Genetic Adaptation of Coxsackievirus B1 during Persistent Infection in Pancreatic Cells

 ,
,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Persistent Infection
Immunofluorescent Labeling
2.2. Next Generation Sequencing
2.3. Analyzing the Sequence Data
2.4. 3D Modelling
3. Results
3.1. Characteristics of Established Persistent Infections
Virus Plaque Morphology
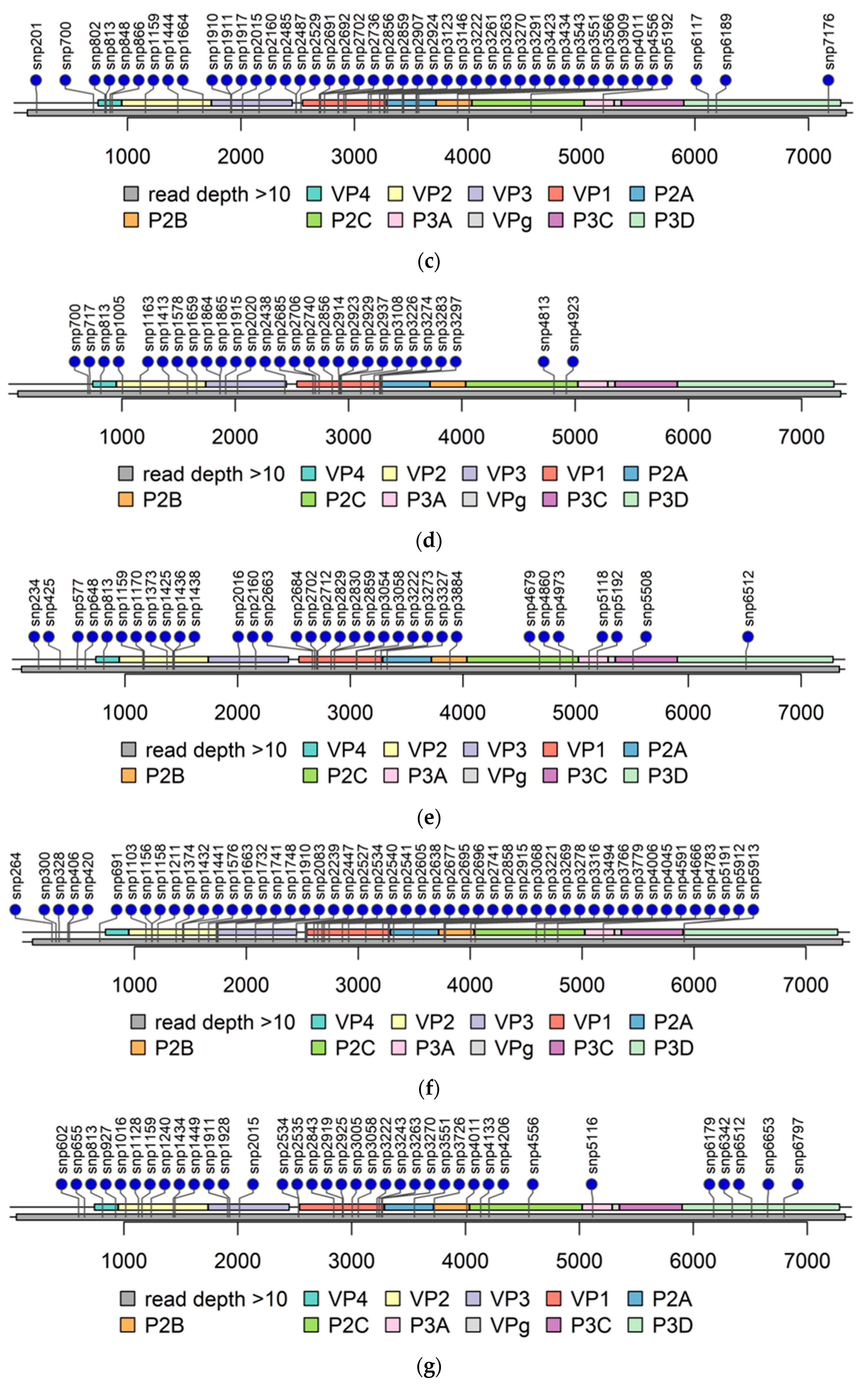
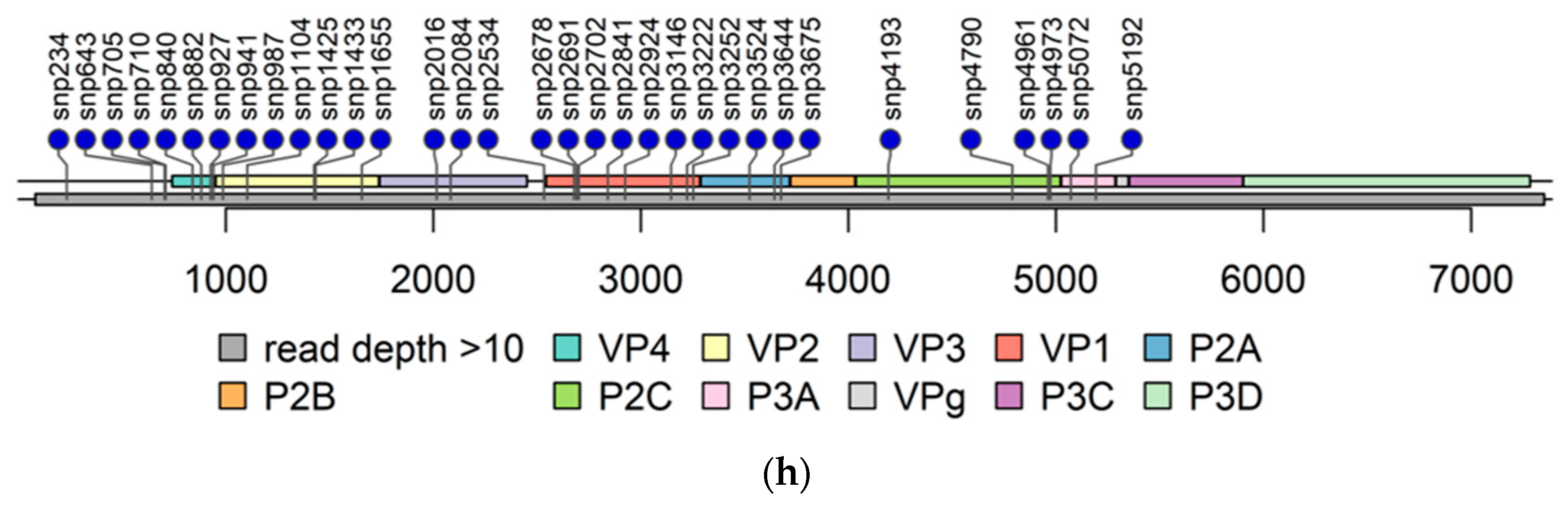
3.2. Determining Genomic Sequences by NGS
3.2.1. Untranslated Regions
3.2.2. Accumulation of Amino Acid Substitutions in Structural Proteins
3.2.3. Accumulation of Amino Acid Substitutions in Non-Structural Viral Proteins
3.2.4. The Second ORF
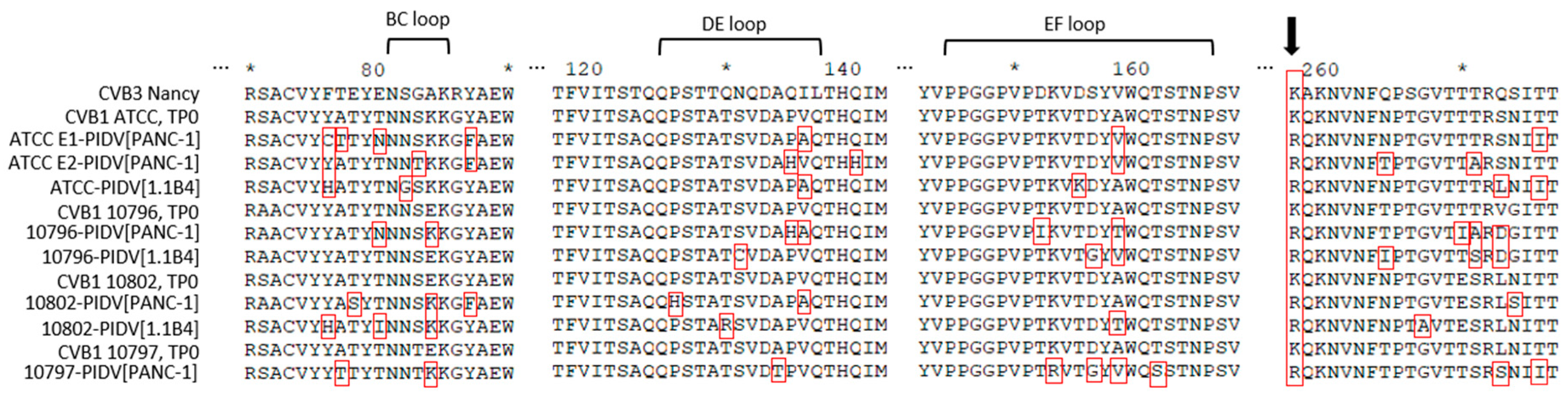
3.2.5. Virus Surface Structure
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tuthill, T.J.; Groppelli, E.; Hogle, J.M.; Rowlands, D.J. Picornaviruses. Curr. Top. Microbiol. Immunol. 2010, 343, 43–89. [Google Scholar]
- Belnap, D.M.; Filman, D.J.; Trus, B.L.; Cheng, N.; Booy, F.P.; Conway, J.F.; Curry, S.; Hiremath, C.N.; Tsang, S.K.; Steven, A.C.; et al. Molecular tectonic model of virus structural transitions: The putative cell entry states of poliovirus. J. Virol. 2000, 74, 1342–1354. [Google Scholar] [CrossRef]
- Olson, N.H.; Kolatkar, P.R.; Oliveira, M.A.; Cheng, R.H.; Greve, J.M.; McClelland, A.; Baker, T.S.; Rossmann, M.G. Structure of a human rhinovirus complexed with its receptor molecule. Proc. Natl. Acad. Sci. USA 1993, 90, 507–511. [Google Scholar] [CrossRef]
- Bostina, M.; Levy, H.; Filman, D.J.; Hogle, J.M. Poliovirus RNA is released from the capsid near a twofold symmetry axis. J. Virol. 2011, 85, 776–783. [Google Scholar] [CrossRef]
- Guo, H.; Li, Y.; Liu, G.; Jiang, Y.; Shen, S.; Bi, R.; Huang, H.; Cheng, T.; Wang, C.; Wei, W. A second open reading frame in human enterovirus determines viral replication in intestinal epithelial cells. Nat. Commun. 2019, 10, 4066–4069. [Google Scholar] [CrossRef]
- Lulla, V.; Dinan, A.M.; Hosmillo, M.; Chaudhry, Y.; Sherry, L.; Irigoyen, N.; Nayak, K.M.; Stonehouse, N.J.; Zilbauer, M.; Goodfellow, I.; et al. An upstream protein-coding region in enteroviruses modulates virus infection in gut epithelial cells. Nat. Microbiol. 2019, 4, 280–292. [Google Scholar] [CrossRef]
- Lozano, G.; Martinez-Salas, E. Structural insights into viral IRES-dependent translation mechanisms. Curr. Opin. Virol. 2015, 12, 113–120. [Google Scholar] [CrossRef]
- Steil, B.P.; Barton, D.J. Conversion of VPg into VPgpUpUOH before and during poliovirus negative-strand RNA synthesis. J. Virol. 2009, 83, 12660–12670. [Google Scholar] [CrossRef]
- Tapparel, C.; Siegrist, F.; Petty, T.J.; Kaiser, L. Picornavirus and enterovirus diversity with associated human diseases. Infect. Genet. Evol. 2013, 14, 282–293. [Google Scholar] [CrossRef]
