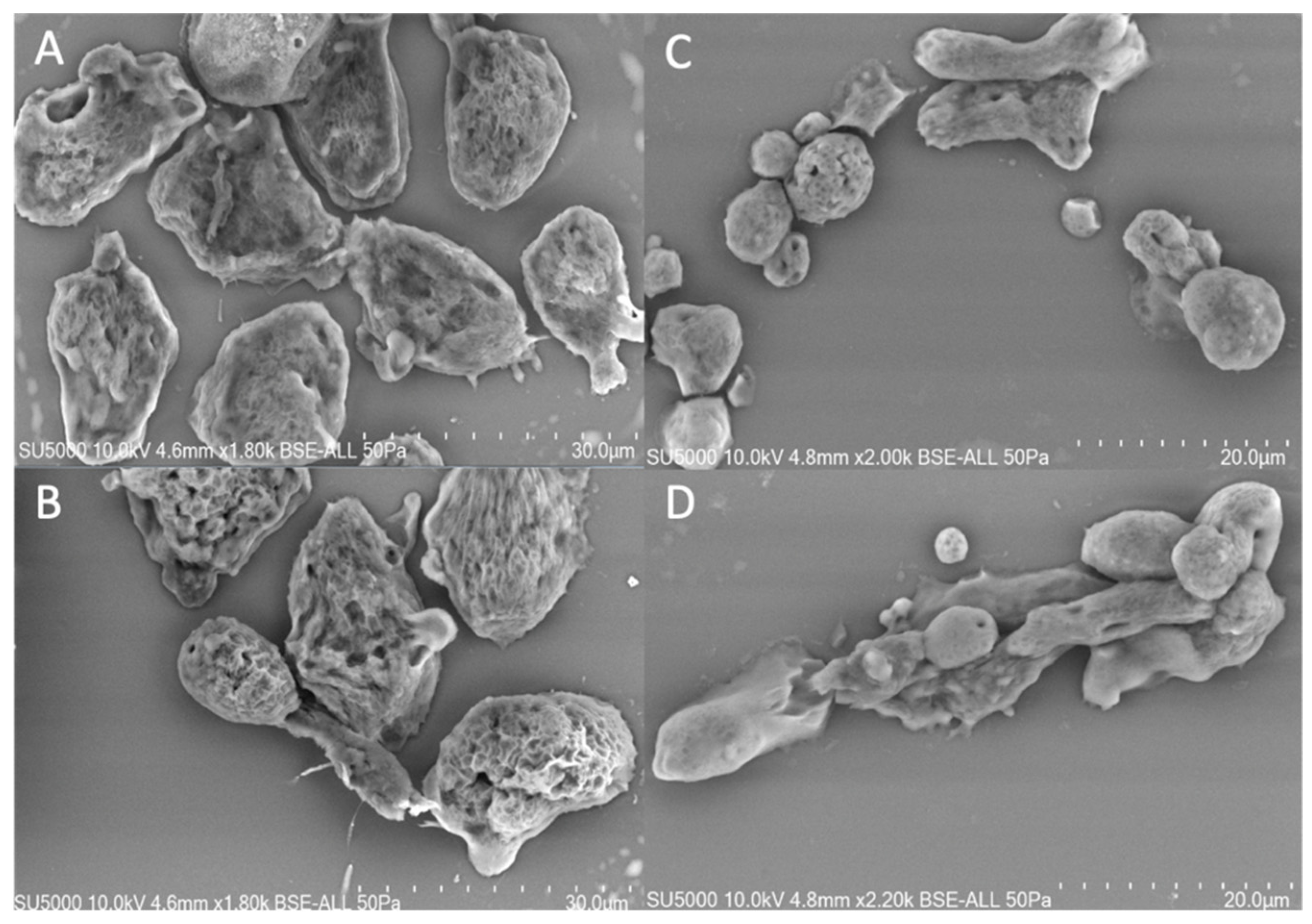
3.1. Phenotype of Willaertia magna Cultivated under Two Different Conditions
3.2. Identification of Differentially Regulated Proteins
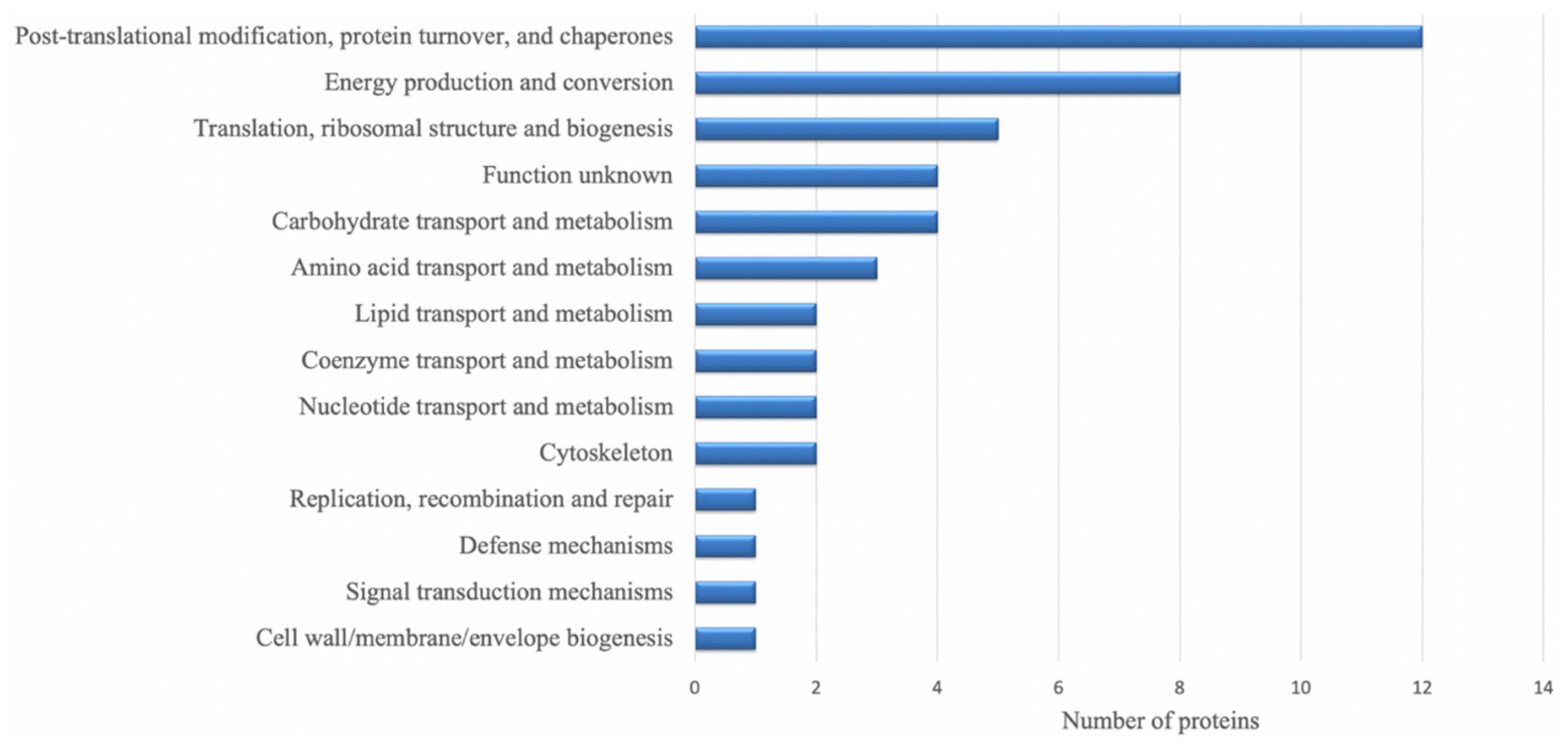
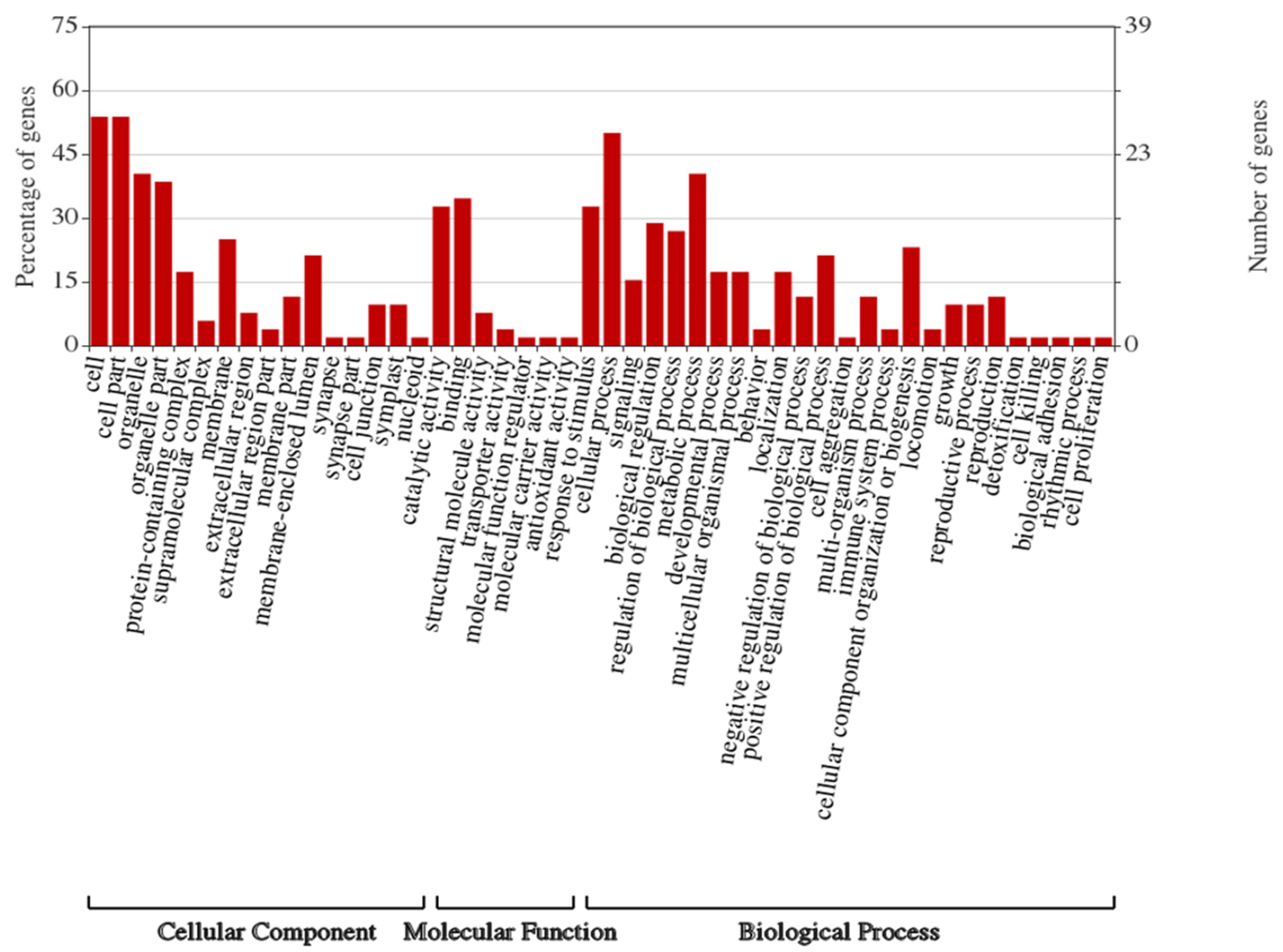
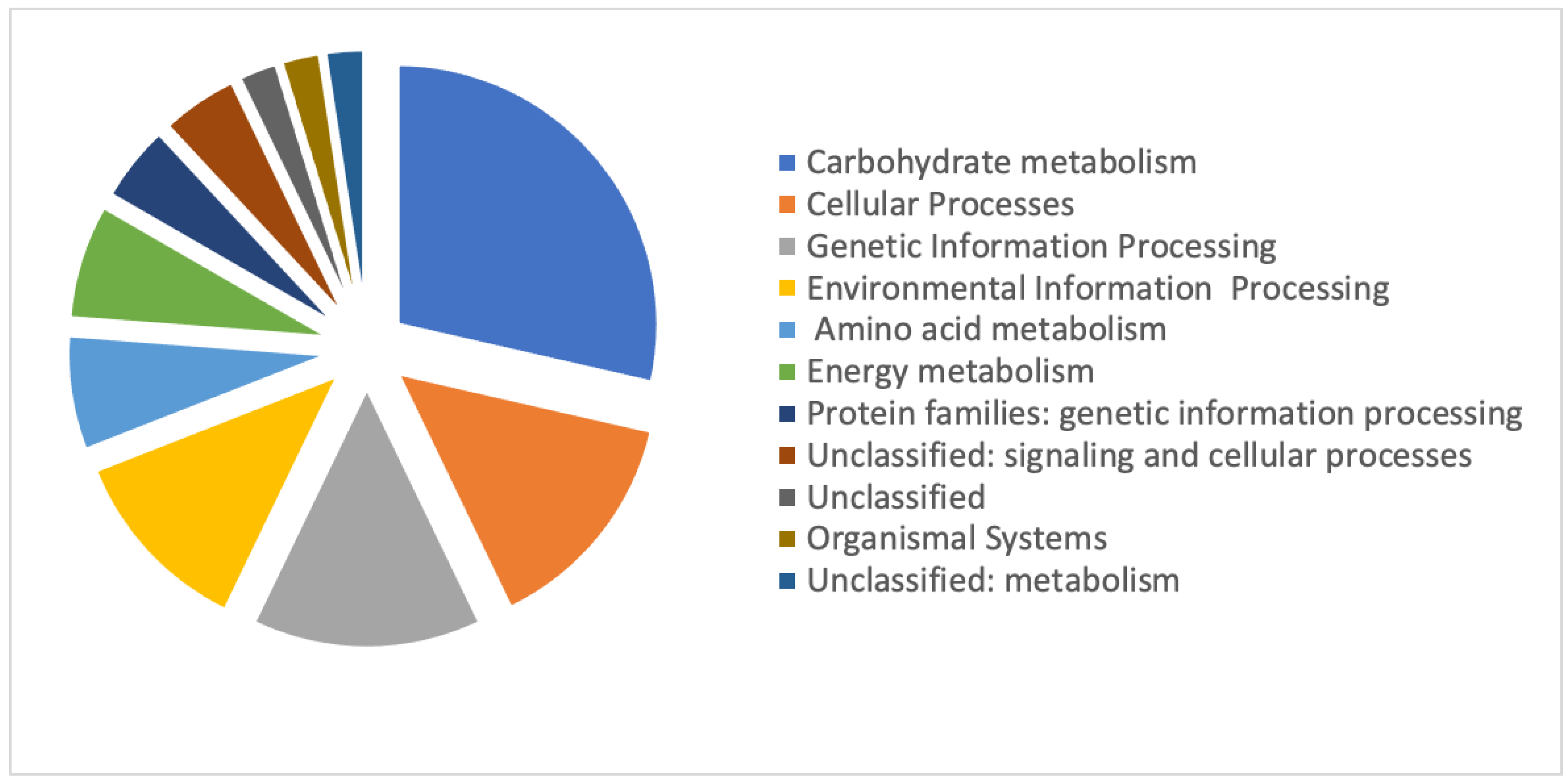
3.3. Identities of Differentially Regulated Proteins
3.4. Analysis of Lipid Classes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pánek, T.; Čepička, I. Diversity of Heterolobosea. In Generic Diversity in Microorgisms; Caliskan, M., Ed.; IntechOpen: London, UK, 2012; ISBN 978-953-51-0064-5. [Google Scholar] [CrossRef]
- Adl, S.M.; Bass, D.; Lane, C.E.; Lukeš, J.; Schoch, C.L.; Smirnov, A.; Agatha, S.; Berney, C.; Brown, M.W.; Burki, F.; et al. Revisions to the Classification, Nomenclature, and Diversity of Eukaryotes. J. Eukaryot. Microbiol. 2019, 66, 4–119. [Google Scholar] [CrossRef] [PubMed]
- Conza, L.; Pagani, S.C.; Gaia, V. Presence of Legionella and Free-Living Amoebae in Composts and Bioaerosols from Composting Facilities. PLoS ONE 2013, 8, e68244. [Google Scholar] [CrossRef] [PubMed]
- De Jonckheere, J.F.; Dive, D.G.; Pussard, M.; Vickerman, K. Willaertia magna gen. nov., sp. nov. (Vahlkampfiidae), a thermophilic amoeba found in different habitats. Protistologica 1984, 201, 5–13. [Google Scholar]
- Dobson, P.J.; Robinson, B.S.; Christy, P.; Hayes, S.J. Low Genetic Diversity in Willaertia Magna From Wide Geographical Sources, and Characterisation of Willaertia Minor N. Sp. (Heterolobosea, Vahlkampfidae). J. Eukaryot. Microbiol. 1993, 40, 298–304. [Google Scholar] [CrossRef]
- Robinson, B.; Christy, P.; De Jonckheere, J. A temporary flagellate (mastigote) stage in the vahlkampfiid amoeba Willaertia magna and its possible evolutionary significance. Biosystems 1989, 23, 75–86. [Google Scholar] [CrossRef]
- Rivera, F.; Lares, F.; Gallegos, E.; Ramírez, E.; Bonilla, P.; Calderon, A.; Martinez, J.J.; Rodríguez, S.; Alcocer, J. Pathogenic amoebae in natural thermal waters of three resorts of Hidalgo, Mexico. Environ. Res. 1989, 50, 289–295. [Google Scholar] [CrossRef]
