Identification of Down-Regulated Proteome in Saccharomyces cerevisiae with the Deletion of Yeast Cathepsin D in Response to Nitrogen Stress



Abstract
1. Introduction
2. Materials and Methods
2.1. Yeast Strains and Chemostat Cultivation Conditions
2.2. Preparation of Intracellular Protein Extracts
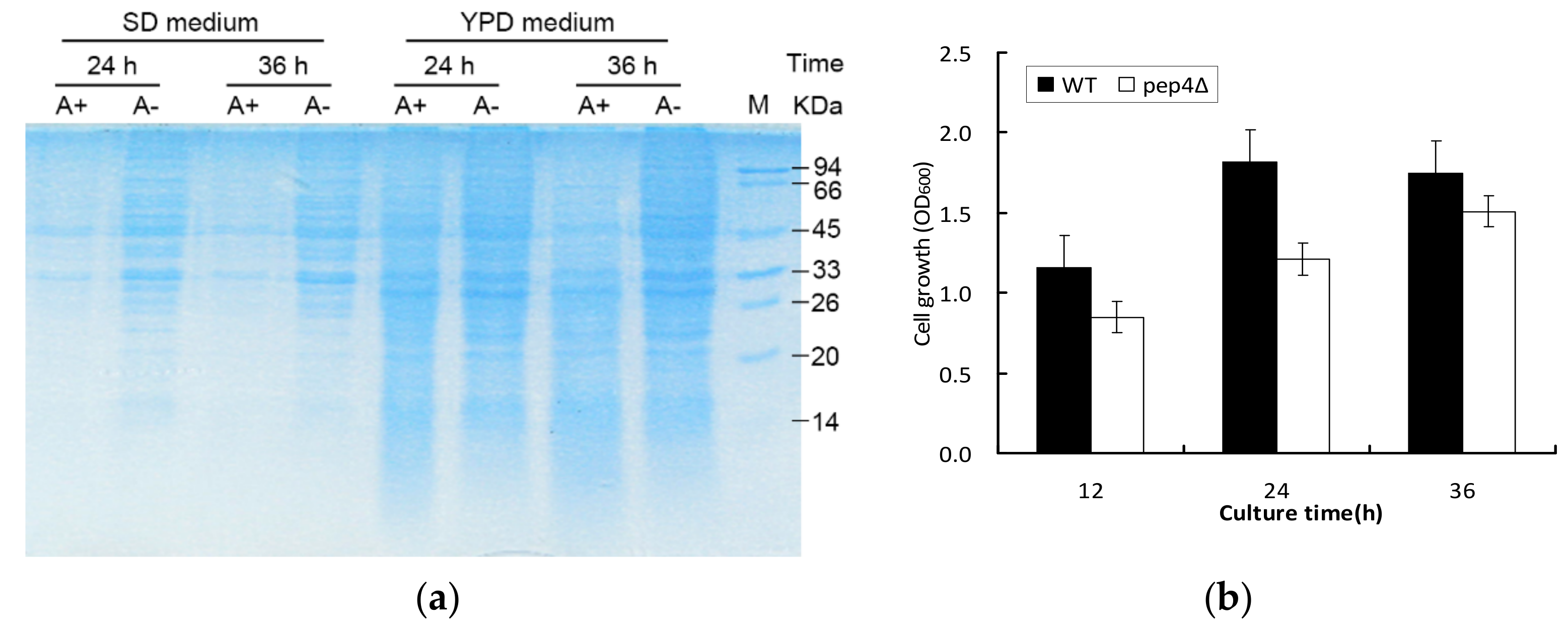
2.3. SDS-PAGE Gel Electrophoresis
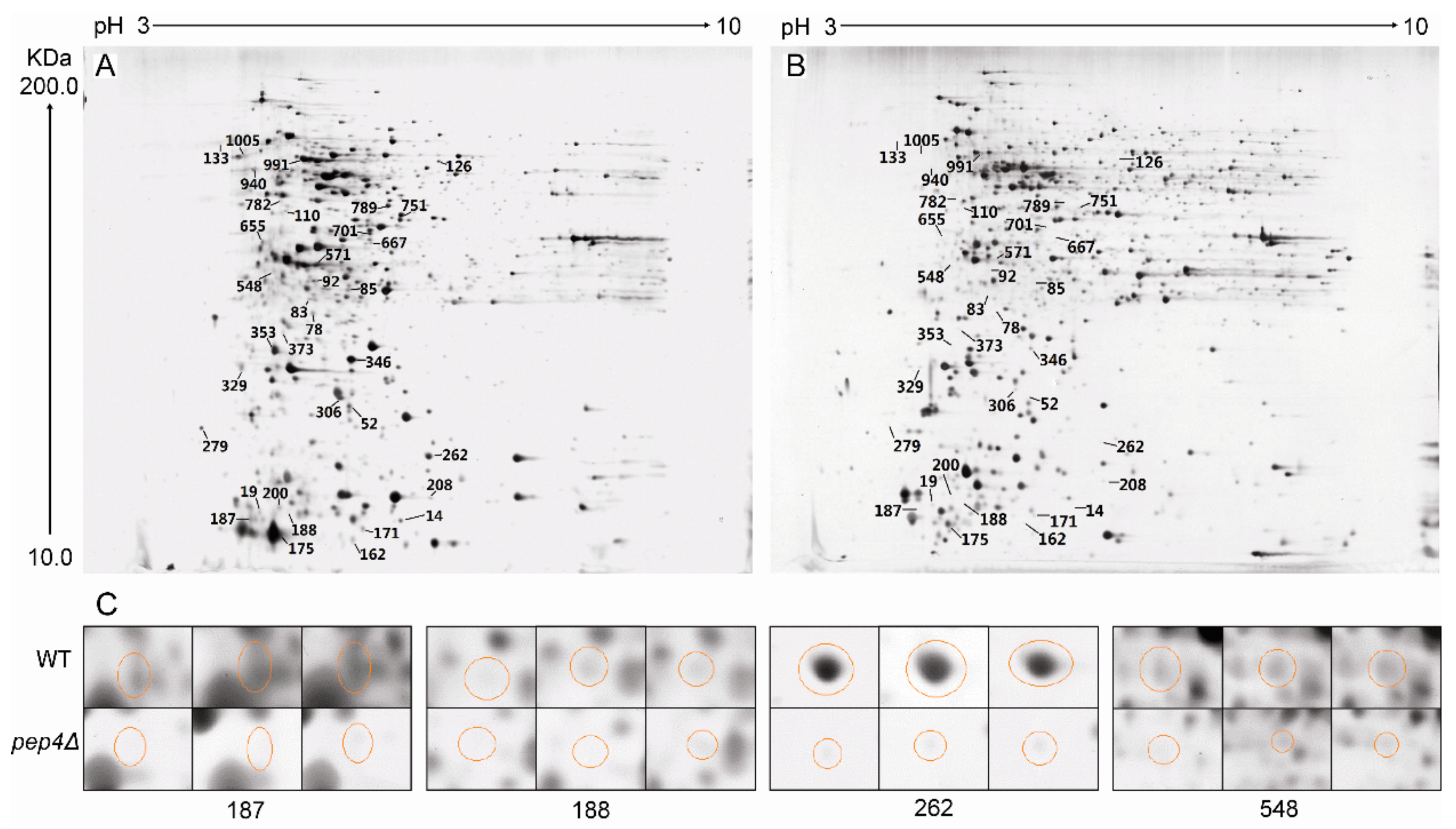
2.4. Two-Dimensional Polyacrylamide Gel Electrophoresis
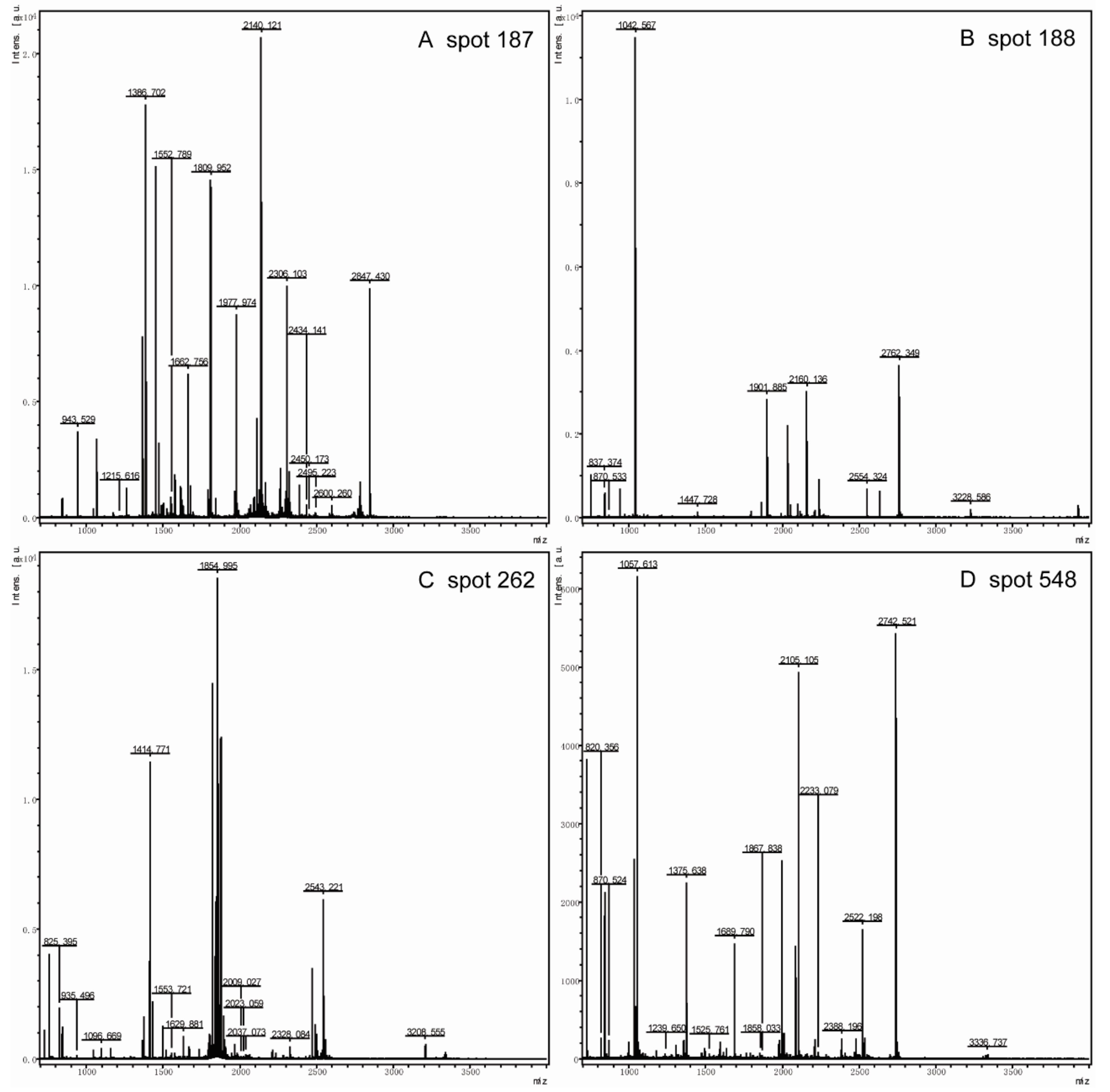
2.5. Mass Spectrometry Analysis and Database Search
2.6. Real-Time Quantitative PCR (RT-qPCR) Assay
3. Results
3.1. Pep4p Proteolyticeffect on Intracellular Proteins and Cell Growth under Different Culture Conditions
3.2. 2-DE Profiling of Wild-Type S. cerevisiae and Pep4p-Deficient Strain
3.2.1. Identification of Down-Regulated Proteins and Gene Ontology (GO) Analysis
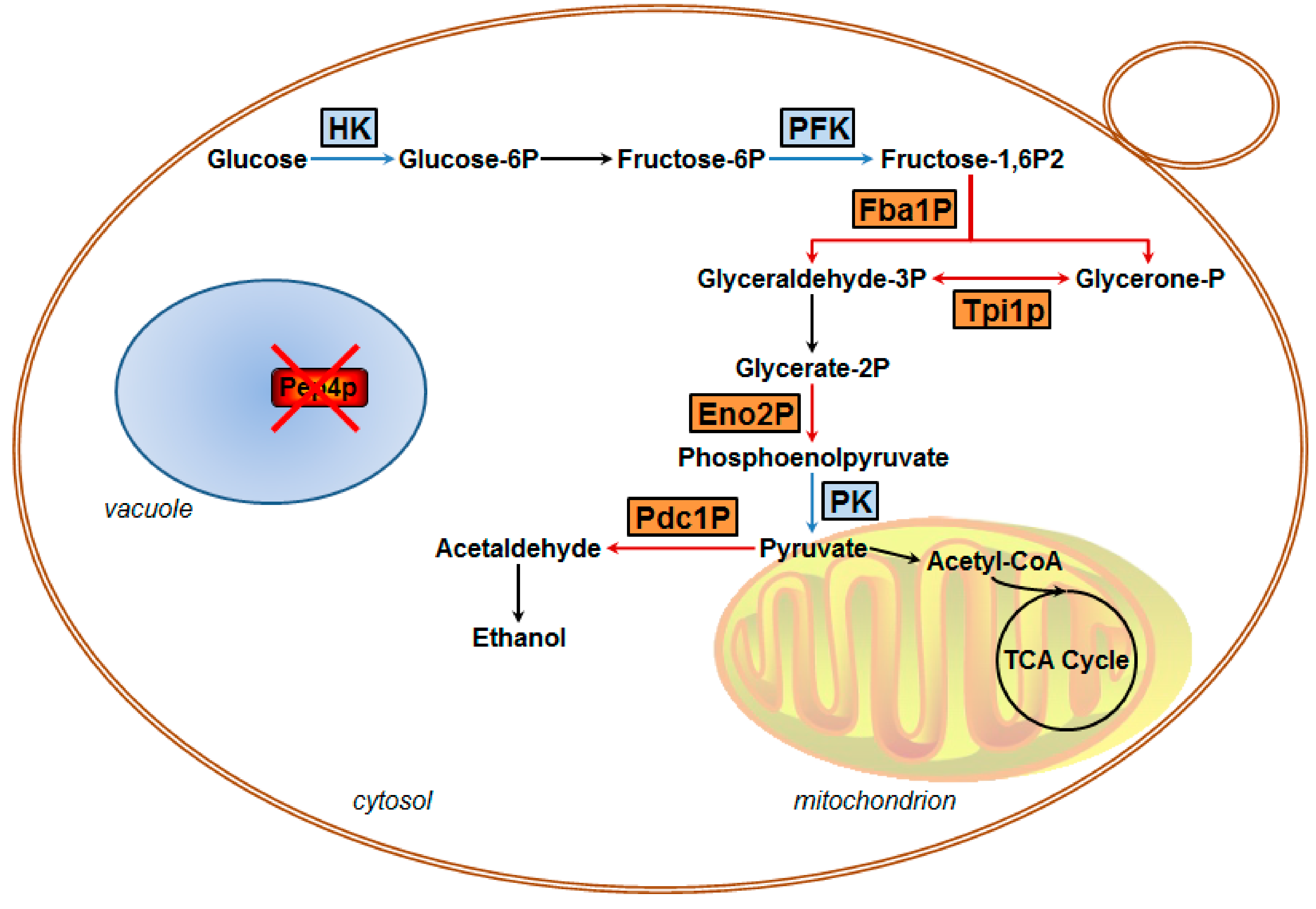
3.2.2. Identification of Key Down-Regulated Proteins Involved in Yeast Cell Metabolic Pathways
3.3. Verification of mRNA Levels of Key Down-Regulated Genes Relating to Glycolytic Flux
4. Discussion
4.1. Glycolytic Metabolism is Influenced by Pep4p Deficiency
4.2. Pep4p Deletion Plays a Systematic Role in Cellular Physiology
4.3. Knockout of Pep4p Interferes in Post-Translational Modification and Transcriptional Regulation
4.4. Depletion of Pep4 Gene Negatively Impacts Stress Proteins Expression
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wang, Z.-Y.; He, G.-Q.; Liu, Z.-S.; Ruan, H.; Chen, Q.-H.; Xiong, H.-P. Purification of yeast proteinase A from fresh beer and its specificity on foam proteins. Int. J. Food Sci. Technol. 2005, 40, 835–840. [Google Scholar] [CrossRef]
- Lu, J.; Dong, J.; Wu, D.; Chen, Y.; Guo, X.; Shi, Y.; Sun, X.; Xiao, D. Construction of recombinant industrial brewer’s yeast with lower diacetyl production and proteinase A activity. Eur. Food Res. Technol. 2012, 235, 951–961. [Google Scholar] [CrossRef]