- Sane, F.; Caloone, D.; Gmyr, V.; Engelmann, I.; Belaich, S.; Kerr-Conte, J.; Pattou, F.; Desailloud, R.; Hober, D. Coxsackievirus B4 can infect human pancreas ductal cells and persist in ductal-like cell cultures which results in inhibition of Pdx1 expression and disturbed formation of islet-like cell aggregates. Cell. Mol. Life Sci. 2013, 70, 4169–4180. [Google Scholar] [CrossRef]
- Pinkert, S.; Klingel, K.; Lindig, V.; Dorner, A.; Zeichhardt, H.; Spiller, O.B.; Fechner, H. Virus-host coevolution in a persistently coxsackievirus B3-infected cardiomyocyte cell line. J. Virol. 2011, 85, 13409–13419. [Google Scholar] [CrossRef]
- Chapman, N.M.; Kim, K.S. Persistent coxsackievirus infection: Enterovirus persistence in chronic myocarditis and dilated cardiomyopathy. Curr. Top. Microbiol. Immunol. 2008, 323, 275–292. [Google Scholar]
- Frisk, G. Mechanisms of chronic enteroviral persistence in tissue. Curr. Opin. Infect. Dis. 2001, 14, 251–256. [Google Scholar] [CrossRef]
- Nurminen, N.; Oikarinen, S.; Hyoty, H. Virus infections as potential targets of preventive treatments for type 1 diabetes. Rev. Diabet. Stud. 2012, 9, 260–271. [Google Scholar] [CrossRef]
- Hyoty, H. Viruses in type 1 diabetes. Pediatr. Diabetes 2016, 17 (Suppl. 22), 56–64. [Google Scholar] [CrossRef]
- Massilamany, C.; Gangaplara, A.; Reddy, J. Intricacies of cardiac damage in coxsackievirus B3 infection: Implications for therapy. Int. J. Cardiol. 2014, 177, 330–339. [Google Scholar] [CrossRef]
- Baj, A.; Colombo, M.; Headley, J.L.; McFarlane, J.R.; Liethof, M.A.; Toniolo, A. Post-poliomyelitis syndrome as a possible viral disease. Int. J. Infect. Dis. 2015, 35, 107–116. [Google Scholar] [CrossRef]
- Sioofy-Khojine, A.B.; Lehtonen, J.; Nurminen, N.; Laitinen, O.H.; Oikarinen, S.; Huhtala, H.; Pakkanen, O.; Ruokoranta, T.; Hankaniemi, M.M.; Toppari, J.; et al. Coxsackievirus B1 infections are associated with the initiation of insulin-driven autoimmunity that progresses to type 1 diabetes. Diabetologia 2018, 61, 1193–1202. [Google Scholar] [CrossRef]
- Laitinen, O.H.; Honkanen, H.; Pakkanen, O.; Oikarinen, S.; Hankaniemi, M.M.; Huhtala, H.; Ruokoranta, T.; Lecouturier, V.; Andre, P.; Harju, R.; et al. Coxsackievirus B1 is associated with induction of beta-cell autoimmunity that portends type 1 diabetes. Diabetes 2014, 63, 446–455. [Google Scholar] [CrossRef]
- Oikarinen, S.; Martiskainen, M.; Tauriainen, S.; Huhtala, H.; Ilonen, J.; Veijola, R.; Simell, O.; Knip, M.; Hyoty, H. Enterovirus RNA in blood is linked to the development of type 1 diabetes. Diabetes 2011, 60, 276–279. [Google Scholar] [CrossRef]
- Vehik, K.; Lynch, K.F.; Wong, M.C.; Tian, X.; Ross, M.C.; Gibbs, R.A.; Ajami, N.J.; Petrosino, J.F.; Rewers, M.; Toppari, J.; et al. TEDDY Study Group Prospective virome analyses in young children at increased genetic risk for type 1 diabetes. Nat. Med. 2019, 25, 1865–1872. [Google Scholar] [CrossRef]
