Geographical Origin Has a Greater Impact on Grape Berry Fungal Community than Grape Variety and Maturation State

 ,
,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Vineyard and Harvesting
2.2. DNA Extraction
2.3. Data Analysis
2.4. Statistical Analysis
3. Results
3.1. Sequences Analysis Results
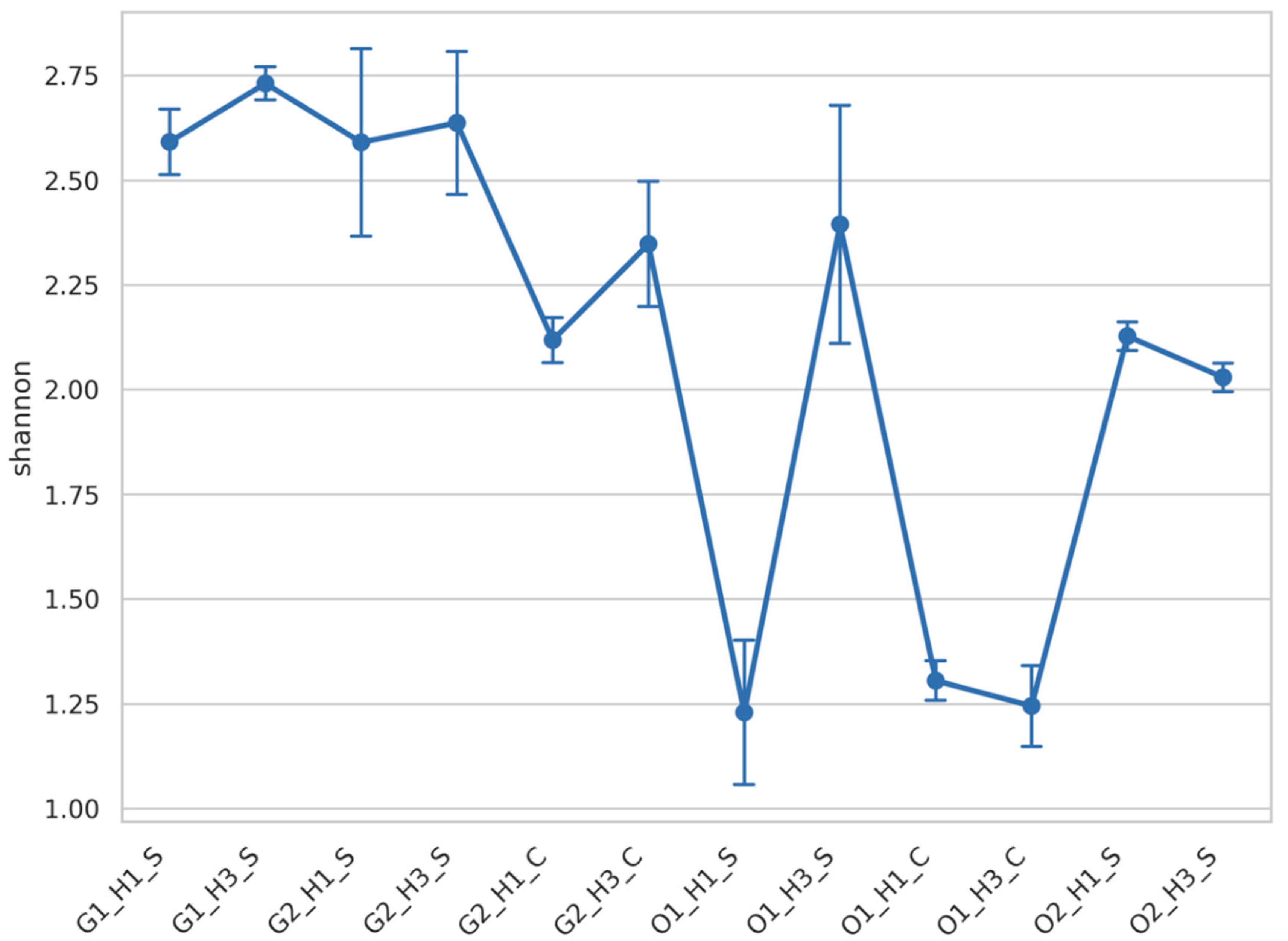
3.2. Fungal Diversity Was Mainly Impacted by the Wine Region
3.3. Fungal Community Clustered Distinctly According to Wine Region, Variety and Harvest
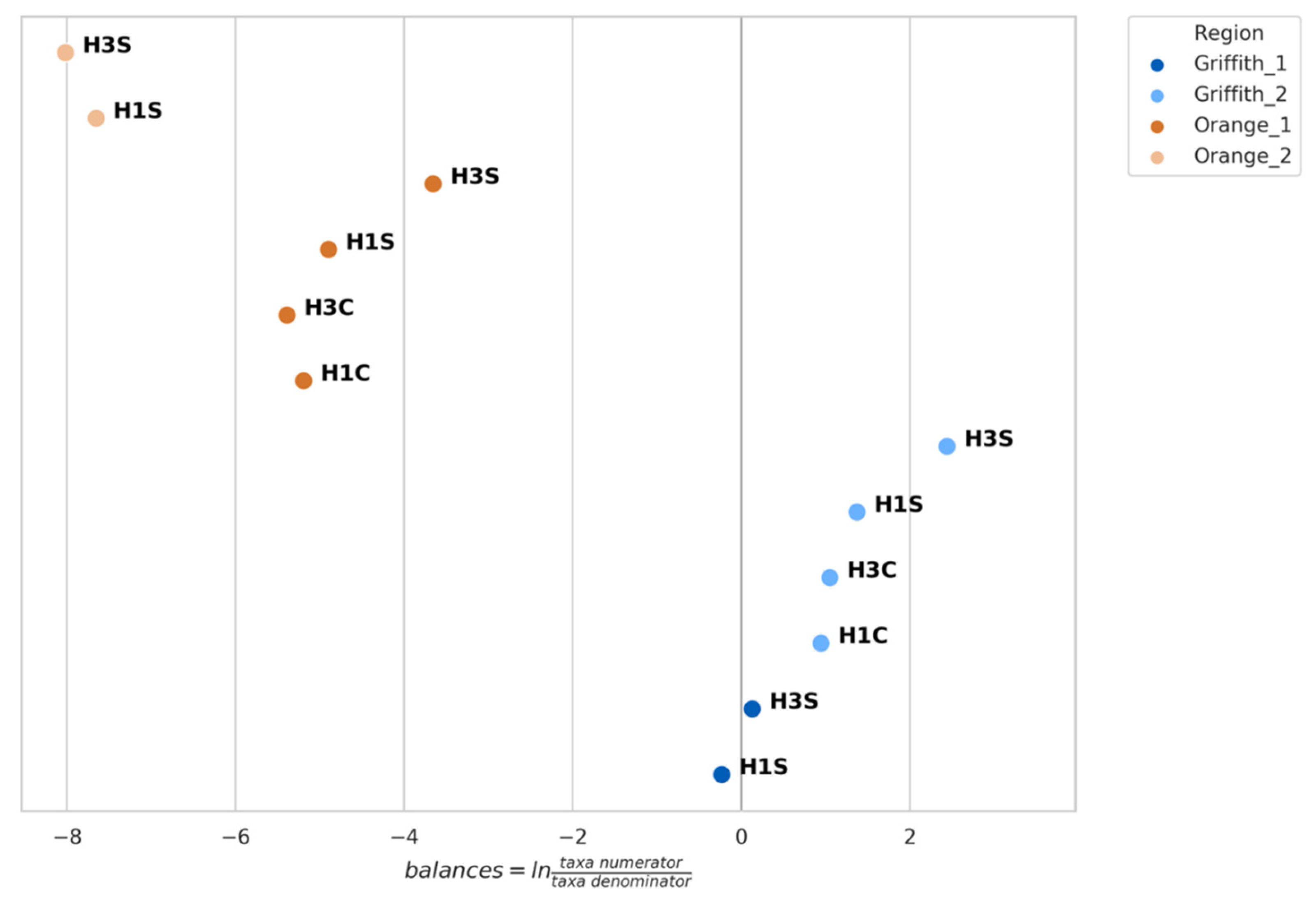
3.4. Genera Balances Affected the Fungal Community Structure
4. Discussion
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vacher, C.; Hampe, A.; Porté, A.J.; Sauer, U.; Compant, S.; Morris, C.E. The Phyllosphere: Microbial Jungle at the Plant–Climate Interface. Annu. Rev. Ecol. Evol. Syst. 2016, 47, 1–24. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Thorngate, J.H.; Richardson, P.M.; Mills, D.A. PNAS Plus: From the Cover: Microbial biogeography of wine grapes is conditioned by cultivar, vintage, and climate. Proc. Natl. Acad. Sci. 2014, 111, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Pinto, C.; Pinho, D.; Sousa, S.; Pinheiro, M.; Egas, C.; Gomes, A.C. Unravelling the diversity of grapevine microbiome. PLoS ONE 2014, 9, e85622. [Google Scholar] [CrossRef]
- Zarraonaindia, I.; Owens, S.M.; Weisenhorn, P.; West, K.; Hampton-Marcell, J.; Lax, S.; Bokulich, N.A.; Mills, D.A.; Martin, G.; Taghavi, S.; et al. The soil microbiome influences grapevine-associated microbiota. MBio 2015, 6, e02527. [Google Scholar] [CrossRef]
- Knight, S.; Klaere, S.; Fedrizzi, B.; Goddard, M.R. Regional microbial signatures positively correlate with differential wine phenotypes: Evidence for a microbial aspect to terroir. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Collins, T.S.; Masarweh, C.; Allen, G.; Heymann, H.; Ebeler, S.E.; Millsa, D.A. Associations among wine grape microbiome, metabolome, and fermentation behavior suggest microbial contribution to regional wine characteristics. MBio 2016, 7, e00631. [Google Scholar] [CrossRef]