- Zhang, H.-B.; Ruan, H.; Li, W.-F.; Zhang, W.; Su, Z.-R.; He, G.-Q.; Chen, Q.-H. Construction of recombinant industrial S. cerevisiae strain with barley lipid-transfer protein 1 secretion capability and lower PrA activity. Eur. Food Res. Technol. 2011, 233, 707–716. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, Q.-H.; Fu, M.-L.; Wang, J.-L.; Zhang, H.-B.; He, G.-Q. Construction of recombinant industrial Saccharomyces cerevisiae strain with bglS gene insertion into PEP4 locus by homologous recombination. J. Zhejiang Univ. Sci. B 2008, 9, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; He, G.Q.; Ruan, H.; Liu, Z.S.; Yang, L.F.; Zhang, B.R. Construction of proteinase A deficient transformant of industrial brewing yeast. Eur. Food Res. Technol. 2007, 225, 831–835. [Google Scholar] [CrossRef]
- Parr, C.L.; Keates, R.A.B.; Bryksa, B.C.; Ogawa, M.; Yada, R.Y. The structure and function of Saccharomyces cerevisiae proteinase A. Yeast 2007, 24, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Aminopeptidase I of Saccharomyces cerevisiae is localized to the vacuole independent of the secretory pathway. J. Cell Boil. 1992, 119, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Teichert, U.; Mechler, B.; Müller, H.; Wolf, D.H. Lysosomal (vacuolar) proteinases of yeast are essential catalysts for protein degradation, differentiation, and cell survival. J. Boil. Chem. 1989, 264, 16037–16045. [Google Scholar]
- Spedale, G.; Mischerikow, N.; Heck, A.J.R.; Timmers, H.T.M.; Pijnappel, W.W.M.P. Identification of Pep4p as the Protease Responsible for Formation of the SAGA-related SLIK Protein Complex. J. Boil. Chem. 2010, 285, 22793–22799. [Google Scholar] [CrossRef]
- Koutelou, E.; Hirsch, C.L.; Dent, S.Y.R. Multiple faces of the SAGA complex. Curr. Opin. Cell Biol. 2010, 22, 374–382. [Google Scholar] [CrossRef]
- Pereira, H.; Azevedo, F.; Rego, A.; Sousa, M.J.; Chaves, S.R.; Côrte-Real, M. The protective role of yeast Cathepsin D in acetic acid-induced apoptosis depends on ANT (Aac2p) but not on the voltage-dependent channel (Por1p). FEBS Lett. 2013, 587, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.; Mojzita, D.; Amorim, M.A.; Almeida, T.; Hohmann, S.; Moradas-Ferreira, P.; Costa, V. The Pep4p vacuolar proteinase contributes to the turnover of oxidized proteins but PEP4 overexpression is not sufficient to increase chronological lifespan in Saccharomyces cerevisiae. Microbiology 2006, 152, 3595–3605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-B.; Zhang, H.