Pet-Human Gut Microbiome Host Classifier Using Data from Different Studies



Abstract
1. Introduction
2. Materials and Methods
2.1. Data
2.2. QIIME2
- DADA2 [49] denoising quality parameter value was set to 10 (--p-trunc-q 10),
- taxonomy assignment using QIIME2 feature-classifier [50],
- a random subsample of 5000 reads was used to calculate feature tables,
- alpha-diversity was calculated by sampling 5000 random reads five times from the whole sample to decrease the impact of low-abundance bacteria; the resulting chao index is the mean of these iterations results,
2.3. Grouping Features at Different Taxonomic Levels
2.4. Filtering Rare Features
2.5. Mann-Whitney Test
2.6. t-SNE
2.7. Balanced Class Dataset
2.8. CLR (Center Log Ratio) Transformation
2.9. RF Implementation
- max_features in the range from 2 to the ‘number of features’,
- max_depth in the range from 2 to 52,
- min_samples_split in the range from 2 to 52,
- n_estimators from the set {1,5,10,50,100,500,1000}.
2.10. The Project Learning Curve
3. Results
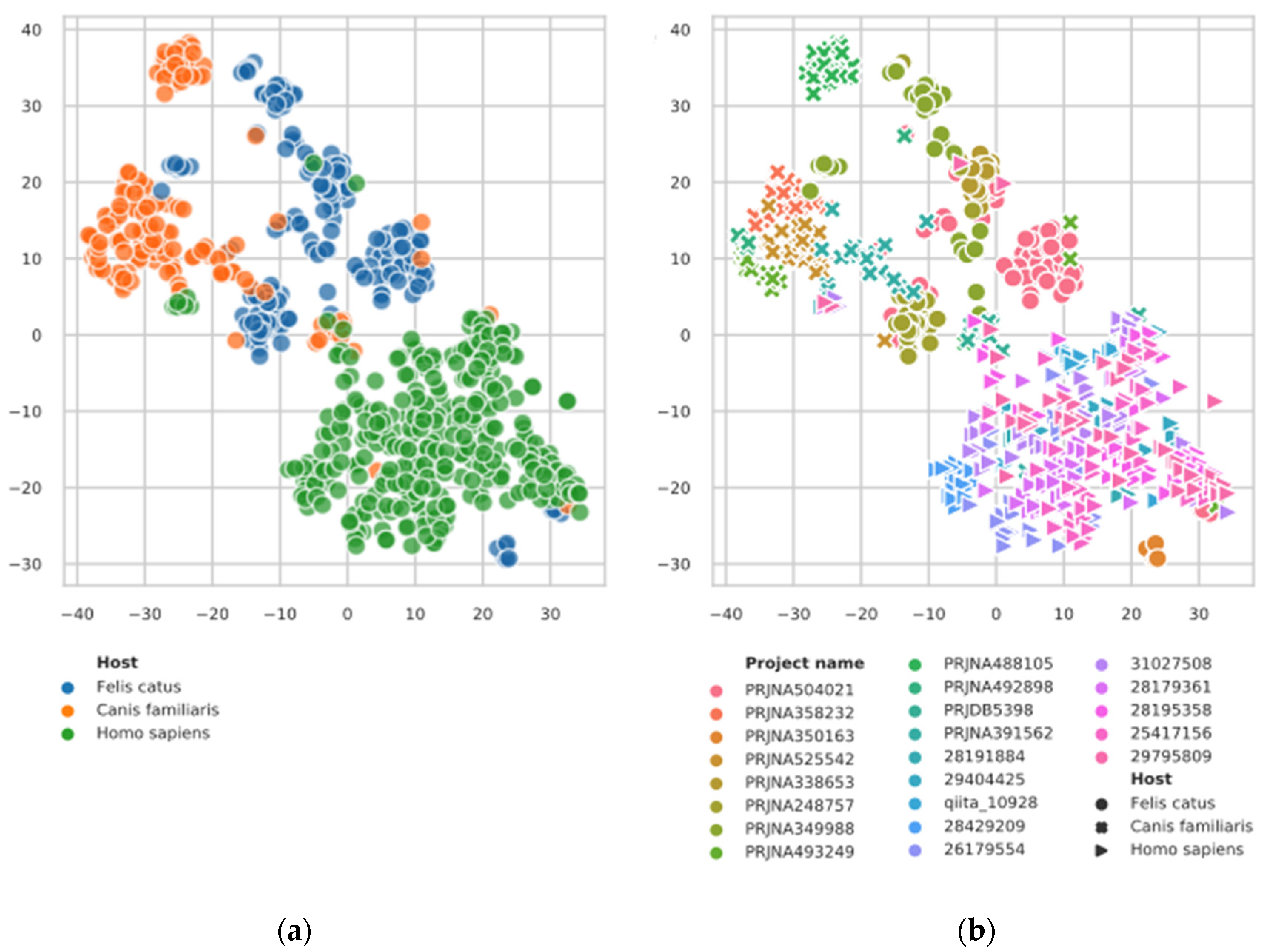
3.1. t-SNE Plot
3.2. Taxa Differentially Abundant in Pets and Humans
3.3. Random Forest Models
3.4. Random Forest Feature Importance
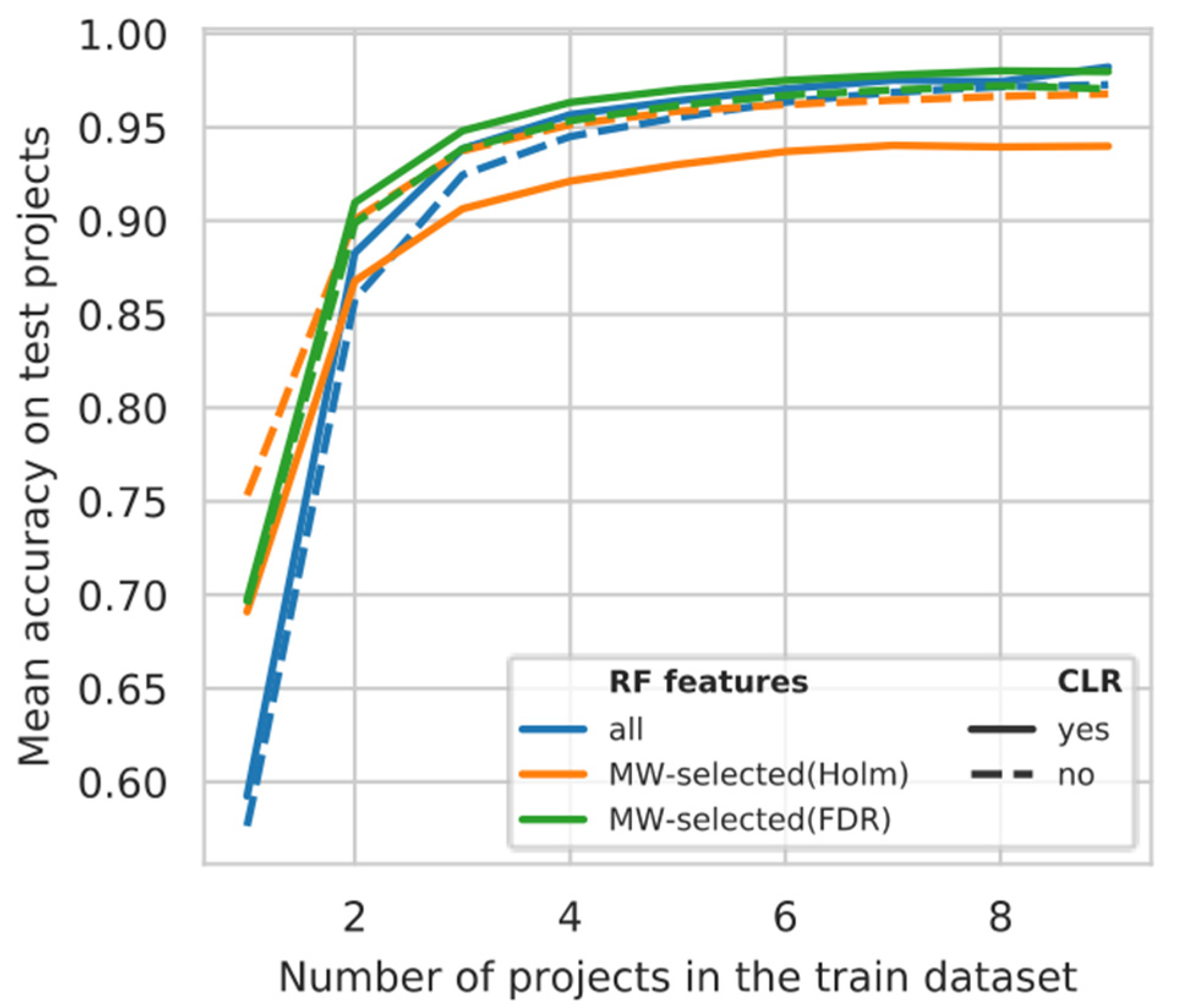
3.5. Projects Learning Curve
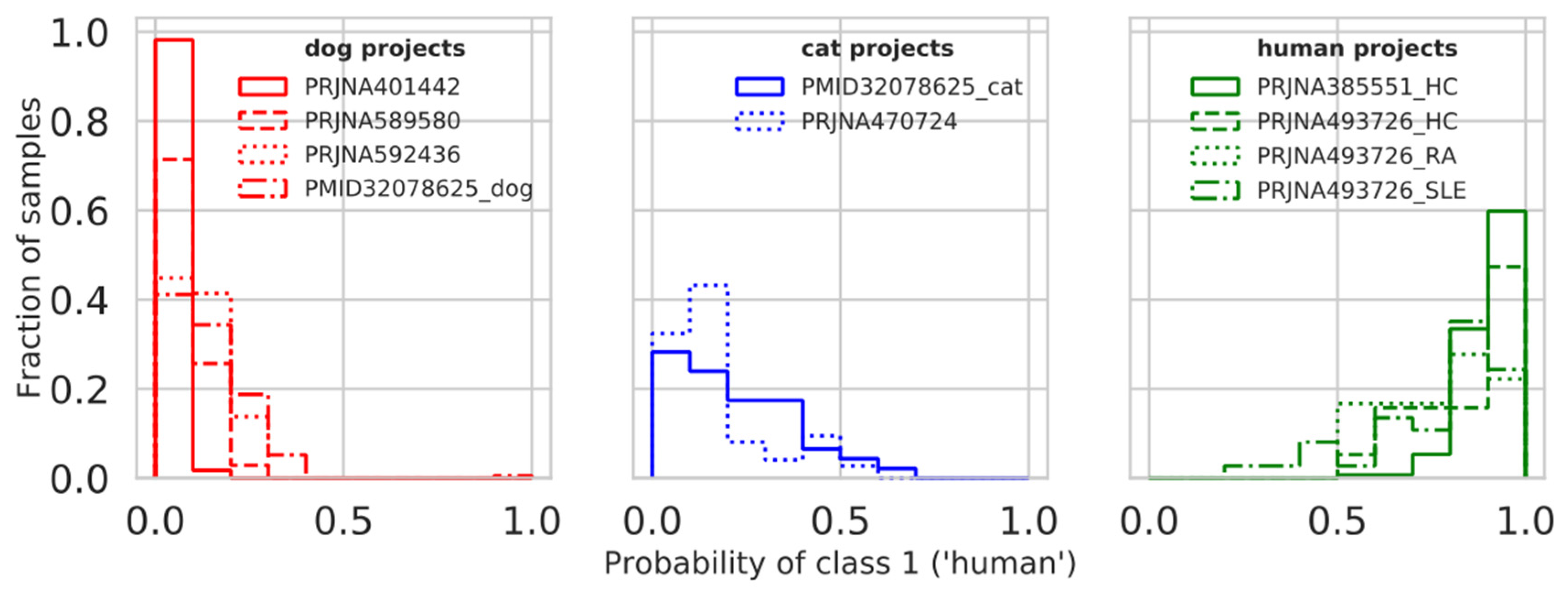
3.6. Model Testing on Additional Projects
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Huang, S.; Haiminen, N.; Carrieri, A.-P.; Hu, R.; Jiang, L.; Parida, L.; Russell, B.; Allaband, C.; Zarrinpar, A.; Vázquez-Baeza, Y.; et al. Human Skin, Oral, and Gut Microbiomes Predict Chronological Age. mSystems 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Tsukimi, T.; Yoshikawa, M.; Suzuki, K.; Takeda, T.; Tomita, M.; Fukuda, S. Cutibacterium acnes (Propionibacterium acnes) 16S rRNA Genotyping of Microbial Samples from Possessions Contributes to Owner Identification. mSystems 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Labus, J.S.; Hollister, E.B.; Jacobs, J.; Kirbach, K.; Oezguen, N.; Gupta, A.; Acosta, J.; Luna, R.A.; Aagaard, K.M.; Versalovic, J.; et al. Differences in gut microbial composition correlate with regional brain volumes in irritable bowel syndrome. Microbiome 2017, 5, 49. [Google Scholar] [CrossRef] [PubMed]