- Oikarinen, M.; Tauriainen, S.; Oikarinen, S.; Honkanen, T.; Collin, P.; Rantala, I.; Maki, M.; Kaukinen, K.; Hyoty, H. Type 1 diabetes is associated with enterovirus infection in gut mucosa. Diabetes 2012, 61, 687–691. [Google Scholar] [CrossRef]
- Tracy, S.; Smithee, S.; Alhazmi, A.; Chapman, N. Coxsackievirus can persist in murine pancreas by deletion of 5′ terminal genomic sequences. J. Med. Virol. 2015, 87, 240–247. [Google Scholar] [CrossRef]
- Jaidane, H.; Sauter, P.; Sane, F.; Goffard, A.; Gharbi, J.; Hober, D. Enteroviruses and type 1 diabetes: Towards a better understanding of the relationship. Rev. Med. Virol. 2010, 20, 265–280. [Google Scholar] [CrossRef]
- Jaidane, H.; Hober, D. Role of coxsackievirus B4 in the pathogenesis of type 1 diabetes. Diabetes Metab. 2008, 34, 537–548. [Google Scholar] [CrossRef]
- Yin, H.; Berg, A.K.; Westman, J.; Hellerstrom, C.; Frisk, G. Complete nucleotide sequence of a Coxsackievirus B-4 strain capable of establishing persistent infection in human pancreatic islet cells: Effects on insulin release, proinsulin synthesis, and cell morphology. J. Med. Virol. 2002, 68, 544–557. [Google Scholar] [CrossRef]
- Kim, K.S.; Chapman, N.M.; Tracy, S. Replication of coxsackievirus B3 in primary cell cultures generates novel viral genome deletions. J. Virol. 2008, 82, 2033–2037. [Google Scholar] [CrossRef]
- Alidjinou, E.K.; Engelmann, I.; Bossu, J.; Villenet, C.; Figeac, M.; Romond, M.B.; Sane, F.; Hober, D. Persistence of Coxsackievirus B4 in pancreatic ductal-like cells results in cellular and viral changes. Virulence 2017, 8, 1229–1244. [Google Scholar] [CrossRef]
- Kim, K.S.; Tracy, S.; Tapprich, W.; Bailey, J.; Lee, C.K.; Kim, K.; Barry, W.H.; Chapman, N.M. 5′-Terminal deletions occur in coxsackievirus B3 during replication in murine hearts and cardiac myocyte cultures and correlate with encapsidation of negative-strand viral RNA. J. Virol. 2005, 79, 7024–7041. [Google Scholar] [CrossRef]
- Lietzen, N.; Hirvonen, K.; Honkimaa, A.; Buchacher, T.; Laiho, J.E.; Oikarinen, S.; Mazur, M.A.; Flodstrom-Tullberg, M.; Dufour, E.; Sioofy-Khojine, A.B.; et al. Coxsackievirus B Persistence Modifies the Proteome and the Secretome of Pancreatic Ductal Cells. iScience 2019, 19, 340–357. [Google Scholar] [CrossRef]
- Hamalainen, S.; Nurminen, N.; Ahlfors, H.; Oikarinen, S.; Sioofy-Khojine, A.B.; Frisk, G.; Oberste, M.S.; Lahesmaa, R.; Pesu, M.; Hyoty, H. Coxsackievirus B1 reveals strain specific differences in plasmacytoid dendritic cell mediated immunogenicity. J. Med. Virol. 2014, 86, 1412–1420. [Google Scholar] [CrossRef]
- Honkimaa, A.; Sioofy-Khojine, A.B.; Oikarinen, S.; Bertin, A.; Hober, D.; Hyoty, H. Eradication of persistent coxsackievirus B infection from a pancreatic cell line with clinically used antiviral drugs. J. Clin. Virol. 2020, 128, 104334. [Google Scholar] [CrossRef]