-F.; Chen, Q.-H.; Ruan, H.; Fu, M.-L.; He, G.-Q. Effects of proteinase A on cultivation and viability characteristics of industrial Saccharomyces cerevisiae WZ65. J. Zhejiang Univ. Sci. B 2009, 10, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.H.; Liu, X.J.; Fu, M.L.; Zhang, H.B. Effect of PrA encoding gene-PEP4 deletion in industrial S. cerevisiae WZ65 on key enzymes in relation to the glycolytic pathway. Eur. Food Res. Technol. 2010, 231, 943–950. [Google Scholar]
- Liu, X.-J.; Feng, Y.; Fu, M.-L.; Dong, Y.-C.; Chen, Q.-H.; Jiao, Y.-C. The shock of vacuolar PrA on glycolytic flux, oxidative phosphorylation, and cell morphology by industrial Saccharomyces cerevisiae WZ65. Eur. Food Res. Technol. 2011, 233, 941–949. [Google Scholar] [CrossRef]
- Pereira, C.; Chaves, S.; Alves, S.; Salin, B.; Camougrand, N.; Manon, S.; Sousa, M.J.; Côrte-Real, M.; Côrte-Real, M. Mitochondrial degradation in acetic acid-induced yeast apoptosis: The role of Pep4 and the ADP/ATP carrier. Mol. Microbiol. 2010, 76, 1398–1410. [Google Scholar] [CrossRef] [PubMed]
- Mason, D.A.; Shulga, N.; Undavai, S.; Ferrando-May, E.; Rexach, M.F.; Goldfarb, D.S. Increased nuclear envelope permeability and Pep4p-dependent degradation of nucleoporins during hydrogen peroxide-induced cell death. FEMS Yeast Res. 2005, 5, 1237–1251. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.J.; Azevedo, F.; Pedras, A.; Marques, C.; Coutinho, O.P.; Preto, A.; Geros, H.; Chaves, S.R.; Côrte-Real, M. Vacuole–mitochondrial cross-talk during apoptosis in yeast: A model for understanding lysosome–mitochondria-mediated apoptosis in mammals. Biochem. Soc. Trans. 2011, 39, 1533–1537. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Diezel, W.; Kopperschläger, G.; Hofmann, E. An improved procedure for protein staining in polyacrylamide gels with a new type of Coomassie Brilliant Blue. Anal. Biochem. 1972, 48, 617–620. [Google Scholar] [CrossRef]