- Sprockett, D.; Fischer, N.; Boneh, R.S.; Turner, D.; Kierkus, J.; Sladek, M.; Escher, J.C.; Wine, E.; Yerushalmi, B.; Dias, J.A.; et al. Treatment-Specific Composition of the Gut Microbiota Is Associated With Disease Remission in a Pediatric Crohn’s Disease Cohort. Inflamm. Bowel Dis. 2019, 25, 1927–1938. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.M.A.; Connors, J.; Dunn, K.A.; Bielawski, J.P.; Comeau, A.M.; Langille, M.G.I.; Van Limbergen, J. Bacterial Taxa and Functions Are Predictive of Sustained Remission Following Exclusive Enteral Nutrition in Pediatric Crohn’s Disease. Inflamm. Bowel Dis. 2020, 26, 1026–1037. [Google Scholar] [CrossRef]
- Qu, K.; Gao, F.; Guo, F.; Zou, Q. Taxonomy dimension reduction for colorectal cancer prediction. Comput. Biol. Chem. 2019, 83, 107160. [Google Scholar] [CrossRef]
- Zheng, Y.-Y.; Wu, T.-T.; Liu, Z.-Q.; Li, A.; Guo, Q.-Q.; Ma, Y.-Y.; Zhang, Z.-L.; Xun, Y.-L.; Zhang, J.-C.; Wang, W.-R.; et al. Gut Microbiome-Based Diagnostic Model to Predict Coronary Artery Disease. J. Agric. Food Chem. 2020, 68, 3548–3557. [Google Scholar] [CrossRef]
- Dong, T.S.; Katzka, W.; Lagishetty, V.; Luu, K.; Hauer, M.; Pisegna, J.; Jacobs, J. A Microbial Signature Identifies Advanced Fibrosis in Patients with Chronic Liver Disease Mainly Due to NAFLD. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Zeng, Q.; Li, N.; He, Y.; Li, Y.; Yang, Z.; Zhao, X.; Liu, Y.; Wang, Y.; Sun, J.; Feng, X.; et al. Discrepant gut microbiota markers for the classification of obesity-related metabolic abnormalities. Sci. Rep. 2019, 9, 13424. [Google Scholar] [CrossRef]
- Liu, B.; Lin, W.; Chen, S.; Xiang, T.; Yang, Y.; Yin, Y.; Xu, G.; Liu, Z.; Liu, L.; Pan, J.; et al. Gut Microbiota as an Objective Measurement for Auxiliary Diagnosis of Insomnia Disorder. Front. Microbiol. 2019, 10, 1770. [Google Scholar] [CrossRef]
- Hu, S.; Li, A.; Huang, T.; Lai, J.; Li, J.; Sublette, M.E.; Lu, H.; Lu, Q.; Du, Y.; Hu, Z.; et al. Gut Microbiota Changes in Patients with Bipolar Depression. Adv. Sci. 2019, 6, 1900752. [Google Scholar] [CrossRef]
- Roguet, A.; Eren, A.M.; Newton, R.J.; McLellan, S.L. Fecal source identification using random forest. Microbiome 2018, 6, 185. [Google Scholar] [CrossRef]
- Eren, A.M.; Sogin, M.L.; Morrison, H.G.; Vineis, J.H.; Fisher, J.C.; Newton, R.J.; McLellan, S.L. A single genus in the gut microbiome reflects host preference and specificity. ISME J. 2014, 9, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Knights, D.; Kuczynski, J.; Charlson, E.S.; Zaneveld, J.; Mozer, M.C.; Collman, R.G.; Bushman, F.D.; Knight, R.; Kelley, S.T. Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 2011, 8, 761–763. [Google Scholar] [CrossRef]