- Lonnrot, M.; Sjoroos, M.; Salminen, K.; Maaronen, M.; Hyypia, T.; Hyoty, H. Diagnosis of enterovirus and rhinovirus infections by RT-PCR and time-resolved fluorometry with lanthanide chelate labeled probes. J. Med. Virol. 1999, 59, 378–384. [Google Scholar] [CrossRef]
- Honkanen, H.; Oikarinen, S.; Pakkanen, O.; Ruokoranta, T.; Pulkki, M.M.; Laitinen, O.H.; Tauriainen, S.; Korpela, S.; Lappalainen, M.; Vuorinen, T.; et al. Human enterovirus 71 strains in the background population and in hospital patients in Finland. J. Clin. Virol. 2013, 56, 348–353. [Google Scholar] [CrossRef]
- Saarinen, N.V.V.; Laiho, J.E.; Richardson, S.J.; Zeissler, M.; Stone, V.M.; Marjomaki, V.; Kantoluoto, T.; Horwitz, M.S.; Sioofy-Khojine, A.; Honkimaa, A.; et al. A novel rat CVB1-VP1 monoclonal antibody 3A6 detects a broad range of enteroviruses. Sci. Rep. 2018, 8, 33–34. [Google Scholar] [CrossRef]
- Schonborn, J.; Oberstrass, J.; Breyel, E.; Tittgen, J.; Schumacher, J.; Lukacs, N. Monoclonal antibodies to double-stranded RNA as probes of RNA structure in crude nucleic acid extracts. Nucleic Acids Res. 1991, 19, 2993–3000. [Google Scholar] [CrossRef]
- Lin, J.; Kimura, B.Y.; Oikarinen, S.; Nykter, M. Bioinformatics Assembling and Assessment of Novel Coxsackievirus B1 Genome. Methods Mol. Biol. 2018, 1838, 261–272. [Google Scholar] [CrossRef]
- Kramna, L.; Kolarova, K.; Oikarinen, S.; Pursiheimo, J.P.; Ilonen, J.; Simell, O.; Knip, M.; Veijola, R.; Hyoty, H.; Cinek, O. Gut virome sequencing in children with early islet autoimmunity. Diabetes Care 2015, 38, 930–933. [Google Scholar] [CrossRef]
- A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 10 January 2018).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- BBMap. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 9 October 2020).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup the Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Deatherage, D.E.; Barrick, J.E. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol. Biol. 2014, 1151, 165–188. [Google Scholar] [CrossRef]
- Ou, J.; Zhu, L.J. trackViewer: A Bioconductor package for interactive and integrative visualization of multi-omics data. Nat. Methods 2019, 16, 453–454. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef]
- Nicholas, K.B.; Nicholas, H.B., Jr.; Deerfield, D.W. GeneDoc: Analysis and Visualization of Genetic Variation. Embnew. News 1997, 4, 14. [Google Scholar]
- Muckelbauer, J.K.; Kremer, M.; Minor, I.; Diana, G.; Dutko, F.J.; Groarke, J.; Pevear, D.C.; Rossmann, M.G. The structure of coxsackievirus B3 at 3.5 Å resolution. Structure 1995, 3, 653–667. [Google Scholar] [CrossRef]
- Yoder, J.D.; Cifuente, J.O.; Pan, J.; Bergelson, J.M.; Hafenstein, S. The crystal structure of a coxsackievirus B3-RD variant and a refined 9-angstrom cryo-electron microscopy reconstruction of the virus complexed with decay-accelerating factor (DAF) provide a new footprint of DAF on the virus surface. J. Virol. 2012, 86, 12571–12581. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Shen, M.Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef]