- Belda, I.; Zarraonaindia, I.; Perisin, M.; Palacios, A.; Acedo, A. Corrigendum: From vineyard soil to wine fermentation: Microbiome approximations to explain the “terroir” concept. Microbiology 2017. [Google Scholar] [CrossRef]
- Morgan, H.H.; du Toit, M.; Setati, M.E. The grapevine and wine microbiome: Insights from high-throughput amplicon sequencing. Front. Microbiol. 2017. [Google Scholar] [CrossRef]
- Setati, M.E.; Jacobson, D.; Andong, U.C.; Bauer, F. The Vineyard Yeast Microbiome, a Mixed Model Microbial Map. PLoS ONE 2012, 7, e52609. [Google Scholar] [CrossRef]
- Gilbert, J.A.; van der Lelie, D.; Zarraonaindia, I. Microbial terroir for wine grapes. Proc. Natl. Acad. Sci. 2014. [Google Scholar] [CrossRef]
- Taylor, M.W.; Tsai, P.; Anfang, N.; Ross, H.A.; Goddard, M.R. Pyrosequencing reveals regional differences in fruit-associated fungal communities. Environ. Microbiol. 2014, 16, 2848–2858. [Google Scholar] [CrossRef]
- Morrison-Whittle, P.; Lee, S.A.; Goddard, M.R. Fungal communities are differentially affected by conventional and biodynamic agricultural management approaches in vineyard ecosystems. Agric. Ecosyst. Environ. 2017, 246, 306–313. [Google Scholar] [CrossRef]
- Burns, K.N.; Bokulich, N.A.; Cantu, D.; Greenhut, R.F.; Kluepfel, D.A.; O’Geen, A.T.; Strauss, S.L.; Steenwerth, K.L. Vineyard soil bacterial diversity and composition revealed by 16S rRNA genes: Differentiation by vineyard management. Soil Biol. Biochem. 2016, 103, 337–348. [Google Scholar] [CrossRef]
- Portillo Mdel, C.; Franquès, J.; Araque, I.; Reguant, C.; Bordons, A. Bacterial diversity of Grenache and Carignan grape surface from different vineyards at Priorat wine region (Catalonia, Spain). Int. J. Food Microbiol. 2016, 219, 56–63. [Google Scholar] [CrossRef]