- Hasni, I.; Chelkha, N.; Baptiste, E.; Mameri, M.R.; Lachuer, J.; Plasson, F.; Colson, P.; La Scola, B. Investigation of potential pathogenicity of Willaertia magna by investigating the transfer of bacteria pathogenicity genes into its genome. Sci. Rep. 2019, 9, 18318. [Google Scholar] [CrossRef]
- Stout, J.E.; Yu, V.L. Legionellosis. N. Engl. J. Med. 1997, 337, 682–687. [Google Scholar] [CrossRef]
- Borella, P.; Guerrieri, E.; Marchesi, I.; Bondi, M.; Messi, P. Water ecology of Legionella and protozoan: Environmental and public health perspectives. Biotechnol. Annu. Rev. 2005, 11, 355–380. [Google Scholar] [CrossRef]
- Hamilton, K.A.; Prussin, A.J.; Ahmed, W.; Haas, C.N. Outbreaks of Legionnaires’ Disease and Pontiac Fever 2006–2017. Curr. Environ. Heal. Rep. 2018, 5, 263–271. [Google Scholar] [CrossRef]
- Yamamoto, H.; Sugiura, M.; Kusunoki, S.; Ezaki, T.; Ikedo, M.; Yabuuchi, E. Factors stimulating propagation of legionellae in cooling tower water. Appl. Environ. Microbiol. 1992, 58, 1394–1397. [Google Scholar] [CrossRef] [PubMed]
- Rowbotham, T.J. Preliminary report on the pathogenicity of Legionella pneumophila for freshwater and soil amoebae. J. Clin. Pathol. 1980, 33, 1179–1183. [Google Scholar] [CrossRef] [PubMed]
- Kilvington, S.; Price, J. Survival of Legionella pneumophila within cysts of Acanthamoeba polyphagafollowing chlorine exposure. J. Appl. Bacteriol. 1990, 68, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Winiecka-Krusnell, E.L.J. Free-living Amoebae Protecting Legionella in Water: The Tip of an Iceberg? Scand. J. Infect. Dis. 1999, 31, 383–385. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, J.D.; Falkow, S.; Tompkins, L.S. Growth of Legionella pneumophila in Acanthamoeba castellanii enhances invasion. Infect. Immun. 1994, 62, 3254–3261. [Google Scholar] [CrossRef] [PubMed]
- Greub, G.; Raoult, D. Microorganisms Resistant to Free-Living Amoebae. Clin. Microbiol. Rev. 2004, 17, 413–433. [Google Scholar] [CrossRef] [PubMed]
- Rowbotham, T.J. Isolation of Legionella pneumophila from clinical specimens via amoebae, and the interaction of those and other isolates with amoebae. J. Clin. Pathol. 1983, 36, 978–986. [Google Scholar] [CrossRef]