- Jaidane, H.; Caloone, D.; Lobert, P.E.; Sane, F.; Dardenne, O.; Naquet, P.; Gharbi, J.; Aouni, M.; Geenen, V.; Hober, D. Persistent infection of thymic epithelial cells with coxsackievirus B4 results in decreased expression of type 2 insulin-like growth factor. J. Virol. 2012, 86, 11151–11162. [Google Scholar] [CrossRef]
- Mahmud, B.; Horn, C.M.; Tapprich, W.E. Structure of the 5′ Untranslated Region of Enteroviral Genomic RNA. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Shi, J.; Huang, X.; Liu, Q.; Huang, Z. Identification of conserved neutralizing linear epitopes within the VP1 protein of coxsackievirus A16. Vaccine 2013, 31, 2130–2136. [Google Scholar] [CrossRef]
- Sauter, P.; Chehadeh, W.; Lobert, P.E.; Lazrek, M.; Goffard, A.; Soumillon, M.; Caloone, D.; Vantyghem, M.C.; Weill, J.; Fajardy, I.; et al. A part of the VP4 capsid protein exhibited by coxsackievirus B4 E2 is the target of antibodies contained in plasma from patients with type 1 diabetes. J. Med. Virol. 2008, 80, 866–878. [Google Scholar] [CrossRef]
- Chehadeh, W.; Lobert, P.E.; Sauter, P.; Goffard, A.; Lucas, B.; Weill, J.; Vantyghem, M.C.; Alm, G.; Pigny, P.; Hober, D. Viral protein VP4 is a target of human antibodies enhancing coxsackievirus B4- and B3-induced synthesis of alpha interferon. J. Virol. 2005, 79, 13882–13891. [Google Scholar] [CrossRef]
- Goffard, A.; Sane, F.; Soumillon, M.; Hober, D. Specificity of the coxsackievirus B4 VP4 capsid protein investigated in silico. Discov. Med. 2011, 12, 153–158. [Google Scholar]
- Smithee, S.; Tracy, S.; Chapman, N.M. Mutational Disruption of cis-Acting Replication Element 2C in Coxsackievirus B3 Leads to 5′-Terminal Genomic Deletions. J. Virol. 2015, 89, 11761–11772. [Google Scholar] [CrossRef]
- Van der Schaar, H.M.; van der Linden, L.; Lanke, K.H.; Strating, J.R.; Purstinger, G.; de Vries, E.; de Haan, C.A.; Neyts, J.; van Kuppeveld, F.J. Coxsackievirus mutants that can bypass host factor PI4KIIIbeta and the need for high levels of PI4P lipids for replication. Cell Res. 2012, 22, 1576–1592. [Google Scholar] [CrossRef]
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 1997, 275, 1320–1323. [Google Scholar] [CrossRef]
- Shafren, D.R.; Williams, D.T.; Barry, R.D. A decay-accelerating factor-binding strain of coxsackievirus B3 requires the coxsackievirus-adenovirus receptor protein to mediate lytic infection of rhabdomyosarcoma cells. J. Virol. 1997, 71, 9844–9848. [Google Scholar] [CrossRef]