- Nishida, A.H.; Ochman, H. Rates of gut microbiome divergence in mammals. Mol. Ecol. 2018, 27, 1884–1897. [Google Scholar] [CrossRef] [PubMed]
- Groussin, M.; Mazel, F.; Sanders, J.G.; Smillie, C.S.; Lavergne, S.; Thuiller, W.; Alm, E.J. Unraveling the processes shaping mammalian gut microbiomes over evolutionary time. Nat. Commun. 2017, 8, 14319. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Stombaugh, J.; Gonzalez, A.; Ackermann, G.; Wendel, D.; Vázquez-Baeza, Y.; Jansson, J.K.; Gordon, J.I.; Knight, R. Meta-analyses of studies of the human microbiota. Genome Res. 2013, 23, 1704–1714. [Google Scholar] [CrossRef]
- Shah, M.S.; DeSantis, T.Z.; Weinmaier, T.; McMurdie, P.J.; Cope, J.; Altrichter, A.; Yamal, J.-M.; Hollister, E.B. Leveraging sequence-based faecal microbial community survey data to identify a composite biomarker for colorectal cancer. Gut 2017, 67, 882–891. [Google Scholar] [CrossRef]
- Gorzelak, M.A.; Gill, S.K.; Tasnim, N.; Ahmadi-Vand, Z.; Jay, M.; Gibson, D.L. Methods for Improving Human Gut Microbiome Data by Reducing Variability through Sample Processing and Storage of Stool. PLoS ONE 2015, 10, e0134802. [Google Scholar] [CrossRef]
- Yuan, S.; Cohen, D.B.; Ravel, J.; Abdo, Z.; Forney, L.J. Evaluation of Methods for the Extraction and Purification of DNA from the Human Microbiome. PLoS ONE 2012, 7, e33865. [Google Scholar] [CrossRef]
- Mao, D.-P.; Zhou, Q.; Chen, C.-Y.; Quan, Z.-X. Coverage evaluation of universal bacterial primers using the metagenomic datasets. BMC Microbiol. 2012, 12, 66. [Google Scholar] [CrossRef] [PubMed]
- Ratan, A.; Miller, W.; Guillory, J.; Stinson, J.; Seshagiri, S.; Schuster, S.C. Comparison of Sequencing Platforms for Single Nucleotide Variant Calls in a Human Sample. PLoS ONE 2013, 8, e55089. [Google Scholar] [CrossRef] [PubMed]
- Rintala, A.; Pietilä, S.; Munukka, E.; Eerola, E.; Pursiheimo, J.-P.; Laiho, A.; Pekkala, S.; Huovinen, P. Gut Microbiota Analysis Results Are Highly Dependent on the 16S rRNA Gene Target Region, Whereas the Impact of DNA Extraction Is Minor. J. Biomol. Tech. JBT 2017, 28, 19–30. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Marsilio, S.; Pilla, R.; Sarawichitr, B.; Chow, B.; Hill, S.L.; Ackermann, M.R.; Estep, J.S.; Lidbury, J.A.; Steiner, J.M.; Suchodolski, J.S. Characterization of the fecal microbiome in cats with inflammatory bowel disease or alimentary small cell lymphoma. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.M.; Jenkins, T.P.; Latrofa, M.S.; Giannelli, A.; Papadopoulos, E.; De Carvalho, L.M.; Nolan, M.J.; Otranto, D.; Cantacessi, C. Helminth infections and gut microbiota—A feline perspective. Parasites Vectors 2016, 9, 625. [Google Scholar] [CrossRef]