- Dey, R.; Bodennec, J.; Mameri, M.O.; Pernin, P. Free-living freshwater amoebae differ in their susceptibility to the pathogenic bacterium Legionella pneumophila. FEMS Microbiol. Lett. 2009, 290, 10–17. [Google Scholar] [CrossRef]
- Hasni, I.; Jarry, A.; Quelard, B.; Carlino, A.; Eberst, J.-B.; Abbe, O.; Demanèche, S. Intracellular Behaviour of Three Legionella pneumophila Strains within Three Amoeba Strains, Including Willaertia magna C2c Maky. Pathogens 2020, 9, 105. [Google Scholar] [CrossRef]
- Abu Khweek, A.; Amer, A.O. Factors Mediating Environmental Biofilm Formation by Legionella pneumophila. Front. Cell. Infect. Microbiol. 2018, 8, 38. [Google Scholar] [CrossRef]
- Beshay, U.; Friehs, K.; Azzam, A.-E.-M.; Flaschel, E. Analysis of the behaviour of Dictyostelium discoideum in immobilised state by means of continuous cultivation. Bioprocess Biosyst. Eng. 2003, 26, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Weekers, P.H.H.; Wijen, J.P.H.; Lomans, B.P.; Vogels, G.D. Axenic mass cultivation of the free-living soil amoeba, Acanthamoeba castellanii in a laboratory fermentor. Antonie Leeuwenhoek 1996, 69, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Weekers, P.H.; Vogels, G.D. Axenic cultivation of the free-living amoebae, Acanthamoeba castellanii and Hartmannella vermiformis in a chemostat. J. Microbiol. Methods 1994, 19, 13–18. [Google Scholar] [CrossRef]
- Tian, J.; Ishibashi, K.; Honda, S.; Boylan, S.A.; Hjelmeland, L.M.; Handa, J.T. The expression of native and cultured human retinal pigment epithelial cells grown in different culture conditions. Br. J. Ophthalmol. 2005, 89, 1510–1517. [Google Scholar] [CrossRef] [PubMed]
- Plain, N.; Largeau, C.; Derenne, S.; Couté, A. Variabilité morphologique de Botryococcus braunii (Chlorococcales, Chlorophyta): Corrélations avec les conditions de croissance et la teneur en lipides. Phycologia 1993, 32, 259–265. [Google Scholar] [CrossRef]
- Ponec, M.; Weerheim, A.; Kempenaar, J.; Elias, P.M.; Williams, M.L. Differentiation of cultured human keratinocytes: Effect of culture conditions on lipid composition of normal vs. malignant cells. Vitr. Cell. Dev. Biol. Anim. 1989, 25, 689–696. [Google Scholar] [CrossRef]