- He, Y.; Chipman, P.R.; Howitt, J.; Bator, C.M.; Whitt, M.A.; Baker, T.S.; Kuhn, R.J.; Anderson, C.W.; Freimuth, P.; Rossmann, M.G. Interaction of coxsackievirus B3 with the full length coxsackievirus-adenovirus receptor. Nat. Struct. Biol. 2001, 8, 874–878. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Lin, F.; Chipman, P.R.; Bator, C.M.; Baker, T.S.; Shoham, M.; Kuhn, R.J.; Medof, M.E.; Rossmann, M.G. Structure of decay-accelerating factor bound to echovirus 7: A virus-receptor complex. Proc. Natl. Acad. Sci. USA 2002, 99, 10325–10329. [Google Scholar] [CrossRef] [PubMed]
- Hafenstein, S.; Bowman, V.D.; Chipman, P.R.; Bator Kelly, C.M.; Lin, F.; Medof, M.E.; Rossmann, M.G. Interaction of decay-accelerating factor with coxsackievirus B3. J. Virol. 2007, 81, 12927–12935. [Google Scholar] [CrossRef]
- Gnadig, N.F.; Beaucourt, S.; Campagnola, G.; Borderia, A.V.; Sanz-Ramos, M.; Gong, P.; Blanc, H.; Peersen, O.B.; Vignuzzi, M. Coxsackievirus B3 mutator strains are attenuated in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 2294. [Google Scholar] [CrossRef]
- Paananen, A.; Ylipaasto, P.; Smura, T.; Lempinen, M.; Galama, J.; Roivainen, M. A single amino acid substitution in viral VP1 protein alters the lytic potential of clone-derived variants of echovirus 9 DM strain in human pancreatic islets. J. Med. Virol. 2013, 85, 1267–1273. [Google Scholar] [CrossRef]
- Wang, M.; Li, J.; Yao, M.X.; Zhang, Y.W.; Hu, T.; Carr, M.J.; Duchene, S.; Zhang, X.C.; Zhang, Z.J.; Zhou, H.; et al. Genome Analysis of Coxsackievirus A4 Isolates from Hand, Foot, and Mouth Disease Cases in Shandong, China. Front. Microbiol. 2019, 10, 1001. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Blake, N.W.; Ouyang, X.; Pandolfino, Y.A.; Morgan-Capner, P.; Archard, L.C. A single amino acid substitution in the capsid protein VP1 of coxsackievirus B3 (CVB3) alters plaque phenotype in Vero cells but not cardiovirulence in a mouse model. Arch. Virol. 1995, 140, 959–966. [Google Scholar] [CrossRef]
- Halim, S.; Ramsingh, A.I. A point mutation in VP1 of coxsackievirus B4 alters antigenicity. Virology 2000, 269, 86–94. [Google Scholar] [CrossRef]
- Smura, T.; Natri, O.; Ylipaasto, P.; Hellman, M.; Al-Hello, H.; Piemonti, L.; Roivainen, M. Enterovirus strain and type-specific differences in growth kinetics and virus-induced cell destruction in human pancreatic duct epithelial HPDE cells. Virus Res. 2015, 210, 188–197. [Google Scholar] [CrossRef]
- Cohen, C.J.; Shieh, J.T.; Pickles, R.J.; Okegawa, T.; Hsieh, J.T.; Bergelson, J.M. The coxsackievirus and adenovirus receptor is a transmembrane component of the tight junction. Proc. Natl. Acad. Sci. USA 2001, 98, 15191–15196. [Google Scholar] [CrossRef]
- Shafren, D.R.; Bates, R.C.; Agrez, M.V.; Herd, R.L.; Burns, G.F.; Barry, R.D. Coxsackieviruses B1, B3, and B5 use decay accelerating factor as a receptor for cell attachment. J. Virol. 1995, 69, 3873–3877. [Google Scholar] [CrossRef] [PubMed]