- Jouber, R.; Strub, J.-M.; Zugmeyer, S.; Kobi, D.; Carte, N.; Van Dorsselaer, A.; Boucherie, H.; Jaquet-Gutfreund, L.; Joubert, R.; Jaquet-Guffreund, L. Identification by mass spectrometry of two-dimensional gel electrophoresis-separated proteins extracted from lager brewing yeast. Electrophoresis 2001, 22, 2969–2982. [Google Scholar] [CrossRef]
- Hierro, N.; Esteve-Zarzoso, B.; González, Á.; Mas, A.; Guillamón, J.M. Real-time quantitative PCR (QPCR) and reverse transcription-QPCR for detection and enumeration of total yeasts in wine. Appl. Environ. Microb. 2006, 72, 7148–7155. [Google Scholar] [CrossRef] [PubMed]
- Vaudano, E.; Costantini, A.; Cersosimo, M.; Delprete, V.; Garciamoruno, E. Application of real-time RT–PCR to study gene expression in active dry yeast (ADY) during the rehydration phase. Int. J. Food Microbiol. 2009, 129, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Woolford, C.A.; Daniels, L.B.; Park, F.J.; Jones, E.W.; Van Arsdell, J.N.; A Innis, M. The PEP4 gene encodes an aspartyl protease implicated in the posttranslational regulation of Saccharomyces cerevisiae vacuolar hydrolases. Mol. Cell. Boil. 1986, 6, 2500–2510. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Gutierrez, D.; A Bauer, M.; Ring, J.; Knauer, H.; Eisenberg, T.; Büttner, S.; Ruckenstuhl, C.; Reisenbichler, A.; Magnes, C.; Rechberger, G.N.; et al. The propeptide of yeast cathepsin D inhibits programmed necrosis. Cell Death Dis. 2011, 2, e161. [Google Scholar] [CrossRef] [PubMed]
- Lundin, M.; Baltscheffsky, H.; Ronne, H. Yeast PPA2 gene encodes a mitochondrial inorganic pyrophosphatase that is essential for mitochondrial function. J. Boil. Chem. 1991, 266, 12168–12172. [Google Scholar]
- Wang, K.; Yang, Z.; Liu, X.; Mao, K.; Nair, U.; Klionsky, D.J. Phosphatidylinositol 4-Kinases Are Required for Autophagic Membrane Trafficking. J. Boil. Chem. 2012, 287, 37964–37972. [Google Scholar] [CrossRef]
- Sekito, T.; Liu, Z.; Thornton, J.; Butow, R.A. RTG-dependent Mitochondria-to-Nucleus Signaling Is Regulated byMKS1 and Is Linked to Formation of Yeast Prion [URE3]. Mol. Boil. Cell 2002, 13, 795–804. [Google Scholar] [CrossRef]
- Prakash, S.; Prakash, L. Nucleotide excision repair in yeast. Mutat. Res. Mol. Mech. Mutagen. 2000, 451, 13–24. [Google Scholar] [CrossRef]