- Gallagher, J.E.; Zheng, W.; Rong, X.; Miranda, N.; Lin, Z.; Dunn, B.; Zhao, H.; Snyder, M.P. Divergence in a master variator generates distinct phenotypes and transcriptional responses. Genes Dev. 2014, 28, 409–421. [Google Scholar] [CrossRef][Green Version]
- Britto, J.A.; Evans, R.D.; Hayward, R.D.; Jones, B.M. From Genotype to Phenotype: The Differential Expression of FGF, FGFR, and TGFβ Genes Characterizes Human Cranioskeletal Development and Reflects Clinical Presentation in FGFR Syndromes. Plast. Reconstr. Surg. 2001, 108, 2026–2039. [Google Scholar] [CrossRef]
- Yang, K.; Han, X. Lipidomics: Techniques, Applications, and Outcomes Related to Biomedical Sciences. Trends Biochem. Sci. 2016, 41, 954–969. [Google Scholar] [CrossRef]
- O’Donnell, S.T.; Ross, R.P.; Stanton, C. The Progress of Multi-Omics Technologies: Determining Function in Lactic Acid Bacteria Using a Systems Level Approach. Front. Microbiol. 2020, 10, 3084. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights 2020, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- Hasni, I.; Decloquement, P.; Demanèche, S.; Mameri, R.M.; Abbe, O.; Colson, P.; La Scola, B. Insight into the Lifestyle of Amoeba Willaertia magna during Bioreactor Growth Using Transcriptomics and Proteomics. Microorganisms 2020, 8, 771. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.; Lohan, A.J.; Liu, B.; Lagkouvardos, I.; Roy, S.; Zafar, N.; Bertelli, C.; Schilde, C.; Kianianmomeni, A.; Buerglin, T.R.; et al. Genome of Acanthamoeba castellanii highlights extensive lateral gene transfer and early evolution of tyrosine kinase signaling. Genome Biol. 2013, 14, R11. [Google Scholar] [CrossRef] [PubMed]
- Fritz-Laylin, L.K.; Prochnik, S.E.; Ginger, M.L.; Dacks, J.B.; Carpenter, M.L.; Field, M.C.; Kuo, A.; Paredez, A.; Chapman, J.; Pham, J.; et al. The Genome of Naegleria gruberi Illuminates Early Eukaryotic Versatility. Cell 2010, 140, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Liechti, N.; Schürch, N.; Bruggmann, R.; Wittwer, M. The genome of Naegleria lovaniensis, the basis for a comparative approach to unravel pathogenicity factors of the human pathogenic amoeba N. fowleri. BMC Genom. 2018, 19, 1–11. [Google Scholar] [CrossRef]