- Renouf, V.; Claisse, O.; Lonvaud-Funel, A. Understanding the microbial ecosystem on the grape berry surface through numeration and identification of yeast and bacteria. Aust. J. Grape Wine Res. 2005, 11, 316–327. [Google Scholar] [CrossRef]
- Martins, G.; Vallance, J.; Mercier, A.; Albertin, W.; Stamatopoulos, P.; Rey, P.; Lonvaud, A.; Masneuf-Pomarède, I. Influence of the farming system on the epiphytic yeasts and yeast-like fungi colonizing grape berries during the ripening process. Int. J. Food Microbiol. 2014, 177, 21–28. [Google Scholar] [CrossRef]
- Pérez-Magariño, S.; González-San José, M.L. Polyphenols and colour variability of red wines made from grapes harvested at different ripeness grade. Food Chem. 2006, 96, 197–208. [Google Scholar] [CrossRef]
- Jolly, N.P.; Augustyn, O.P.H.; Pretorius, I.S. The Occurrence of Non-Saccharomyces cerevisiae Yeast Species Over Three Vintages in Four Vineyards and Grape Musts From Four Production Regions of the Western Cape, South Africa. South African J. Enol. Vitic. 2003, 24, 8–10. [Google Scholar] [CrossRef]
- Prakitchaiwattana, C.J.; Fleet, G.H.; Heard, G.M. Application and evaluation of denaturing gradient gel electrophoresis to analyse the yeast ecology of wine grapes. FEMS Yeast Res. 2004, 4, 865–877. [Google Scholar] [CrossRef]
- Barata, A.; Malfeito-Ferreira, M.; Loureiro, V. The microbial ecology of wine grape berries. Int. J. Food Microbiol. 2012, 153, 243–259. [Google Scholar] [CrossRef]
- Martins, G.; Miot-Sertier, C.; Lauga, B.; Claisse, O.; Lonvaud-Funel, A.; Soulas, G.; Masneuf-Pomarède, I. Grape berry bacterial microbiota: Impact of the ripening process and the farming system. Int. J. Food Microbiol. 2012, 158, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Amann, R.I.; Ludwig, W.; Schleifer, K.H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 1995, 59, 143–169. [Google Scholar] [PubMed]