- Bell, E.T.; Suchodolski, J.S.; Isaiah, A.; Fleeman, L.M.; Cook, A.K.; Steiner, J.M.; Mansfield, C.S. Faecal Microbiota of Cats with Insulin-Treated Diabetes Mellitus. PLoS ONE 2014, 9, e108729. [Google Scholar] [CrossRef]
- Whittemore, J.C.; Stokes, J.E.; Price, J.M.; Suchodolski, J.S. Effects of a synbiotic on the fecal microbiome and metabolomic profiles of healthy research cats administered clindamycin: A randomized, controlled trial. Gut Microbes 2019, 10, 521–539. [Google Scholar] [CrossRef]
- Vientós-Plotts, A.I.; Ericsson, A.C.; Rindt, H.; Grobman, M.E.; Graham, A.; Bishop, K.; Cohn, L.A.; Reinero, C. Dynamic changes of the respiratory microbiota and its relationship to fecal and blood microbiota in healthy young cats. PLoS ONE 2017, 12, e0173818. [Google Scholar] [CrossRef]
- Jarett, J.K.; Carlson, A.; Serao, M.R.; Strickland, J.; Serfilippi, L.; Ganz, H.H. Diets with and without edible cricket support a similar level of diversity in the gut microbiome of dogs. PeerJ 2019, 7, e7661. [Google Scholar] [CrossRef]
- Herstad, K.M.V.; Moen, A.E.F.; Gaby, J.C.; Moe, L.; Skancke, E. Characterization of the fecal and mucosa-associated microbiota in dogs with colorectal epithelial tumors. PLoS ONE 2018, 13, e0198342. [Google Scholar] [CrossRef] [PubMed]
- Fujishiro, M.A.; Lidbury, J.; Pilla, R.; Steiner, J.M.; Lappin, M.R.; Suchodolski, J.S. Evaluation of the effects of anthelmintic administration on the fecal microbiome of healthy dogs with and without subclinical Giardia spp. and Cryptosporidium canis infections. PLoS ONE 2020, 15, e0228145. [Google Scholar] [CrossRef] [PubMed]
- Omatsu, T.; Omura, M.; Katayama, Y.; Kimura, T.; Okumura, M.; Okumura, A.; Murata, Y.; Mizutani, T. Molecular diversity of the faecal microbiota of Toy Poodles in Japan. J. Veter Med. Sci. 2018, 80, 749–754. [Google Scholar] [CrossRef]
- McDonald, D.; Hyde, E.; Debelius, J.W.; Morton, J.T.; Gonzalez, A.; Ackermann, G.; Aksenov, A.A.; Behsaz, B.; Brennan, C.; Chen, Y.; et al. American Gut: An Open Platform for Citizen Science Microbiome Research. mSystems 2018, 3, e00031-18. [Google Scholar] [CrossRef]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human Genetics Shape the Gut Microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef]