- Zysset-Burri, D.C.; Müller, N.; Beuret, C.M.; Heller, M.; Schürch, N.; Gottstein, B.; Wittwer, M. Genome-wide identification of pathogenicity factors of the free-living amoeba Naegleria fowleri. BMC Genom. 2014, 15, 496. [Google Scholar] [CrossRef]
- Liechti, N.; Schürch, N.; Bruggmann, R.; Wittwer, M. Nanopore sequencing improves the draft genome of the human pathogenic amoeba Naegleria fowleri. Sci. Rep. 2019, 9, 16040. [Google Scholar] [CrossRef]
- Chelkha, N.; Hasni, I.; Louazani, A.C.; Levasseur, A.; La Scola, B.; Colson, P. Vermamoeba vermiformis CDC-19 draft genome sequence reveals considerable gene trafficking including with candidate phyla radiation and giant viruses. Sci. Rep. 2020, 10, 5928. [Google Scholar] [CrossRef]
- Hasni, I.; Andreani, J.; Colson, P.; La Scola, B. Description of Virulent Factors and Horizontal Gene Transfers of Keratitis-Associated Amoeba Acanthamoeba Triangularis by Genome Analysis. Pathogens 2020, 9, 217. [Google Scholar] [CrossRef]
- Detering, H.; Aebischer, T.; Dabrowski, P.W.; Radonic, A.; Nitsche, A.; Renard, B.Y.; Kiderlen, A.F. First Draft Genome Sequence ofBalamuthia mandrillaris, the Causative Agent of Amoebic Encephalitis. Genome Announc. 2015, 3, e01013-15. [Google Scholar] [CrossRef]
- Greninger, A.L.; Messacar, K.; Dunnebacke, T.; Naccache, S.N.; Federman, S.; Bouquet, J.; Mirsky, D.M.; Nomura, Y.; Yagi, S.; Glaser, C.A.; et al. Clinical metagenomic identification of Balamuthia mandrillaris encephalitis and assembly of the draft genome: The continuing case for reference genome sequencing. Genome Med. 2015, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- De Jonckheere, J. Use of an axenic medium for differentiation between pathogenic and nonpathogenic Naegleria fowleri isolates. Appl. Environ. Microbiol. 1977, 33, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Bioréacteur Pilote GPC. Available online: http://www.gpcbio.com/bioracteurpilote.html (accessed on 21 March 2020).