- Hoyt, M.A.; Macke, J.P.; Roberts, B.T.; Geiser, J.R. Saccharomyces Cerevisiae Pac2 Functions with Cin1, 2 and 4 in a Pathway Leading to Normal Microtubule Stability. Genetics 1997, 146, 849–857. [Google Scholar] [PubMed]
- Roelants, F.M.; Torrance, P.D.; Bezman, N.; Thorner, J. Pkh1 and Pkh2 Differentially Phosphorylate and Activate Ypk1 and Ykr2 and Define Protein Kinase Modules Required for Maintenance of Cell Wall Integrity. Mol. Boil. Cell 2002, 13, 3005–3028. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.P.; Sauer, U. The importance of post-translational modifications in regulating Saccharomyces cerevisiae metabolism. FEMS Yeast Res. 2012, 12, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.-S.; Kim, Y.-S.; Yoon, H.-S. Rice ASR1 protein with reactive oxygen species scavenging and chaperone-like activities enhances acquired tolerance to abiotic stresses in Saccharomyces cerevisiae. Mol. Cells 2012, 33, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Norbeck, J.; Blomberg, A. The level of cAMP-dependent protein kinase A activity strongly affects osmotolerance and osmo-instigated gene expression changes in Saccharomyces cerevisiae. Yeast 2000, 16, 121–137. [Google Scholar] [CrossRef]
- Morano, K.A.; Grant, C.M.; Moye-Rowley, W.S. The response to heat shock and oxidative stress in Saccharomyces cerevisiae. Genetics 2012, 190, 1157–1195. [Google Scholar] [CrossRef] [PubMed]
- Hoehamer, C.F.; Cummings, E.D.; Hilliard, G.M.; Rogers, P.D. Changes in the Proteome of Candida albicans in Response to Azole, Polyene, and Echinocandin Antifungal Agents. Antimicrob. Agents Chemother. 2010, 54, 1655–1664. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spotno a | Accession No. in NCBI | Protein Identity | Mr (kDa)/pI | Coverage (%)b | Fold Change (C/M)c | Protein Score | Function/Annotation |
|---|---|---|---|---|---|---|---|
| Glycolysis/Gluconeogenesis | |||||||
| 133 | gi|6320255 | Tpi1p | 26.89/5.75 | 38 | 16.22 ± 1.81 | 74 | Triose phosphate isomerase. |
| 188 | gi|6322790 | Fba1p | 39.88/5.51 | 67 | 11.53 ± 4.69 | 242 | Fructose 1,6-bisphosphate aldolase. |