- Hill-Burns, E.M.; Debelius, J.W.; Bs, J.T.M.; Ba, W.T.W.; Ms, M.R.L.; Ms, Z.D.W.; Peddada, S.D.; Do, S.A.F.; Molho, E.; Zabetian, C.P.; et al. Parkinson’s disease and Parkinson’s disease medications have distinct signatures of the gut microbiome. Mov. Disord. 2017, 32, 739–749. [Google Scholar] [CrossRef]
- Pascal, V.; Pozuelo, M.; Borruel, N.; Casellas, F.; Campos, D.; Santiago, A.; Martinez, X.; Varela, E.; Sarrabayrouse, G.; Machiels, K.; et al. A microbial signature for Crohn’s disease. Gut 2017, 66, 813–822. [Google Scholar] [CrossRef]
- Liu, H.; Chen, X.; Hu, X.; Niu, H.; Tian, R.; Wang, H.; Pang, H.; Jiang, L.; Qiu, B.; Chen, X.; et al. Alterations in the gut microbiome and metabolism with coronary artery disease severity. Microbiome 2019, 7, 68. [Google Scholar] [CrossRef]
- Keshavarzian, A.; Green, S.J.; Engen, P.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 2015, 30, 1351–1360. [Google Scholar] [CrossRef]
- Petrov, V.A.; Saltykova, I.V.; Zhukova, I.A.; Alifirova, V.M.; Zhukova, N.G.; Dorofeeva, Y.B.; Tyakht, A.V.; Kovarsky, B.A.; Alekseev, D.G.; Kostryukova, E.S.; et al. Analysis of Gut Microbiota in Patients with Parkinson’s Disease. Bull. Exp. Biol. Med. 2017, 162, 734–737. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, Z.Z.; He, Y.; Yang, Y.; Liu, L.; Lin, Q.; Nie, Y.; Li, M.; Zhi, F.; Liu, S.; et al. Gut Microbiota Offers Universal Biomarkers across Ethnicity in Inflammatory Bowel Disease Diagnosis and Infliximab Response Prediction. mSystems 2018, 3, e00188-17. [Google Scholar] [CrossRef] [PubMed]
- Halfvarson, J.; Brislawn, C.J.; Lamendella, R.; Vázquez-Baeza, Y.; Walters, W.A.; Bramer, L.M.; D’Amato, M.; Bonfiglio, F.; McDonald, D.; Gonzalez, A.; et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2017, 2, 17004. [Google Scholar] [CrossRef] [PubMed]
- Bermingham, E.N.; Young, W.; Butowski, C.F.; Moon, C.D.; MacLean, P.H.; Rosendale, D.; Cave, N.J.; Thomas, D.G. The Fecal Microbiota in the Domestic Cat (Felis catus) Is Influenced by Interactions Between Age and Diet; A Five Year Longitudinal Study. Front. Microbiol. 2018, 9, 1231. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.R.; Shmalberg, J.; Tanprasertsuk, J.; Perry, L.; Massey, D.; Honaker, R.W. Characterization of gut microbiomes of household pets in the United States using a direct-to-consumer approach. PLoS ONE 2020, 15, e0227289. [Google Scholar] [CrossRef]
- Bian, G.; Gloor, G.B.; Gong, A.; Jia, C.; Zhang, W.; Hu, J.; Zhang, H.; Zhang, Y.; Zhou, Z.; Zhang, J.; et al. The Gut Microbiota of Healthy Aged Chinese Is Similar to That of the Healthy Young. mSphere 2017, 2, e00327-17. [Google Scholar] [CrossRef]