- Riabi, S.; Harrath, R.; Gaaloul, I.; Bouslama, L.; Nasri, D.; Aouni, M.; Pillet, S.; Pozzetto, B. Study of Coxsackie B viruses interactions with Coxsackie Adenovirus receptor and Decay-Accelerating Factor using Human CaCo-2 cell line. J. Biomed. Sci. 2014, 21, 50. [Google Scholar] [CrossRef] [PubMed]
- Milstone, A.M.; Petrella, J.; Sanchez, M.D.; Mahmud, M.; Whitbeck, J.C.; Bergelson, J.M. Interaction with coxsackievirus and adenovirus receptor, but not with decay-accelerating factor (DAF), induces A-particle formation in a DAF-binding coxsackievirus B3 isolate. J. Virol. 2005, 79, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, J.M.; Modlin, J.F.; Wieland-Alter, W.; Cunningham, J.A.; Crowell, R.L.; Finberg, R.W. Clinical coxsackievirus B isolates differ from laboratory strains in their interaction with two cell surface receptors. J. Infect. Dis. 1997, 175, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, J.M.; Mohanty, J.G.; Crowell, R.L.; St John, N.F.; Lublin, D.M.; Finberg, R.W. Coxsackievirus B3 adapted to growth in RD cells binds to decay-accelerating factor (CD55). J. Virol. 1995, 69, 1903–1906. [Google Scholar] [CrossRef] [PubMed]
- Cifuente, J.O.; Ferrer, M.F.; Jaquenod de Giusti, C.; Song, W.C.; Romanowski, V.; Hafenstein, S.L.; Gomez, R.M. Molecular determinants of disease in coxsackievirus B1 murine infection. J. Med. Virol. 2011, 83, 1571–1581. [Google Scholar] [CrossRef]
- Stadnick, E.; Dan, M.; Sadeghi, A.; Chantler, J.K. Attenuating mutations in coxsackievirus B3 map to a conformational epitope that comprises the puff region of VP2 and the knob of VP3. J. Virol. 2004, 78, 13987–14002. [Google Scholar] [CrossRef]
- Sherry, B.; Rueckert, R. Evidence for at least two dominant neutralization antigens on human rhinovirus 14. J. Virol. 1985, 53, 137–143. [Google Scholar] [CrossRef]
- Sherry, B.; Mosser, A.G.; Colonno, R.J.; Rueckert, R.R. Use of monoclonal antibodies to identify four neutralization immunogens on a common cold picornavirus, human rhinovirus 14. J. Virol. 1986, 57, 246–257. [Google Scholar] [CrossRef]
- Minor, P.D.; Ferguson, M.; Evans, D.M.; Almond, J.W.; Icenogle, J.P. Antigenic structure of polioviruses of serotypes 1, 2 and 3. J. Gen. Virol. 1986, 67, 1283–1291. [Google Scholar] [CrossRef]
- Page, G.S.; Mosser, A.G.; Hogle, J.M.; Filman, D.J.; Rueckert, R.R.; Chow, M. Three-dimensional structure of poliovirus serotype 1 neutralizing determinants. J. Virol. 1988, 62, 1781–1794. [Google Scholar] [CrossRef]
- Knowlton, K.U.; Jeon, E.S.; Berkley, N.; Wessely, R.; Huber, S. A mutation in the puff region of VP2 attenuates the myocarditic phenotype of an infectious cDNA of the Woodruff variant of coxsackievirus B3. J. Virol. 1996, 70, 7811–7818. [Google Scholar] [CrossRef] [PubMed]
- Dan, M.; Chantler, J.K. A genetically engineered attenuated coxsackievirus B3 strain protects mice against lethal infection. J. Virol. 2005, 79, 9285–9295. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pfeiffer, J.K. Emergence of a Large-Plaque Variant in Mice Infected with Coxsackievirus B3. mBio 2016, 7, 119. [Google Scholar] [CrossRef]