- Rantsiou, K.; Urso, R.; Iacumin, L.; Cantoni, C.; Cattaneo, P.; Comi, G.; Cocolin, L. Culture-dependent and -independent methods to investigate the microbial ecology of Italian fermented sausages. Appl. Environ. Microbiol. 2005, 71, 1977–1986. [Google Scholar] [CrossRef]
- Tsilimigras, M.C.B.; Fodor, A.A. Compositional data analysis of the microbiome: fundamentals, tools, and challenges. Ann. Epidemiol. 2016, 26, 330–335. [Google Scholar] [CrossRef]
- Weiss, S.; Xu, Z.Z.; Peddada, S.; Amir, A.; Bittinger, K.; Gonzalez, A.; Lozupone, C.; Zaneveld, J.R.; Vázquez-Baeza, Y.; Birmingham, A.; et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 2017, 5, 1–18. [Google Scholar] [CrossRef]
- Morton, J.T.; Sanders, J.; Quinn, R.A.; Mcdonald, D.; Gonzalez, A.; Vázquez-baeza, Y.; Navas-molina, J.A.; Song, S.J.; Metcalf, J.L.; Hyde, E.R.; et al. crossm Differentiation. mSystems 2017, 2, 1–11. [Google Scholar]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. PeerJ 2018, 6, e27295v2. [Google Scholar] [CrossRef]
- Tonietto, J.; Carbonneau, A. A multicriteria climatic classification system for grape-growing regions worldwide. Agric. For. Meteorol. 2004, 124, 81–97. [Google Scholar] [CrossRef]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Kõljalg, U.; Larsson, K.H.; Abarenkov, K.; Nilsson, R.H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Høiland, K.; Kjøller, R.; Larsson, E.; et al. UNITE: A database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytol. 2005, 166, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2006, 26, 32–46. [Google Scholar]
- Seabold, S.; Perktold, J. Statsmodels: Econometric and Statistical Modeling with Python. Proc. 9th Python Sci. Conf. 2010, 57. [Google Scholar] [CrossRef]
- Jones, E.; Oliphant, T.; Peterson, P. Scipy: Open Source Scientific Tools for Python. Available online: http://www.scipy.org (accessed on 18 October 2019).