- Renesto, P.; Abergel, C.; Decloquement, P.; Moinier, D.; Azza, S.; Ogata, H.; Fourquet, P.; Gorvel, J.-P.; Claverie, J.-M. Mimivirus Giant Particles Incorporate a Large Fraction of Anonymous and Unique Gene Products. J. Virol. 2006, 80, 11678–11685. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; Von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef]
- Ye, J.; Zhang, Y.; Cui, H.; Liu, J.; Wu, Y.; Cheng, Y.; Xu, H.; Huang, X.; Li, S.; Zhou, A.; et al. WEGO 2.0: A web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 2018, 46, W71–W75. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L.; et al. InterPro: The integrative protein signature database. Nucleic Acids Res. 2009, 37, D211–D215. [Google Scholar] [CrossRef]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.J.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015, 43, D222–D226. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ding, X.; Han, S.; He, T.; Zhang, H.; Yang, L.; Yang, S.; Gai, J. Differential proteomics analysis to identify proteins and pathways associated with male sterility of soybean using iTRAQ-based strategy. J. Proteom. 2016, 138, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A Rapid Method of Total Lipid Extraction and Purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Isaac, G.; McDonald, S.; Astarita, G. Lipid Separation Using UPLC with Charged Surface Hybrid Technology, Milfort: Waters Application Note. 2011. Available online: https://www.waters.com/webassets/cms/library/docs/720004107en.pdf (accessed on 9 September 2020).
- Čabovska, B. Application of Multivariate Analysis and LC-MS for the Detection of Counterfeit Cosmetics. Waters Application Note. 2015. Available online: https://www.waters.com/webassets/cms/library/docs/720005402en.pdf (accessed on 9 September 2020).
- Dione, N.; Sankar, S.; Lagier, J.-C.; Khelaifia, S.; Michele, C.; Armstrong, N.; Richez, M.; Abrahão, J.; Raoult, D.; Fournier, P.-E. Genome sequence and description of Anaerosalibacter massiliensis sp. nov. New Microbes New Infect. 2016, 10, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Ngom, I.; Hasni, I.; Lo, C.; Traore, S.; Fontanini, A.; Raoult, D.; Fenollar, F. Taxono-genomics and description of Gordonibacter massiliensis sp. nov., a new bacterium isolated from stool of healthy patient. New Microbes New Infect. 2020, 33, 100624. [Google Scholar] [CrossRef]
- Sasser, M. Bacterial Identification by Gas Chromatographic Analysis of Fatty Acid Methyl Esters (GC-FAME); Technical Note 101; MIDI Inc.: Newark, DE, USA, 2006; Available online: http://midi-inc.com/pdf/MIS_Technote_101.pdf (accessed on 9 September 2020).