- Ramsingh, A.I.; Collins, D.N. A point mutation in the VP4 coding sequence of coxsackievirus B4 influences virulence. J. Virol. 1995, 69, 7278–7281. [Google Scholar] [CrossRef] [PubMed]
- Ramsingh, A.I.; Caggana, M.; Ronstrom, S. Genetic mapping of the determinants of plaque morphology of coxsackievirus B4. Arch. Virol. 1995, 140, 2215–2226. [Google Scholar] [CrossRef] [PubMed]
- Reimann, B.Y.; Zell, R.; Kandolf, R. Mapping of a neutralizing antigenic site of Coxsackievirus B4 by construction of an antigen chimera. J. Virol. 1991, 65, 3475–3480. [Google Scholar] [CrossRef] [PubMed]
- Rohll, J.B.; Percy, N.; Ley, R.; Evans, D.J.; Almond, J.W.; Barclay, W.S. The 5′-untranslated regions of picornavirus RNAs contain independent functional domains essential for RNA replication and translation. J. Virol. 1994, 68, 4384–4391. [Google Scholar] [CrossRef]
- Sharma, N.; Ogram, S.A.; Morasco, B.J.; Spear, A.; Chapman, N.M.; Flanegan, J.B. Functional role of the 5′ terminal cloverleaf in Coxsackievirus RNA replication. Virology 2009, 393, 238–249. [Google Scholar] [CrossRef]
- Sean, P.; Nguyen, J.H.; Semler, B.L. Altered interactions between stem-loop IV within the 5′ noncoding region of coxsackievirus RNA and poly(rC) binding protein 2: Effects on IRES-mediated translation and viral infectivity. Virology 2009, 389, 45–58. [Google Scholar] [CrossRef]
- Zhong, Z.; Li, X.; Zhao, W.; Tong, L.; Liu, J.; Wu, S.; Lin, L.; Zhang, Z.; Tian, Y.; Zhang, F. Mutations at nucleotides 573 and 579 within 5′-untranslated region augment the virulence of coxsackievirus B1. Virus Res. 2008, 135, 255–259. [Google Scholar] [CrossRef] [PubMed]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Honkimaa, A.; Kimura, B.; Sioofy-Khojine, A.-B.; Lin, J.; Laiho, J.; Oikarinen, S.; Hyöty, H. Genetic Adaptation of Coxsackievirus B1 during Persistent Infection in Pancreatic Cells. Microorganisms 2020, 8, 1790. https://doi.org/10.3390/microorganisms8111790
Honkimaa A, Kimura B, Sioofy-Khojine A-B, Lin J, Laiho J, Oikarinen S, Hyöty H. Genetic Adaptation of Coxsackievirus B1 during Persistent Infection in Pancreatic Cells. Microorganisms. 2020; 8(11):1790. https://doi.org/10.3390/microorganisms8111790
Chicago/Turabian StyleHonkimaa, Anni, Bryn Kimura, Amir-Babak Sioofy-Khojine, Jake Lin, Jutta Laiho, Sami Oikarinen, and Heikki Hyöty. 2020. "Genetic Adaptation of Coxsackievirus B1 during Persistent Infection in Pancreatic Cells" Microorganisms 8, no. 11: 1790. https://doi.org/10.3390/microorganisms8111790
APA StyleHonkimaa, A., Kimura, B., Sioofy-Khojine, A.-B., Lin, J., Laiho, J., Oikarinen, S., & Hyöty, H. (2020). Genetic Adaptation of Coxsackievirus B1 during Persistent Infection in Pancreatic Cells. Microorganisms, 8(11), 1790. https://doi.org/10.3390/microorganisms8111790