- McKinney, W. Data Structures for Statistical Computing in Python. Proc. 9th Python Sci. Conf. 2010, 1697900, 51–56. [Google Scholar]
- Čadež, N.; Zupan, J.; Raspor, P. The effect of fungicides on yeast communities associated with grape berries. FEMS Yeast Res. 2010, 10, 619–630. [Google Scholar] [CrossRef]
- Šuklje, K.; Carlin, S.; Antalick, G.; Blackman, J.W.; Deloire, A.; Vrhovsek, U.; Schmidtke, L.M. Regional Discrimination of Australian Shiraz Wine Volatome by Two-Dimensional Gas Chromatography Coupled to Time-of-Flight Mass Spectrometry. J. Agric. Food Chem. 2019, 67, 10273–10284. [Google Scholar] [CrossRef]
- Šuklje, K.; Carlin, S.; Stanstrup, J.; Antalick, G.; Blackman, J.W.; Meeks, C.; Deloire, A.; Schmidtke, L.M.; Vrhovsek, U. Unravelling wine volatile evolution during Shiraz grape ripening by untargeted HS-SPME-GC × GC-TOFMS. Food Chem. 2019, 277, 753–765. [Google Scholar] [CrossRef]
- Young, P.R.; Eyeghe-Bickong, H.A.; du Plessis, K.; Alexandersson, E.; Jacobson, D.A.; Coetzee, Z.; Deloire, A.; Vivier, M.A. Grapevine plasticity in response to an altered microclimate: Sauvignon Blanc modulates specific metabolites in response to increased berry exposure. Plant Physiol. 2016, 170, 1235–1254. [Google Scholar] [CrossRef]
- Gao, F.; Chen, J.; Xiao, J.; Cheng, W.; Zheng, X.; Wang, B.; Shi, X. Microbial community composition on grape surface controlled by geographical factors of different wine regions in Xinjiang, China. Food Res. Int. 2019, 122, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; García-Fernández, D.; Mas, A.; Esteve-Zarzoso, B. Fungal diversity in grape must and wine fermentation assessed by massive sequencing, quantitative PCR and DGGE. Front. Microbiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Setati, M.E.; Jacobson, D.; Bauer, F.F. Sequence-based analysis of the Vitis vinifera L. cv cabernet sauvignon grape must mycobiome in three South African vineyards employing distinct agronomic systems. Front. Microbiol. 2015, 6, 1–12. [Google Scholar] [CrossRef]
- Ding, S.; Li, N.; Cao, M.; Huang, Q.; Chen, G.; Xie, S.; Zhang, J.; Cheng, G.; Li, W. Diversity of epiphytic fungi on the surface of Kyoho grape berries during ripening process in summer and winter at Nanning region, Guangxi, China. Fungal Biol. 2019, 123, 283–289. [Google Scholar] [CrossRef]
- Dissanayake, A.; Purahong, W.; Wubet, T.; Hyde, K.; Zhang, W.; Xu, H.; Zhang, G.; Fu, C.; Liu, M.; Xing, Q.; et al. Direct comparison of culture-dependent and culture-independent molecular approaches reveal the diversity of fungal endophytic communities in stems of grapevine (Vitis vinifera). Fungal Divers. 2018, 90, 85–107. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor | DF | R2 | F | Pr (>F) |