| 208/262 | gi|6321968 | Eno2p | 46.94/5.67 | 36/47 | 23.53 ± 9.15/44.79 ± 6.47 | 94/130 | Enolase II, catalyzes conversion of 2-phosphoglycerate to phosphoenolpyruvate during glycolysis and the reverse reaction during gluconeogenesis. |
| 353/548 | gi|6323073 | Pdc1p | 61.69/5.80 | 36/53 | 40.91 ± 8.23/12.26 ± 1.64 | 91/159 | Major of three pyruvate decarboxylase isozymes; decarboxylates pyruvate to acetaldehyde; involved in amino acid catabolism. |
| Intracellular transport | |||||||
| 92 | gi|323349761 | Arl1p | 19.51/5.13 | 48 | 37.88 ± 5.01 | 150 | Soluble GTPase with a role in regulation of membrane traffic; regulates potassium influx; Gprotein of the Ras superfamily, similar to ADP-ribosylation factor. |
| 701 | gi|323337062 | Kha1p | 68.02/5.05 | 33 | 33.39 ± 10.78 | 165 | Putative K+/H+ antiporter with a probable role in intracellular cation homeostasis. |
| 782 | gi|207347902 | Nup170-like protein | 87.97/5.25 | 40 | 32.31 ± 9.57 | 278 | Nucleocytoplasmic transport, structural constituent of nuclear pore. |
| Stress response proteins | |||||||
| 162 | gi|110590736 | Hsp82p | 26.99/5.53 | 49 | 11.66 ± 4.18 | 190 | Hsp90 chaperone; chain A, yeast Hsp82 in complex with the novel Hsp90 inhibitor 8-(6-bromo-benzo[1,3]dioxol-5-ylsulfanyl)-9-(3-isopropylamino-propyl)-adenine. |
| 171 | gi|6320560 | Trr1p | 34.45/5.69 | 31 | 11.92 ± 2.30 | 131 | Cytoplasmic thioredoxin reductase. |
| 187 | gi|172713 | Ssa1Ssa1 | 37.59/5.24 | 63 | 68.05 ± 13.55 | 181 | 70kDa heat shock protein. |
| Transcriptional regulation | |||||||
| 19 | gi|323307055 | Ino4p | 11.83/6.59 | 92 | 14.42 ± 8.18 | 245 | Transcription factor required for derepression of inositol-choline-regulated genes involved in phospholipid synthesis. |
| 78 | gi|323334029 | Lsm6p | 14.66/9.40 | 100 | 10.37 ± 1.77 | 305 | One of the Sm-like proteins; part of heteroheptameric complexes (Lsm2p-7p and either Lsm1p or 8p), involved in RNA degradation. |
| 110 | gi|7546208 | Prp18p | 19.65/9.27 | 80 | 13.79 ± 5.11 | 246 | Splicing factor involved in the positioning of the 3′ splice site during the second catalytic step of splicing, part of snRNP U5, interacts with Slu7p. |
| 306 | gi|207344589 | Ndt80-like protein | 53.66/7.60 | 45 | 11.45 ± 1.23 | 141 | DNA binding, sequence-specific DNA binding transcription factor activity. |