- Ahsan, K.; Lekli, I.; Ray, D.; Yodoi, J.; Das, D.K. Redox Regulation of Cell Survival by the Thioredoxin Superfamily: An Implication of Redox Gene Therapy in the Heart. Antioxid. Redox Signal. 2009, 11, 2741–2758. [Google Scholar] [CrossRef]
- Netto, L.E.S.; Antunes, A.F. The Roles of Peroxiredoxin and Thioredoxin in Hydrogen Peroxide Sensing and in Signal Transduction. Mol. Cells 2016, 39, 65–71. [Google Scholar] [CrossRef]
- Zigmond, S.H. How Wasp Regulates Actin Polymerization. J. Cell Biol. 2000, 150, F117–F120. [Google Scholar] [CrossRef]
- Hagopian, K.; Ramsey, J.J.; Weindruch, R. Enzymes of glycerol and glyceraldehyde metabolism in mouse liver: Effects of caloric restriction and age on activities. Biosci. Rep. 2008, 28, 107–115. [Google Scholar] [CrossRef]
- Wendel, A.A.; Lewin, T.M.; Coleman, R.A. Glycerol-3-phosphate acyltransferases: Rate limiting enzymes of triacylglycerol biosynthesis. Biochim. et Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2009, 1791, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.-L.; Chen, H.-H.; Jiang, J.-G. Implications of glycerol metabolism for lipid production. Prog. Lipid Res. 2017, 68, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H. Choline, Other Methyl-Donors and Epigenetics. Nutrients 2017, 9, 445. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, K.; Marth, J.D. Glycosylation in Cellular Mechanisms of Health and Disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Kopitz, J. Lipid glycosylation: A primer for histochemists and cell biologists. Histochem. Cell Biol. 2016, 147, 175–198. [Google Scholar] [CrossRef]
- Van Meer, G.; Wolthoorn, J.; DeGroote, S. The fate and function of glycosphingolipid glucosylceramide. Philos. Trans. R. Soc. B Biol. Sci. 2003, 358, 869–873. [Google Scholar] [CrossRef]
- DeGroote, S.; Wolthoorn, J.; Van Meer, G. The cell biology of glycosphingolipids. Semin. Cell Dev. Biol. 2004, 15, 375–387. [Google Scholar] [CrossRef]
- Rivera, R.; Chun, J. Biological effects of lysophospholipids. Rev. Physiol. Biochem. Pharmacol. Biochem. Pharmacol. 2006, 160, 25–46. [Google Scholar]
- Ailte, I.; Lingelem, A.B.D.; Kavaliauskiene, S.; Bergan, J.; Kvalvaag, A.S.; Myrann, A.-G.; Skotland, T.; Sandvig, K. Addition of lysophospholipids with large head groups to cells inhibits Shiga toxin binding. Sci. Rep. 2016, 6, 30336. [Google Scholar] [CrossRef]
- Speranza, F.; Mahankali, M.; Henkels, K.M.; Gómez-Cambronero, J. The Molecular Basis of Leukocyte Adhesion Involving Phosphatidic Acid and Phospholipase D. J. Biol. Chem. 2014, 289, 28885–28897. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Sequence Accession of W. magna | Anova (p) | Fold Change | Protein Function |
|---|---|---|---|
| protein 17463 | 1.45 × 10−14 | 410.39 | predicted protein |
| protein 8919 | 4.00 × 10−8 | 54.12 | uncharacterized protein |
| protein 2287 | 5.55 × 10−16 | 44.36 | cathepsin B-like protein |
| protein 8084 | 5.68 × 10−14 | 7.2 | malate dehydrogenase |
| protein 15939 | 4.35 × 10−13 | 6.48 | Ras GTP-binding protein RhoL |
| protein 4754 | 1.81 × 10−9 | 5.96 | Hemerythrin-like protein |
| protein 15039 | 1.81 × 10−9 | 5.96 | Hemerythrin-like protein |
| protein 17074 | 2.62 × 10−6 | 4.88 | Unknown function (ORFan) |
| protein 15365 | 1.43 × 10−12 | 4.8 | predicted protein |
| protein 13743 | 3.72 × 10−6 | 4.7 | ubiquinone biosynthesis protein |
| protein 8031 | 3.75 × 10−8 | 4.11 | cathepsin B-like protein |
| protein 4072 | 1.84 × 10−8 | 4.02 | glyceraldehyde-3-phosphate dehydrogenase |
| protein 17615 | 6.17 × 10−6 | 3.95 | V-type H+-transporting ATPase subunit F |
| protein 2899 | 1.20 × 10−9 | 3.24 | beta-tubulin |
| protein 3274 | 7.59 × 10−12 | 3.16 | adenylate kinase |
| protein 7439 | 4.31 × 10−8 | 2.99 | triosephosphate isomerase |
| protein 7923 | 2.67 × 10−4 | 2.82 | protein domain specific binding |
| protein 12867 | 1.20 × 10−5 | 2.78 | arsonoacetate metabolic process |
| protein 2947 | 1.27 × 10−11 | 2.65 | 3-oxoacid CoA-transferase activity |
| protein 8114 | 1.69 × 10−3 | 2.47 | cathepsin B-like protein |
| protein 7780 | 4.69 × 10−4 | 2.41 | pyruvate dehydrogenase |
| protein 5985 | 2.67 × 10−6 | 2.4 | alpha-actinin |
| protein 3614 | 1.27 × 10−10 | 2.39 | malate dehydrogenase |
| protein 885 | 2.48 × 10−7 | 2.39 | RRM domain-containing protein |
| protein 13554 | 2.66 × 10−10 | 2.39 | glutamate dehydrogenase |
| protein 3571 | 5.22 × 10−5 | 2.38 | Profilin |
| protein 8014 | 5.79 × 10−8 | 2.25 | phosphoenolpyruvate carboxykinase |
| protein 6333 | 3.93 × 10−3 | 2.21 | predicted protein |
| protein 3628 | 1.13 × 10−5 | 2.2 | cathepsin D |