|---|---|---|---|---|
| Variety | 1.0 | 0.25 | 5.218 | 0.048 |
| Harvest | 1.0 | 0.29 | 5.522 | 0.043 |
| Factor | DF | R2 | F | Pr (>F) |
|---|---|---|---|---|
| Region | 2.0 | 0.53 | 33.575 | 1.744e-8 |
| Variety | 1.0 | 0.20 | 25.710 | 1.754e-5 |
| Harvest | 1.0 | 0.02 | 2.807 | 0.103 |
| Factor | DF | R2 | F | Pr (>F) |
|---|---|---|---|---|
| Region | 3.0 | 0.532 | 20.694 | 0.001 |
| Variety | 1.0 | 0.079 | 9.267 | 0.001 |
| Harvest | 1.0 | 0.027 | 3.229 | 0.029 |
| Taxonomy | G1_H1_S | G1_H3_S | G2_H1_S | G2_H3_S | G2_H1_C | G2_H3_C | O1_H1_S | O1_H3_S | O1_H1_C | O1_H3_C | O2_H1_S | O2_H3_S |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diplodia | 25 | 52 | 21 | 45 | 14 | 11 | 9 | 119 | 2 | 3 | 1 | 4 |
| 1–46 | 0–128 | 12 –33 | 26–80 | 6–18 | 10–11 | 3–12 | 66–163 | 1–6 | 2–3 | 0–1 | 2–5 | |
| Cladosporium | 2 | 1 | 2 | 2 | 3 | 2 | 54 | 98 | 53 | 21 | 6 | 7 |
| 1–4 | 0–3 | 0–4 | 1–7 | 3–6 | 1–7 | 21–54 | 91–135 | 41–58 | 19–25 | 6–7 | 7–7 | |
| Mycosphaerella | 709 | 630 | 2046 | 1554 | 3876 | 2754 | 2536 | 3406 | 1750 | 1780 | 1784 | 1068 |
| 578–1041 | 467–796 | 1568–3104 | 852–2780 | 3511–4283 | 2105–2764 | 1845–2783 | 3073–3512 | 1695–1840 | 1519–2026 | 1782–2349 | 1011–1500 | |
| Aureobasidium | 4190 | 3492 | 3783 | 2390 | 5384 | 4333 | 8178 | 5257 | 8545 | 8712 | 3393 | 2939 |
| 3373–5254 | 2849–4175 | 1996–4907 | 1502–5085 | 5237–5559 | 4122–4625 | 7963–9262 | 3293–5927 | 8513–8696 | 8331–9090 | 3289–4590 | 2868–3802 | |
| Alternaria | 354 | 284 | 508 | 304 | 517 | 399 | 53 | 536 | 64 | 61 | 47 | 62 |
| 286–440 | 206–366 | 268–804 | 227–580 | 440–531 | 287–428 | 53–55 | 415–833 | 53–64 | 47–79 | 44–80 | 42–72 | |
| Stemphylium | 196 | 162 | 376 | 282 | 346 | 300 | 86 | 188 | 49 | 58 | 27 | 18 |
| 107–263 | 126–232 | 226–479 | 207–409 | 303–404 | 211–337 | 86–139 | 176–214 | 42–65 | 35–61 | 23–48 | 13–32 | |
| Phoma | 198 | 147 | 138 | 67 | 279 | 304 | 150 | 118 | 88 | 92 | 341 | 14 |
| 183–327 | 118–180 | 60–319 | 41–150 | 266–331 | 226–316 | 142–186 | 89–144 | 78–92 | 76–116 | 70–349 | 14–23 | |
| Aspergillus | 1567 | 2263 | 1634 | 3734 | 44 | 32 | 6 | 14 | 1 | 2 | 3 | 0 |
| 465–2626 | 1826–3485 | 234–5548 | 811–6248 | 38–67 | 6–36 | 5–93 | 6–35 | 0–6 | 1–12 | 2–4 | 0–1 | |
| Penicillium | 520 | 1320 | 685 | 460 | 34 | 17 | 35 | 164 | 25 | 2 | 2 | 1 |
| 360–943 | 324–2727 | 304–1414 | 268–933 | 21–54 | 5–19 | 16–129 | 106–307 | 10–406 | 1–44 | 0–4 | 0–4 | |
| Erysiphe | 4 | 9 | 17 | 30 | 0 | 9 | 6 | 9 | 1 | 8 | 44 | 260 |
| 2–8 | 3–15 | 7–22 | 14–83 | 0–1 | 5–9 | 5–12 | 2–13 | 0–12 | 2–9 | 41–89 | 251–310 | |