| 473 | gi|256273812 | Dbp2p | 61.29/8.90 | 42 | 13.29 ± 4.54 | 163 | ATP-dependent RNA helicase of the DEAD-box protein family; involved in mRNA decay and rRNA processing. |
| 571 | gi|151945667 | SCY_5111 | 64.90/7.90 | 50 | 50.27 ± 16.62 | 195 | Hypothetical protein, regulation of transcription. |
| 655 | gi|323335109 | Aep3p | 65.27/9.71 | 33 | 23.13 ± 2.72 | 134 | Protein that may facilitate use of unformylated tRNA-Met in mitochondrial translation initiation; stabilizes the bicistronic AAP1-ATP6 mRNA. |
| 789 | gi|2131081 | Scp160p | 93.04/5.46 | 30 | 32.14 ± 13.20 | 162 | Essential RNA-binding G protein effector of mating response pathway, mainly associated with nuclear envelope and ER, interacts in mRNA-dependent manner with translating ribosomes via multiple KH domains, similar to vertebrate vigilins. |
| Amino acid metabolism | |||||||
| 667 | gi|6324253 | Mks1p | 65.72/9.36 | 51 | 13.38 ± 2.67 | 284 | Pleiotropic negative transcriptional regulator involved in Ras-CAMP and lysine biosynthetic pathways and nitrogen regulation; involved in retrograde (RTG) mitochondria-to-nucleus signaling. |
| 751 | gi|323308391 | Gsh1p | 77.80/5.87 | 40 | 13.33 ± 3.50 | 187 | Gamma glutamylcysteine synthetase. |
| 1005 | gi|6320332 | Aro1p | 175.94/5.90 | 20 | 10.41 ± 4.22 | 74 | Pentafunctional arom protein, catalyzes steps 2 through 6 in the biosynthesis of chorismate, which is a precursor to aromatic amino acids |
| Other proteins | |||||||
| 14 | gi|323333563 | Rts3p | 11.62/10.83 | 99 | 32.83 ± 5.19 | 298 | Putative component of the protein phosphatase type 2A complex. |
| 52 | gi|259150352 | EC1118_1P2_5424p | 12.12/9.41 | 68 | 18.76 ± 3.51 | 151 | Similar to YPR195C Dubious open reading frame. |
| 83 | gi|323305205 | Pac2p | 55.98/9.17 | 25 | 10.76 ± 3.74 | 70 | Microtubule effector required for tubulin heterodimer formation, binds alpha-tubulin, required for normal microtubule function. |
| 85 | gi|74583851 | Irc11p | 17.98/8.76 | 67 | 10.48 ± 0.46 | 256 | Putative increased recombination centers protein 11. |
| 126 | gi|151945809 | SCY_4911 | 21.99/5.62 | 34 | 18.11 ± 7.27 | 111 | Hypothetical protein, participates in carbohydrate metabolic process. |