- Li, Y.; Wang, H.-F.; Li, X.; Li, H.-X.; Zhang, Q.; Zhou, H.-W.; He, Y.; Li, P.; Fu, C.; Zhang, X.-H.; et al. Disordered intestinal microbes are associated with the activity of Systemic Lupus Erythematosus. Clin. Sci. 2019, 133, 821–838. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.; Caporaso, J.G. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2′s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Ritari, J.; Salojärvi, J.; Lahti, L.; De Vos, W.M. Improved taxonomic assignment of human intestinal 16S rRNA sequences by a dedicated reference database. BMC Genom. 2015, 16, 1056. [Google Scholar] [CrossRef]
- Holm, S. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 1979, 6, 65–70. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome Datasets Are Compositional: And This Is Not Optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef] [PubMed]
- Deng, P.; Swanson, K.S. Gut microbiota of humans, dogs and cats: Current knowledge and future opportunities and challenges. Br. J. Nutr. 2014, 113, S6–S17. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mazcorro, J.F.; Minamoto, Y.; Kawas, J.R.; Suchodolski, J.S.; De Vos, W.M. Akkermansia and Microbial Degradation of Mucus in Cats and Dogs: Implications to the Growing Worldwide Epidemic of Pet Obesity. Veter Sci. 2020, 7, 44. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Project Name | Host | Host Type | Number of Samples | Number of Samples (Train Dataset) | PMID | Author/Year | Ref. |
|---|---|---|---|---|---|---|---|
| PRJNA504021 | Felis catus | pet | 65 | 65 | 31844119 | Marsilio et al. (2019) | [25] |
| PRJNA349988 | Felis catus | pet | 44 | 44 | 27912797 | Duarte et al. (2016) | [26] |
| PRJNA248757 | Felis catus | pet | 30 | 30 | 25279695 | Bell et al. (2014) | [27] |
| PRJNA338653 | Felis catus | pet | 19 | 19 | 30709324 | Whittemore et al. (2019) | [28] |
| PRJNA350163 | Felis catus | pet | 6 | 6 | 28278278 | Vientós-Plotts et al. (2017) | [29] |
| PRJNA488105 | Canis familiaris | pet | 34 | 34 | no paper | ||
| PRJNA525542 | Canis familiaris | pet | 32 | 32 | 31565574 | Jarett et al. (2019) | [30] |
| PRJNA358232 | Canis familiaris | pet | 30 | 30 | no paper | ||
| PRJNA391562 | Canis familiaris | pet | 23 | 23 | 29852000 | Herstad et al. (2018) | [31] |
| PRJNA493249 | Canis familiaris | pet | 19 | 19 | 32027665 | Fujishiro et al. (2020) | [32] |
| PRJDB5398 | Canis familiaris | pet | 13 | 13 | 29643280 | Omatsu et al. (2018) | [33] |