| protein 16124 | 1.07 × 10−9 | 2.19 | ADP/ATP translocase 1 domain protein |
| protein 8489 | 3.94 × 10−6 | 2.19 | actin beta/gamma 1 |
| protein 11280 | 1.15 × 10−3 | 2.14 | 60S ribosomal protein L23 |
| protein 4486 | 6. × 10−11 | 2.13 | cystathionine gamma-lyase |
| protein 2738 | 5.05 × 10−5 | 2.12 | cathepsin B |
| protein 8503 | 3.55 × 10−10 | 2.11 | ATP synthase F1 subunit alpha |
| protein 7955 | 3.57 × 10−6 | 2.1 | large subunit ribosomal protein L24e |
| protein 4073 | 7.58 × 10−9 | 2.05 | fructose-bisphosphate aldolase |
| protein 15452 | 3.15 × 10−4 | 2.02 | malate metabolic process |
| protein 2384 | 6.51 × 10−9 | 2.02 | mitochondrial citrate synthetase |
| protein 18191 | 2.88 × 10−9 | 2.01 | thioredoxin |
| Protein Sequence Accession of W. magna | Anova (p) | Fold Change | Protein Function |
|---|---|---|---|
| protein 5557 | 1.63 × 10−10 | Infinity | Unknown protein (ORFan) |
| protein 3984 | 4.59 × 10−12 | 15.52 | large subunit ribosomal protein L7/L12 |
| protein 2294 | 3.31 × 10−13 | 11.34 | metE; 5-methyltetrahydropteroyltriglutamate–homocysteine methyltransferase |
| protein 16136 | 8.95 × 10−14 | 5.98 | membrane protein |
| protein 2157 | 5.28 × 10−4 | 4.12 | ribosomal protein L7/L12, putative |
| protein 10142 | 4.22 × 10−14 | 3.43 | HSP-20 domain-containing protein |
| protein 18401 | 8.68 × 10−7 | 3.41 | nuclear transport factor 2 |
| protein 18200 | 5.01 × 10−6 | 2.84 | maleylacetoacetate isomerase |
| protein 2831 | 8.88 × 10−16 | 2.68 | HSP20 family protein |
| protein 11490 | 2.19 × 10−4 | 2.59 | predicted protein |
| protein 2849 | 3.97 × 10−8 | 2.49 | predicted protein |
| protein 8080 | 3.66 × 10−6 | 2.46 | long-chain acyl-CoA synthetase |
| protein 11879 | 7.72 × 10−5 | 2.3 | hypothetical protein |
| protein 15002 | 2.04 × 10−10 | 2.21 | membrane protein |
| protein 16271 | 6.39 × 10−12 | 2.18 | heat shock 70kDa protein 5 |
| protein 3173 | 6.20 × 10−6 | 2.18 | mitochondrial chaperonin hsp10 |
| protein 14923 | 5.72 × 10−7 | 2.15 | predicted protein |
| protein 13231 | 5.57 × 10−7 | 2.15 | sugar phosphate isomerase/epimerase |
| protein 13228 | 3.71 × 10−3 | 2.12 | predicted protein |
| protein 6869 | 5.31 × 10−7 | 2.07 | alcohol dehydrogenase |
| protein 2046 | 7.87 × 10−5 | 2.02 | catalase |
| Culture Condition | Suspension | Adhesion |
|---|---|---|
| Positive ionization | 4 apolar lipids (1 Ceramide, 3 Acylglycerols) 4 Glyco lipids (Glycosyldiacylglycerols) 11 phospholipids (6 PCs, 5 unknown) | 2 apolar lipids (Acylglycerols) 5 Glyco lipids (3 Hex Sphingosine, 2 Glycosyldiacylglycerols) 7 Phospholipids (3 PCs, 1 phosphatidyl serine, 3 unknown) 7 Lysophospholipids |
| Negative ionization | 2 apolar lipids (Ceramides) 8 Free Fatty Acids (7 identified) 6 phospholipids (1 PE, 5 PC) 1 lysophospholipid | 5 apolar lipid (1 Ceramide, 1 Wax Ester, 1 Coenzyme A, 2 unknown) 2 Glyco lipids (1 Ceramide, 1 Glycosyldiacylglycerol) 13 Free Fatty Acids (6 identified) 6 Phospholipids (1 phosphatidylinositol, 1 phosphatidic acid, 4 PEs) 14 Lysophospholipids |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasni, I.; Armstrong, N.; Decloquement, P.; Azza, S.; Fontanini, A.; Abbe, O.; Cherif Louazani, A.; Demanèche, S.; Chabrière, E.; Colson, P.; et al. Proteomics and Lipidomics Investigations to Decipher the Behavior of Willaertia magna C2c Maky According to Different Culture Modes. Microorganisms 2020, 8, 1791. https://doi.org/10.3390/microorganisms8111791
Hasni I, Armstrong N, Decloquement P, Azza S, Fontanini A, Abbe O, Cherif Louazani A, Demanèche S, Chabrière E, Colson P, et al. Proteomics and Lipidomics Investigations to Decipher the Behavior of Willaertia magna C2c Maky According to Different Culture Modes. Microorganisms. 2020; 8(11):1791. https://doi.org/10.3390/microorganisms8111791
Chicago/Turabian StyleHasni, Issam, Nicholas Armstrong, Philippe Decloquement, Said Azza, Anthony Fontanini, Olivier Abbe, Amina Cherif Louazani, Sandrine Demanèche, Eric Chabrière, Philippe Colson, and et al. 2020. "Proteomics and Lipidomics Investigations to Decipher the Behavior of Willaertia magna C2c Maky According to Different Culture Modes" Microorganisms 8, no. 11: 1791. https://doi.org/10.3390/microorganisms8111791
APA StyleHasni, I., Armstrong, N., Decloquement, P., Azza, S., Fontanini, A., Abbe, O., Cherif Louazani, A., Demanèche, S., Chabrière, E., Colson, P., & La Scola, B. (2020). Proteomics and Lipidomics Investigations to Decipher the Behavior of Willaertia magna C2c Maky According to Different Culture Modes. Microorganisms, 8(11), 1791. https://doi.org/10.3390/microorganisms8111791