| 175 | gi|151945974 | Ppa2p | 35.79/6.47 | 33 | 10.44 ± 2.10 | 90 | Mitochondrial inorganic pyrophosphatase. |
| 200 | gi|6323857 | Rad14p | 43.47/6.91 | 27 | 14.52 ± 3.73 | 109 | Protein that recognizes and binds damaged DNA during nucleotide excision repair; subunit of Nucleotide Excision Repair Factor 1 (NEF1). |
| 279 | gi|349581962 | Tif3p | 48.52/5.12 | 46 | 11.23 ± 5.06 | 201 | Nucleic acid binding, nucleotide binding. |
| 329 | gi|207343593 | MYO3p-like protein | 16.49/4.12 | 67 | 12.42 ± 4.78 | 252 | Contains one SH3 domain. |
| 346 | gi|256273760 | Hem14p | 60.05/9.33 | 49 | 13.77 ± 2.60 | 202 | Protoporphyrinogen oxidase. |
| 940 | gi|151944281 | Pik1p | 120.39/6.02 | 28 | 11.12 ± 5.13 | 148 | Phosphatidylinositol 4-kinase; may control nonselective autophagy and mitophagy through trafficking of Atg9p |
| 991 | gi|171789 | Ypk2p | 129.51/9.53 | 22 | 11.48 ± 2.06 | 75 | Protein kinase 2, participates in a signaling pathway required for optimal cell wall integrity; homolog of mammalian kinase SGK. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, J.; Yu, L.; Shu, Q.; Chen, Q. Identification of Down-Regulated Proteome in Saccharomyces cerevisiae with the Deletion of Yeast Cathepsin D in Response to Nitrogen Stress. Microorganisms 2019, 7, 214. https://doi.org/10.3390/microorganisms7080214
Hu J, Yu L, Shu Q, Chen Q. Identification of Down-Regulated Proteome in Saccharomyces cerevisiae with the Deletion of Yeast Cathepsin D in Response to Nitrogen Stress. Microorganisms. 2019; 7(8):214. https://doi.org/10.3390/microorganisms7080214
Chicago/Turabian StyleHu, Jingjin, Lingxiao Yu, Qin Shu, and Qihe Chen. 2019. "Identification of Down-Regulated Proteome in Saccharomyces cerevisiae with the Deletion of Yeast Cathepsin D in Response to Nitrogen Stress" Microorganisms 7, no. 8: 214. https://doi.org/10.3390/microorganisms7080214
APA StyleHu, J., Yu, L., Shu, Q., & Chen, Q. (2019). Identification of Down-Regulated Proteome in Saccharomyces cerevisiae with the Deletion of Yeast Cathepsin D in Response to Nitrogen Stress. Microorganisms, 7(8), 214. https://doi.org/10.3390/microorganisms7080214