| Botrytis | 540 | 1121 | 50 | 1235 | 32 | 21 | 18 | 202 | 8 | 6 | 4302 | 5314 |
| 450–3317 | 318–1631 | 10–659 | 130–1723 | 22–54 | 9–25 | 12–21 | 132–508 | 3–9 | 2–7 | 2315–4794 | 4383–5398 | |
| Metschnikowia | 6 | 51 | 2 | 0 | 4 | 1 | 4 | 103 | 5 | 4 | 0 | 0 |
| 0–58 | 0–148 | 0–5 | 0–2 | 2–5 | 1–4 | 1–5 | 16–151 | 2–6 | 3–17 | 0–2 | 0–1 | |
| Acremonium | 12 | 16 | 122 | 232 | 44 | 32 | 0 | 2 | 0 | 0 | 1 | 1 |
| 1–35 | 1–32 | 47–662 | 63–316 | 43–67 | 16–33 | 0–0 | 1–4 | 0–2 | 0–0 | 0–4 | 1–1 | |
| Colletotrichum | 112 | 250 | 1 | 14 | 331 | 2567 | 0 | 0 | 1 | 1 | 0 | 0 |
| 7–575 | 42–509 | 0–2 | 5–17 | 187–428 | 2554–3623 | 0–1 | 0–0 | 0–2 | 0–1 | 0–0 | 0–0 | |
| Rhodotorula | 1638 | 1153 | 8 | 2 | 10 | 8 | 138 | 131 | 763 | 622 | 47 | 60 |
| 30–4445 | 81–2663 | 0–20 | 1–10 | 8–12 | 6–12 | 60–252 | 104–179 | 512–780 | 562–716 | 31–62 | 45–77 | |
| Sporobolomyces | 139 | 90 | 236 | 148 | 184 | 249 | 11 | 288 | 24 | 22 | 12 | 7 |
| 84–264 | 35–175 | 89–361 | 108–311 | 182–228 | 127–287 | 3–14 | 159–434 | 18–32 | 12–41 | 6–31 | 5–12 | |
| Bullera | 19 | 13 | 84 | 86 | 118 | 125 | 5 | 29 | 9 | 10 | 8 | 4 |
| 12–26 | 10–17 | 32–132 | 51–115 | 115–128 | 74–160 | 0–15 | 17–35 | 5–15 | 4–25 | 7–9 | 5 | |
| Cryptococcus | 159 | 104 | 902 | 538 | 327 | 410 | 78 | 1303 | 97 | 136 | 1559 | 1770 |
| 81–256 | 58–170 | 27–1563 | 326–1425 | 277–353 | 234–468 | 34–88 | 691–1739 | 87–101 | 110–199 | 1195–1926 | 1441–1872 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kioroglou, D.; Kraeva-Deloire, E.; Schmidtke, L.M.; Mas, A.; Portillo, M.C. Geographical Origin Has a Greater Impact on Grape Berry Fungal Community than Grape Variety and Maturation State. Microorganisms 2019, 7, 669. https://doi.org/10.3390/microorganisms7120669
Kioroglou D, Kraeva-Deloire E, Schmidtke LM, Mas A, Portillo MC. Geographical Origin Has a Greater Impact on Grape Berry Fungal Community than Grape Variety and Maturation State. Microorganisms. 2019; 7(12):669. https://doi.org/10.3390/microorganisms7120669
Chicago/Turabian StyleKioroglou, Dimitrios, Elena Kraeva-Deloire, Leigh M. Schmidtke, Albert Mas, and Maria C. Portillo. 2019. "Geographical Origin Has a Greater Impact on Grape Berry Fungal Community than Grape Variety and Maturation State" Microorganisms 7, no. 12: 669. https://doi.org/10.3390/microorganisms7120669
APA StyleKioroglou, D., Kraeva-Deloire, E., Schmidtke, L. M., Mas, A., & Portillo, M. C. (2019). Geographical Origin Has a Greater Impact on Grape Berry Fungal Community than Grape Variety and Maturation State. Microorganisms, 7(12), 669. https://doi.org/10.3390/microorganisms7120669