| PRJNA492898 | Canis familiaris | pet | 6 | 6 | 32027665 | Fujishiro et al. (2020) | [32] |
| PMID29795809 | Homo sapiens | human | 681 | 46 | 29795809 | McDonald et al. (2018) | [34] |
| PMID25417156 | Homo sapiens | human | 200 | 45 | 25417156 | Goodrich et al. (2014) | [35] |
| PMID28195358 | Homo sapiens | human | 115 | 45 | 28195358 | Hill-Burns et al. (2017) | [36] |
| PMID28179361 | Homo sapiens | human | 102 | 45 | 28179361 | Pascal et al. (2017) | [37] |
| PMID31027508 | Homo sapiens | human | 49 | 45 | 31027508 | Liu et al. (2019) | [38] |
| PMID26179554 | Homo sapiens | human | 31 | 31 | 26179554 | Keshavarzian et al. (2015) | [39] |
| PMID28429209 | Homo sapiens | human | 22 | 22 | 28429209 | Petrov et al. (2017) | [40] |
| qiita_10928 | Homo sapiens | human | 21 | 21 | no paper | ||
| PMID29404425 | Homo sapiens | human | 12 | 12 | 29404425 | Zhou et al. (2018) | [41] |
| PMID28191884 | Homo sapiens | human | 9 | 9 | 28191884 | Halfvarson et al. (2017) | [42] |
| Additional Projects | |||||||
| PRJNA470724 | Felis catus | pet | 74 | 29971046 | Bermingham et al. (2018) | [43] | |
| PMID32078625 | Felis catus | pet | 46 | 32078625 | Jha et al. (2020) | [44] | |
| PMID32078625 | Canis familiaris | pet | 192 | 32078625 | Jha et al. (2020) | [44] | |
| PRJNA401442 | Canis familiaris | pet | 56 | no paper | |||
| PRJNA589580 | Canis familiaris | pet | 35 | no paper | |||
| PRJNA592436 | Canis familiaris | pet | 29 | no paper | |||
| PRJNA385551 | Homo sapiens | human | 284 | 28959739 | Bian et al. (2017) | [45] | |
| PRJNA493726 | Homo sapiens | human | 74 | 30872359 | Li et al. (2019) | [46] | |
| Model Name | Level | Features Type | Number of Features | CLR | Accuracy | F1 Score | Precision | Recall |
|---|---|---|---|---|---|---|---|---|
| Family_ALL_CLR | Family | all | 56 | yes | 0.981 ± 0.004 | 0.980 ± 0.004 | 0.987 ± 0.004 | 0.974 ± 0.006 |
| Family_ALL | Family | all | 56 | no | 0.983 ± 0.004 | 0.983 ± 0.004 | 0.989 ± 0.004 | 0.977 ± 0.006 |
| Family_MW-FDR_CLR | Family | best_fdr | 14 | yes | 0.966 ± 0.004 | 0.966 ± 0.004 | 0.976 ± 0.005 | 0.955 ± 0.006 |
| Family_MW-FDR | Family | best_fdr | 14 | no | 0.970 ± 0.003 | 0.970 ± 0.003 | 0.986 ± 0.004 | 0.955 ± 0.005 |
| Family_MW-Holm_CLR | Family | best_holm | 6 | yes | 0.954 ± 0.003 | 0.954 ± 0.003 | 0.957 ± 0.004 | 0.951 ± 0.004 |
| Family_MW-Holm | Family | best_holm | 6 | no | 0.953 ± 0.004 | 0.953 ± 0.003 | 0.951 ± 0.006 | 0.955 ± 0.004 |
| Genus_ALL_CLR | Genus | all | 139 | yes | 0.990 ± 0.002 | 0.990 ± 0.002 | 0.999 ± 0.001 | 0.981 ± 0.003 |
| Genus_ALL | Genus | all | 139 | no | 0.992 ± 0.002 | 0.992 ± 0.002 | 0.999 ± 0.002 | 0.985 ± 0.003 |
| Genus_MW-FDR_CLR | Genus | best_fdr | 32 | yes | 0.986 ± 0.002 | 0.985 ± 0.002 | 0.997 ± 0.003 | 0.974 ± 0.004 |
| Genus_MW-FDR | Genus | best_fdr | 32 | no | 0.989 ± 0.002 | 0.989 ± 0.002 | 0.998 ± 0.003 | 0.979 ± 0.003 |
| Genus_MW-Holm_CLR | Genus | best_holm | 12 | yes | 0.972 ± 0.003 | 0.972 ± 0.003 | 0.982 ± 0.004 | 0.963 ± 0.004 |
| Genus_MW-Holm | Genus | best_holm | 12 | no | 0.967 ± 0.003 | 0.967 ± 0.003 | 0.981 ± 0.004 | 0.953 ± 0.005 |
| Host Type | Host | Test Project | Accuracy | Number of Samples |
|---|---|---|---|---|
| human | Homo sapiens | PRJNA385551 | 1 | 284 |
| human | Homo sapiens | PRJNA493726 | 0.932 | 74 |
| human | Homo sapiens | PRJNA493726_HC | 1 | 19 |
| human | Homo sapiens | PRJNA493726_RA | 1 | 18 |
| human | Homo sapiens | PRJNA493726_SLE | 0.865 | 37 |
| pet | Felis catus + Canis familiaris | PMID32078625 | 0.983 | 238 |
| pet | Felis catus | PMID32078625_cat | 0.935 | 46 |
| pet | Canis familiaris | PMID32078625_dog | 0.995 | 192 |
| pet | Canis familiaris | PRJNA401442 | 1 | 56 |
| pet | Felis catus | PRJNA470724 | 0.973 | 74 |
| pet | Canis familiaris | PRJNA589580 | 1 | 35 |
| pet | Canis familiaris | PRJNA592436 | 1 | 29 |
| Metric Name | Total Dataset | Only Healthy Controls |
|---|---|---|
| Accuracy | 0.971 ± 0.010 | 0.988 ± 0.008 |
| Precision | 0.976 ± 0.015 | 0.977 ± 0.015 |
| Recall | 0.966 ± 0.016 | 1.000 ± 0.000 |
| F1 score | 0.971 ± 0.010 | 0.988 ± 0.008 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bykova, N.; Litovka, N.; Popenko, A.; Musienko, S. Pet-Human Gut Microbiome Host Classifier Using Data from Different Studies. Microorganisms 2020, 8, 1591. https://doi.org/10.3390/microorganisms8101591
Bykova N, Litovka N, Popenko A, Musienko S. Pet-Human Gut Microbiome Host Classifier Using Data from Different Studies. Microorganisms. 2020; 8(10):1591. https://doi.org/10.3390/microorganisms8101591
Chicago/Turabian StyleBykova, Nadia, Nikita Litovka, Anna Popenko, and Sergey Musienko. 2020. "Pet-Human Gut Microbiome Host Classifier Using Data from Different Studies" Microorganisms 8, no. 10: 1591. https://doi.org/10.3390/microorganisms8101591
APA StyleBykova, N., Litovka, N., Popenko, A., & Musienko, S. (2020). Pet-Human Gut Microbiome Host Classifier Using Data from Different Studies. Microorganisms, 8(10), 1591. https://doi.org/10.3390/microorganisms8101591